bachelorarbeit nadine friedewald - monami · der abteilung für molekulargenetische forensik der...
TRANSCRIPT

BACHELORARBEIT
Frau Nadine Friedewald
Y-chromosomale STR Analysen an einem historischen Gräberfeld
aus Zentraleuropa
Mittweida, 2014


Fakultät MNI
BACHELORARBEIT
Y-chromosomale STR Analysen an einem historischen Gräberfeld
aus Zentraleuropa
Autor:Frau
Nadine Friedewald
Studiengang:Biotechnologie/ Bioinformatik
Seminargruppe:BI11w1-B
Erstprüfer:Prof. Dr. rer. nat. D. Labudde
Zweitprüfer:OA Dr. med. K. Thiele
Einreichung:Mittweida, 25.08.2014
Verteidigung/Bewertung:Mittweida, 2014

Bibliographische Beschreibung:
Friedewald, Nadine: Y-chromosomal STR Analysen an einem historischen Gräberfeld
aus Zentraleuropa. - 2014. - Seitenzahl Verzeichnisse: 6, Inhalt: 68, Anhänge: 6,
Mittweida, Hochschule Mittweida, Fakultät MNI, Bachelorarbeit, 2014
Kurzbeschreibung: Ziel dieser Arbeit ist es, unter Verwendung forensischer DNA-Analysemethoden auf
populationsgenetische Fragestellungen einzugehen. Es soll eine Untersuchung
molekulargenetischer Marker (Y-STR) des Y-Chromosoms anhand historischen
Knochenmaterials aus der römischen Kaiser- und Völkerwanderungszeit erfolgen sowie
eine Einteilung in Haplogruppen vorgenommen werden. Anhand der erhobenen Daten
soll ein Vergleich zu heutigen Populationen gezogen werden.
Englischer Titel: Y-chromosomal STR analysis of historical skeletal remains from Central Europe. Abstract: The present work should contribute to current population genetic issues using forensic
aDNA analysis. In this study Y-chromosomal STRs should be analysed based on
historical skeletal material from the Roman Empire and the Migration Period. It should
ensure a classification in haplogroups. Based on the collected data, a comparison to
present haplogroup frequencies shall be drawn.

Danksagung
Danksagung
Danken möchte ich an dieser Stelle Herrn OA Dr. med. K. Thiele und Herrn Prof. D.
Labudde, die mir diese Zusammenarbeit ermöglichten. Ebenfalls möchte ich dem Team
der Abteilung für molekulargenetische Forensik der Rechtsmedizin Leipzig und der
Prosektur Chemnitz danken. Großer Dank gilt weiterhin meiner Betreuerin M. Sc. Frau
Anne-Marie Pflugbeil und B. Sc. Frau Maria Harthun für die methodische Einarbeitung
und die tatkräftige Unterstützung in allen Bereichen. Besonderer Dank gilt weiterhin M.
Sc. Herrn Florian Heinke, der durch die Generierung eines auf die Problemstellung
abgestimmten Programms eine präzise Auswertung der Daten ermöglichte.
Meiner Familie und Freunden möchte ich für die Unterstützung und das mir entgegen
gebrachte Verständnis danken.

Inhaltsverzeichnis
I
Inhaltsverzeichnis
Inhaltsverzeichnis ............................................................................................................ I
Abbildungsverzeichnis .................................................................................................. III
Tabellenverzeichnis ........................................................................................................ 1
Formelverzeichnis ........................................................................................................... 2
Abkürzungsverzeichnis .................................................................................................. 3
1 Einleitung ...................................................................................................................... 4
1.1 Das Gräberfeld Görzig - Eine Fallbeschreibung ............................................... 4
1.2 Forensische Populationsgenetik ........................................................................ 6
1.3 Populationsgenetischer Aspekt des Y-Chromosoms ........................................ 8
1.4 Relevante molekulargenetische Marker .......................................................... 12
1.4.1 Gonosomale STRs ...................................................................................... 13
1.4.2 Amplifikation gonosomaler STRs .............................................................. 15
1.4.3 Fragmentlängenanalyse mittels Kapillarelektrophorese ............................. 16
1.5 Formulierung von Haplotypen .................................................................... 18
1.7 aDNA-Isolation und Amplifikation ................................................................ 21
1.6 DNA-Erhalt in historischem Zahnmaterial ..................................................... 24
1.8 Populationsgenetische Datenbanken ............................................................... 26
1.8.1 Y-Search .................................................................................................. 26
1.8.2 YHRD ..................................................................................................... 26
1.8.3 AMOVA-Analyse ................................................................................... 28
2 Zielstellung ................................................................................................................. 30
3 Material ....................................................................................................................... 31
3.1 Geräte und Software ....................................................................................... 31
3.2 Chemikalien und Verbrauchslösungen ........................................................... 33
4 Methoden .................................................................................................................... 35
4.1 Aufarbeitung des Zahnmaterials ..................................................................... 35
4.2 Dekalzifizierung, Zelllyse und Proteinase K-Verdau ..................................... 36
4.3 DNA-Aufreinigung mittels Phenol-Chloroform/Isoamylalkohol ................... 37
4.4 DNA-Quantifizierung ..................................................................................... 39
4.5 Autosomale STR-Analyse mittels PowerPlex® S5 Kit .................................. 41
4.6 Haplotypisierung mittels PowerPlex® Y23 Kit .............................................. 42

Inhaltsverzeichnis
II
4.7 Genealogische Charakterisierung des Datensatzes ......................................... 45
4.7.1 Haplogruppenverteilung ............................................................................. 45
4.7.2 Allelverteilung ............................................................................................ 45
4.7.3 Haplotyp-Diversität, Gen-Diversität und Discrimination-Capacity ........... 46
5 Ergebnisse ................................................................................................................... 47
5.1 Mechanische Aufarbeitung des Zahnmaterials und DNA-Isolation ............... 47
5.2 DNA-Quantifizierung ..................................................................................... 47
5.3 Geschlechtsbestimmung anhand autosomaler STR-Analysen mittels
PowerPlex® S5 Kit ..................................................................................................... 47
5.4 Haplotypisierung mittels PowerPlex® Y23 Kit ............................................. 50
5.5 Haplogruppenverteilung innerhalb der Population ......................................... 52
5.6 Auswertung der genealogischen Daten ........................................................... 55
5.6.1 Allelverteilung ............................................................................................ 55
5.6.2 Haplotyp-Diversität, Gen-Diversität und Discrimination-Capacity ........... 57
6 Diskussion ................................................................................................................... 58
6.1 STR-Analysen zur Geschlechtsbestimmung und Haplotypisierung .............. 58
6.3 Vergleich der molekulargenetischen und morphologischen
Geschlechtsbestimmung ............................................................................................. 60
6.2 Haplogruppenverteilung innerhalb der Population ......................................... 61
6.4 Aussagekraft der biostatistischen Ergebnisse ................................................ 63
7 Zusammenfassung und Ausblick .............................................................................. 64
Literaturverzeichnis ..................................................................................................... 65
Anhang ........................................................................................................................... 69
Selbstständigkeitserklärung ......................................................................................... 75

Abbildungsverzeichnis
III
Abbildungsverzeichnis
Abbildung 1: Fundort Görzig [URL-7, URL-8]. .............................................................. 4
Abbildung 2: Karyogramm des menschlichen Chromosomensatzes [URL-1]. ............... 8
Abbildung 3: Das humane Y-Chromosom [URL-2]. ....................................................... 9
Abbildung 4: Mögliche Motivwiederholung eines STR-Systems. ................................. 14
Abbildung 5: Schematische Darstellung einer PCR [mod. B08]. ................................... 15
Abbildung 6: Schematische Darstellung einer Kapillarelektrophorese [G06]. .............. 17
Abbildung 7: ChrY –Haplogruppen in Europa [URL-3] . ........................................... 18
Abbildung 8: Schematische Darstellung der Haplogruppen-Phylogenie. ...................... 19
Abbildung 9: Aus einem Kiefer herausgelöste Backenzähne (Molare). ........................ 25
Abbildung 10: Schematische Darstellung der Extensionsphase einer TaqMan® PCR
[S06]. ....................................................................................................... 40
Abbildung 11: Elektropherogramme (PowerPlex® S5). ................................................ 48
Abbildung 12: Geschlechtsverteilung der betrachteten Individuen. ............................... 49
Abbildung 13: Morphologische Geschlechtsbestimmung. ............................................. 49
Abbildung 14: Vergleich der molekulargenetischen und morphologischen
Geschlechtsbestimmung. ........................................................................ 50
Abbildung 15: Elektropherogrammausschnitt eines mit PowerPlex® Y23 amplifizierten
DNA-Isolats. ........................................................................................... 51
Abbildung 16: Haplogruppenbestimmung der Individuen mittels Y-Search. ................ 52
Abbildung 17: Haplogruppenbestimmung der Individuen mittels Y-Search. ................ 53
Abbildung 18: Haplogruppenverteilung [URL-6]. ......................................................... 54
Abbildung 19: Säulendiagramm der Allelverteilung für den Lokus DYS576. .............. 55
Abbildung 20: Säulendiagramm der Allelfrequenz des Lokus DYS570. ....................... 55
Abbildung 21: Gen-Diversitäten der Loci. ..................................................................... 57
Abbildung 22: Schematische Darstellung der Haplogruppen-Migration [W08]. ........... 62

Tabellenverzeichnis
1
Tabellenverzeichnis
Tabelle 1: Übersicht über mögliche Schäden an aDNA [H06]. ..................................... 23
Tabelle 2: Reaktionsansatz zur DNA-Quantifizierung. .................................................. 39
Tabelle 3: Reaktionsansatz des PowerPlex® S5 Kits für die Amplifikation. ................. 41
Tabelle 4: Amplifikationsprotokoll PowerPlex® S5. ..................................................... 42
Tabelle 5: Reaktionsansatz der Post-Amplifikation des PowerPlex® S5 Kits. .............. 42
Tabelle 6: Reaktionsansatz des PowerPlex® Y23 Kits für die Amplifikation. .............. 43
Tabelle 7: Amplifikationsprotokoll PowerPlex® Y23. ................................................... 43
Tabelle 8: Reaktionsansatz der Post-Amplifikation des Power-Plex® Y23 Kit. ........... 44
Tabelle 9: Übersicht der ermittelten Allelfrequenzen aller Loci. ................................... 56

Formelverzeichnis
2
Formelverzeichnis
Formel 1: Berechnung der Molekularen Varianz ��. .................................................... 28
Formel 2: Berechnung der genetischen Distanz ΦST. ...................................................... 28
Formel 3: Berechnung der Varianz-Korrelation. ............................................................ 28
Formel 4: Berechnung der molekularen Distanz von Haplotypen. ................................ 29
Formel 5: Berechnung der molekularen Distanz zwischen Populationen. ..................... 29
Formel 6: Berechnung der Allelfrequenz. ...................................................................... 45
Formel 7: Berechnung der Haplotyp-Diversität. ............................................................ 46
Formel 8: Berechnung der Gen-Diversität. ..................................................................... 46
Formel 9: Berechnung der Discrimination-Capacity. .................................................... 46

Abkürzungsverzeichnis
3
Abkürzungsverzeichnis
aDNA ancient Desoxyribonukleinsäure
bp Basenpaare
DNA Desoxyribonukleinsäure
DTT Dithiothreitol
EDTA Ethylendiamintetraessigsäure
ChrX X-Chromosom
ChrY Y-Chromosom
gDNA genomische DNA
INDEL INsertion und DELetion
MRCA most recent common ancestor
MSY male-specific region of the Y
mtDNA mitochondriale DNA
NaCl Natriumchlorid
PAR Pseudoautosomale Region
PCI Phenol-Chloroform/Isoamylalkohol
PCR Polymerasekettenreaktion
RFU Relative Fluorescence Unit
SNP short nucleotide polymorphism
SRY Sex determining region of the Y
STR short tandem repeats
UV ultra violett
VNTR Variable Number of Tandem Repeats

1 Einleitung
4
1 Einleitung
1.1 Das Gräberfeld Görzig - Eine Fallbeschreibung
Die Gemeinde Görzig bei Köthen, liegt östlich des Harzes im Bundesland Sachsen-
Anhalt. Bei den dort geborgenen Skelettfunden handelt es sich um einen birituellen,
vorwiegend aus Körpergräbern bestehenden Bestattungsort (Abbildung 1). Nach
anthropologischer Begutachtung ist bei diesem Fund von einer, in der Nähe von Görzig
ansiedelnden homogenen Bevölkerung auszugehen. Bestätigt wird diese Annahme
durch die Grabbeigaben und Bestattungssitten, die in den Körpergräbern geborgen
wurden sowie der vertretenen Ansicht, dass es sich nicht um Verstorbene
unterschiedlicher Bevölkerungsgruppen handelt [B00, K11].
Abbildung 1: Fundort Görzig [URL-7, URL-8]. In dieser Abbildung wird der Fundort Görzig (v.r. n.l.) durch eine Markierung auf der Deutschlandkarte und mit Hilfe einer Satellitenaufnahme veranschaulicht.
Das Gräberfeld Görzig umfasst 75 Körperbestattungen. Nach ersten anthropologischen
Auswertungen sind 69 Gräber der römischen Kaiserzeit und der beginnenden
Völkerwanderungszeit zuzuordnen. Das Skelettmaterial aus drei Gräbern wurde jedoch
der bronzezeitlichen Epoche zugeordnet. Drei weitere Gräber konnten auf Grund
fehlender Beigaben und fehlenden Zusatzinformationen nicht datiert werden.
Eine Bergung und archäologische Untersuchung des Skelettmaterials begann bereits im
Jahr 1913 durch W. Götze der eine erste Sterbealtersschätzung und
Geschlechtsbestimmung der Individuen vornahm. Er betrachtete jedoch lediglich die
Schädel der Bestatteten und ließ das postcraniale Skelett außen vor. In den Jahren
danach wurden die Grabungsarbeiten vernachlässigt und es kam lediglich zu
Gelegenheitsfunden durch Feldarbeiter. 1959 wurden die Arbeiten unter der Aufsicht
von B. Schmidt wieder aufgenommen und es begann eine planmäßige Ausgrabung, bei
der jedoch eine unzureichende archäologische und anthropologische Begutachtung des
Görzig

1 Einleitung
5
Skelettmaterials erfolgte. Es folgten anschließend lediglich Erwähnungen des
Gräberfeldes in J. Bemmann´s Habilitationsschrift [B00]. Weiterhin finden sich Auszüge
von Bestattungsrieten der Kaiser- und Völkerwanderungszeit und ihrer Beigaben in dem
Katalog von B. Schmidt und J. Bemmann [K11]. Da in vorangegangenen Untersuchungen
die detaillierte Darstellung und Analyse des Gräberfeldes hinsichtlich zeitlicher und
kultureller Abläufe außen vor blieb, versuchte J. Kleinecke in ihrer Magisterarbeit die
archäologischen Aspekte korrekt zu erfassen und auszuwerten. Es folgte eine
nochmalige Betrachtung des Fundguts in Einzeluntersuchungen sowie eine geeignete
Einordnung des Materials mit Hilfe vorgegebener Typologien und Formenkatalogen.
Die detaillierte Vorstellung der Gräber und des Fundmaterials beschränkte sich dabei
nicht nur auf die Grabstätte selbst, sondern weitete sich auf die Umgebung, unter
Einbeziehung von Funden aus diesem Areal, aus. An dem vorhandenen Sekelettmaterial
erfolgte eine Bestandaufnahme sowie eine Geschlechts- und Altersabschätzung der
einzelnen Individuen [K11]. In vorangegangenen Forschungsarbeiten wurden
ausführliche morphognostische und morphometrische Untersuchungen zur
Phänotypisierung der Einzelindividuen durchgeführt und abgeschlossen sowie erste
DNA-Analysen zur Genotypisierung realisiert. Im Rahmen dieser Forschungsarbeit gilt
es weiterführend, die molekulargenetischen Analysen zur Genotypisierung
abzuschließen sowie eine Haplotypisierung der maskulinen Individuen durchzuführen.
Die Haplotypisierung erfolgt mittels 23 Y-chromosomalen STR-Markern, die es zu
detektieren gilt.

1 Einleitung
6
1.2 Forensische Populationsgenetik
Prägend für die Bildung stark räumlich voneinander getrennter Population war das
Abdriften der Subkontinente des großen Urkontinents in verschiedene Klimazonen und
Bereiche der Erde. Die Individuen wurden gezwungen, sich an die neuen
Gegebenheiten anzupassen (Adaption). Grundlage des Evolutionsgeschehens bilden
Veränderungen der genetischen Information. Hierbei spielt die Dynamik der Gene eine
entscheidende Rolle bzw. die genetische Veränderung in Abhängigkeit der Zeit. Da
Gene die einzige Information sind, die von Generation zu Generation weiter vererbt
werden, kann die Evolution im Grunde als Modifikation der genetischen Information
bzw. der Genfrequenz betrachten werden. Eine Änderung der genetischen Information
geschieht durch vorhandene Variationen sowie durch Evolutionsfaktoren wie Selektion,
Mutation und Gendrifts. Die Eigenschaft der Variation ist dabei essentiell für
Lebewesen in einer Population. Diese Variationen sind auf Unterschiede bzw.
Veränderungen in den Genen, der Umwelt und auf ökologische Faktoren sowie deren
Interaktion zurückzuführen. Selektion zwischen den ausgebildeten Variationen bildet
hierbei den Grundstein der Evolution und sichert den Fortbestand sowie die Vielfalt
einer Art [PSH08]. Von großem Interesse ist dabei, im Hinblick auf genetische
Unterschiede zwischen Individuen, die forensische Anwendung der
populationsgenetischen Betrachtung. Mit Hilfe forensischer Analysemethoden ist es
realisierbar, Verteilungsstatistiken spezifisch auftretender Genmuster in diversen
Populationen abzubilden und Spurenmaterial auch über räumliche Grenzen hinweg zu
charakterisieren. Die anthropologisch, forensische Populationsgenetik findet ihre
Anwendung in der Beschreibung historischer Populationen und deren Einordnung in
ihren geschichtlichen Kontext. Anhand der Erhebung von Populationsdaten historischen
DNA-Materials ist eine genaue Charakterisierung eines vorliegenden Datensatzes
möglich. Mit Hilfe der gewonnenen Ergebnisse können, auf Grundlage der Berechnung,
Vergleiche mit bestehenden Datensätzen durchgeführt und Erkenntnisse im Hinblick
auf eine mögliche Herkunft und Migration gewonnen werden. Entsprechende
populationsgenetische Analysen beruhen auf der biomolekularen DNA-Analyse.
Spezifische Eigenschaften polymorpher DNA-Merkmale machen es möglich,
populationsspezifische Rückschlüsse auf Verwandtschaftsgrade oder
Wanderungsbewegungen abzuleiten [L13]. Aufgrund des evolutionären Wertegangs
von Individuen sowie der räumlichen Ausbreitung haben sich die unterschiedlichsten
Polymorphismen (z.B. STRs, SNPs, INDELs) in der DNA ausgeprägt.
Diese populationsgenetische Einordnung anhand molekulargenetischer Analysen steht
zum jetzigen Zeitpunkt noch an ihren Anfängen. Momentan bestehen in diversen
Datenbanken lediglich neuzeitige Populationsdaten die keinen Hinweis zu historischen
Populationen auf molekulargenetischer Ebene liefern. Das Interesse an diesen Daten
wächst jedoch zunehmend, da häufig historisches Knochenmaterial geborgen wird und
es dieses einzuordnen gilt. Darin besteht der Ansatz der anthropologisch, forensischen
Populationsgenetik diese Datenbestände aufzufüllen. Diese Einordnung beinhaltet die

1 Einleitung
7
Typisierung des DNA-Materials sowie den Vergleich der erhaltenen Populationsdaten
mit bestehenden Datensätzen.
Wird die forensische Populationsgenetik näher betrachtet, so beschäftigt sich diese mit
Vererbungsvorgängen und der Weitergabe genetischer Informationen, der Analyse der
natürlich vorkommenden genetischen Unterschiede innerhalb von Populationen sowie
der Abbildung dieser in Verteilungen zur genaueren Charakterisierung. Untersucht
werden dafür Gen- und Allelfrequenzen auf mögliche Rekombination, Mutation,
Selektion oder zufällige Gendrifts. Als Allelfrequenz oder Allelhäufigkeit wird die
relative Häufigkeit auftretender Allelkopien in einer Population bezeichnet. Die
Genfrequenz beschreibt die Häufigkeit des Auftretens eines bestimmten Gens in
identischer oder nahezu identischer Form in einer Population [L09]. Besteht eine
geringe Genfrequenz, also eine geringe Anhäufung an homologen oder ähnlichen
Genen, so kann von einer hohen genetischen Vielfalt ausgegangen werden. Ändern sich
im Laufe von Zeit und Raum die Allelfrequenzen einer Population, so kann von einer
evolutionären Entwicklung ausgegangen werden. Mögliche Ursachen können dabei
natürliche Selektion, sowie auftretende Gendrifts sein, welche eine zufällige
Veränderung der Genfrequenzen innerhalb eines Genpools in einer Population bewirken
[PSH08]. Selektion hat dabei keinen zufälligen Einfluss, sondern ist direkt mit dem
Überlebens- und Reproduktionserfolg eines Individuums verbunden. Es werden dabei
Veränderungen der Vielfalt auf molekularer Ebene untersucht und die Dynamik der
Allelfrequenzen einzelner Generationen differenziert. Im Ganzen betrachtet bilden diese
Unterschiede die genetische Variation, deren Gründe des Vorkommens und der
Veränderung im Laufe von Generationen anhand der Populationsgenetik dargestellt
werden sollen [L09]. Die beschriebene genotypische Variation ist somit essentiell.
Weiterhin können Untersuchungen mit bestimmten Divergenzparametern (Parameter
der Artenbildung) innerhalb von Populationen erfolgen sowie Analysen zwischen
Populationen der gleichen Art und zwischen verschiedenen Arten beschrieben werden.
Forensische Anwendungen der populationsgenetischen Betrachtung beziehen sich auf
eben diese Unterschiede der genetischen Variation. Mittels umfangreicher Analysen
dieser, werden Haplotyp-Frequenzen für entsprechende Populationen abgebildet. So
wird es möglich zum einen besonders „seltene“ und zum anderen häufige Allele in den
Populationen aufzuspüren. Das Zusammenspiel der populationsgenetischen Forschung
auf dem Gebiet der Forensik hat sich in der Vergangenheit stark etabliert und findet
seine Hauptanwendung in der Analyse individueller DNA-Proben von sowohl
historischem als auch rezentem Untersuchungsmaterial. Immer wieder tauchen
gebietsweise Knochengräber auf, deren populationsgenetischer Hintergrund durch
Migrationsereignisse ungeklärt ist. Mit Hilfe molekulargenetischer Marker können diese
signifikanten Stellen detektiert werden und Rückschlüsse auf
Abstammungshintergründe liefern.

1 Einleitung
8
1.3 Populationsgenetischer Aspekt des Y-Chromosoms
Chromosomen befinden sich im Zellkern von Eukaryoten und sind Träger der
Erbinformation. Sie liegen in fadenförmigen Gebilden vor, in denen die DNA um
sogenannte Histone zur Stabilisierung gewickelt ist. Kommt es zur Zellteilung durch
Meiose, verdichten sich die Chromosomen zu dem allgemein bekannten X-förmigen
Gebilde [H12]. Die Gesamtheit des Genoms kann in Geschlechtschromosomen und
geschlechtsunspezifische Chromosomen (Autosomen) unterteilt werden. Der
menschliche, diploide Chromosomensatz beläuft sich auf 44 autosomale Chromosomen,
die aus 22 homologen Paaren bestehen, und den Geschlechtschromosomen XX bei
Frauen und XY bei Männern. Das Y-Chromosom (Abbildung 3) wird somit paternal
(väterlicherseits) vererbt.
Abbildung 2: Karyogramm des menschlichen Chromosomensatzes [URL-1]. Abgebildet wird hier der männliche diploide Chromosomensatz bestehend aus 22 homologen
Chromosomen und den beiden Geschlechtschromosomen XY.
Kommt es zur Zellteilung, oder zur Ausbildung von Geschlechtszellen, halbiert sich der
diploide Chromosomensatz. Es liegt ein haploider Chromosomensatz vor. Zu Beginn
der Meiose werden die homologen Chromosomen parallel zueinander angeordnet und
es kann zu einem sogenannten crossing-over kommen. Dabei kommt es zu einer
Überlappung bzw. zu einem Umschlingen der Chromosomen und DNA-Fragmente
können zwischen maternalem und paternalem Erbgut ausgetauscht werden [L09]. Je
nach dem, welches Gonosom das Spermium des Mannes enthält, verschmelzen ein X
oder ein Y-Chromosom (ChrY) mit dem X-Chromosom (ChrX) der Mutter. Entsteht
eine XX Kombination, so wird das Kind weiblich. Die Kombination von XY bringt,
sofern keine Mutation vorliegt, einen männlichen Nachkommen hervor. Entscheidend
für die Ausprägung des maskulinen Phänotyps ist nicht das Y-Chromosom an sich,
sondern die geschlechtsspezifische sex determining region of Y (SRY) auf dem
Chromosom. Ist diese aufgrund von Deletion oder Inaktivierung, ausgelöst durch

1 Einleitung
9
Mutation, nicht auf dem Y-Chromosom enthalten, wird trotz der Kombination aus XY
der weibliche Phänotyp ausgeprägt (XY-Frau). Eine weitere Möglichkeit ist die durch
Translokation verursachte Übertragung der SRY auf das X-Chromosom. Trotz eines
XX-Chromosomensatzes wird der männliche Phänotyp ausgebildet (XX-Mann).
Abbildung 3: Das humane Y-Chromosom [URL-2]. Zu sehen ist hier der schematische Aufbau des Y-Chromosoms mit den beiden pseudoautosomalen Regionen (PAR) sowie der geschlechtsspezifischen Region (SRY) und der größten Region (MSY) die das männliche Geschlecht definiert.
Im Gegensatz zum ChrX hat das ChrY im diploiden Chromosomensatz keinen
vollkommen homologen Partner. Durch das fehlende Telomer ist das ChrY mit etwa 50-
60 mb kleiner als das ChrX und kann nur auf knapp fünf Prozent seiner Länge, den
Telomerenden, mit dem ChrX rekombinieren. Diese Regionen werden als
pseudoautosomale Regionen PAR1 und PAR2 bezeichnet [L09]. Allerdings weist das
ChrY weitere Gene auf, die auf dem ChrX dupliziert sind. Unter den Genen, welche auf
dem ChrX nicht vorhanden sind, befinden sich auch Gene in der sogenannten male-
specific region of the Y (MSY), die das männliche Geschlecht definieren [R08]. Die
MSY nimmt auf dem gesamten ChrY 95 % ein und besteht aus heterochromatischen
sowie X-degenerierten und repetitiven euchromatischen Sequenzen [R08]. Für die
Generierung von Haplotypen werden vorrangig MSY-Regionen verwendet, da diese
rekombinationsfreie Regionen darstellen und in der Vererbung weitestgehend stabil
weitergegeben werden [S10]. Es wird heute davon ausgegangen, dass ChrX und ChrY
einmal dieselbe Struktur besaßen. Im Laufe der Evolution entstand auf dem längen
ChrX aus dem Gen SOX3 die SRY. Diese führte zur Entwicklung der Hoden im
Embryo. Ein weiterer Faktor, der die Rekombination zwischen ChrX und ChrY
verhinderte, war die Inversion des längeren ChrY-Arms. Diese Veränderungen hatten

1 Einleitung
10
zur Folge, dass die ChrX sich untereinander weiter rekombinieren konnten und sich
daher im Laufe der Evolution nur wenig veränderten [L09]. Durch diese fehlende
Rekombination beim ChrY kam es zu einer Anhäufung von Mutationen und dem
Entstehen defekter Gene, welche im evolutionären Verlauf durch Deletionen verloren
gingen [L09]. Die nun vorherrschende Struktur des ChrY spiegelt das fehlende
crossing-over wieder. Im Vergleich mit anderen Chromosomen ist die Fülle an
genetischen Informationen, die das ChrY enthält, eher gering. Zurückzuführen ist diese
Tatsache auf die bereits beschriebene fehlende Rekombinationsmöglichkeit und die
Abhängigkeit der Frequenzen in der Bevölkerung. Weiterhin stützt sich die Aussage auf
den evolutionären Erklärungssatz, der besagt, dass aufgrund bestimmter
Fortpflanzungsverhalten die Populationsgröße des ChrY reduziert sein kann [L09].
Archäologische und genetische Daten belegen, dass vor ca. 100.000 Jahren eine
Populationsexpansion von Afrika in die übrigen Gebiete der Welt stattfand („Out of
Africa“–Hypothese). Durch diese vermutlich kleine Ausgangspopulation wurde das
ChrY in die unterschiedlichsten Gegenden der Erde getragen, wodurch es zu regionalen
Differenzierungen kam. Diese wurden, in Abhängigkeit des Genflusses und
auftretenden Mutationen, modifiziert und konserviert. Infolge dessen prägten sich
regional charakteristische und populationsspezifische Haplotypen aus, die die Basis der
Y-chromosomalen Populationsgenetik bilden. Einfluss auf die Diversität nehmen neben
dem genetischen Aspekt weiterhin ethnologische und kulturelle Normen wie Kriege,
religiöse Bedingungen oder Polygamie [L09]. Diese Parameter führen zu einer
charakteristischen und regionalen bzw. ethnischen Spezifität der Merkmalsverteilung.
Diese lässt die Y-chromosomalen DNA-Analysen zu einem effektiven Mittel für die
Verbesserung des Verständnisses der Populationsgeschichte verschiedener Epochen
werden. In der Forensik ist es durch die differenzierbare Haplogruppenverteilung
möglich, historische Aussagen hinsichtlich der Herkunft der maskulinen Individuen,
dessen Entwicklungsgeschichte und ihrer verwandtschaftlichen Nähe zueinander sowie
von Populationen und Subpopulationen zu treffen. Das ChrY ist somit ein wichtiges
Instrument um das Verständnis der humanen genetischen Geschichte voran zu treiben
[S08]. Zum Einsatz kommen dabei auf der DNA befindliche polymorphe
Sequenzabschnitte, welche für jedes Individuum spezifisch sind. Ebenso wie
Autosomen besitzen Gonosomen polymorphe und hochpolymorphe Regionen. Diese
auf den Geschlechtschromosomen befindlichen variablen Sequenzabschnitte verhalten
sich in ihrer Struktur und Polymorphie ähnlich zu den autosomalen DNA-Merkmalen
und besitzen in ihrer Vielfalt vor allem im nichtkodierenden Bereich des ChrY große
geografische und kulturelle Aussagekraft von Populationen [R08]. Die o.g. fehlende
Rekombinationsmöglichkeit ist dabei nicht nur negativ zu betrachten. Eben diese
ausbleibende Möglichkeit des crossing-overs sowie die geringe Mutationsrate der
polymorphen Sequenzabschnitte ermöglichen die Betrachtung der Phylogenie bis hin zu
den vorväterlichen Wurzeln. Dadurch können ChrY Haplotypen bzw. Haplogruppen
generiert werden, deren evolutionärer Hintergrund in einem einzigen Stammbaum
dargestellt werden kann [R08]. Da im männlichen Geschlecht nur ein ChrX vorhanden

1 Einleitung
11
ist, kann die Allelkonstellation unmittelbar als Haplotyp verwendet werden. Das ChrY
kann so phylogeografisch analysiert werden und es lassen sich zeitliche und regionale
Rückschlüsse einer männlichen Population ziehen, Wanderungsbewegungen
rekonstruieren sowie Vorfahren einer Art bestimmen.

1 Einleitung
12
1.4 Relevante molekulargenetische Marker
Etwa 99,9 % des menschlichen Genoms aller Individuen sind identisch. Aufgrund der
enormen Größe des Genoms ist der variable Teil der DNA trotzdem beträchtlich. Solche
Variationen auf der DNA-Sequenz zwischen Individuen werden als Polymorphismen
bezeichnet. Individuen mit einem hohen Grad an Polymorphie stehen somit in keinem
verwandtschaftlichen Verhältnis zueinander. Der Grad an Polymorphie kann in DNA-
Analysemethoden von großem Nutzen sein. Werkzeug zum Studium der genetischen
Variation ist eine sogenannte Mustersuche mit spezifischen Markern [B04].
Dabei kommen sogenannte repetitive Sequenzwiederholungen zum Einsatz, die sich im
kodierenden und nicht kodierenden Bereich des Kerngenoms befinden [L13]. Ihr Ort im
Genom muss somit bekannt sein. Das Hauptsaugenmerk liegt auf sogenannten Short
Tandem Repeats (STRs). Diese bilden eine Untergruppe der Variable Number Tandem
Repeats (VNTRs) und bezeichnen polymorphe Abschnitte einer DNA-Sequenz, die sich
durch eine bestimmte Anzahl an Wiederholungen des gleichen Motivs auszeichnen.
Von forensischer Relevanz sind hauptsächlich STR-Systeme, die in den nicht
kodierenden Regionen (Introns) der DNA liegen. Aufgrund gesetzlicher Richtlinien in
Deutschland ist es untersagt eine Analyse kodierender DNA-Bereiche und somit die
Untersuchung phänotypischer STR-Marker durchzuführen [BL+14]. Die Analyse von
STR-Motiven hat sich zu einem der leistungsfähigsten Hilfsmittel der DNA-
Typisierung in der forensischen Fallarbeit entwickelt. Sie ermöglichen eine zuverlässige
Identifikation von Personen sowie die Charakterisierung und Einordnung von
biologischen Spuren und repräsentieren ein maßgebliches Verfahren in
anthropologischen Studien.
Eine weitere Gruppe relevanter molekulargenetischer Marker stellen die Single
Nucleotide Polymorphisms (SNPs) dar. Sie hingegen sind Variationen einzelner
Basenpaare auf dem DNA-Strang. Es kommt hierbei zu einem Austausch einer
einzelnen Base und ihrem Komplement auf der DNA-Sequenz, welche über
Generationen hinweg weiter vererbt werden. SNPs sind zufällige, über das gesamte
Genom verteilte Punktmutationen eines Individuums, die ungefähr alle 500-1000
Nukleotide auftreten. Diese Punktmutationen kommen als autosomale, mitochondriale
und als gonosomale SNPs vor. So finden sich im menschlichen Genom ca. 38 Mio.
SNPs. Die häufigsten Austausche die vorkommen, sind der Ersatz des Nukleotids
Cytosin mit dem Nukleotid Thymin [F00]. Je nachdem wo ein solcher Austausch im
Genom stattfindet, kann er keine bis schwere Folgen haben. Um die Auswirkungen
dieser Basenwechsel besser darstellen zu können, unterteilt man die SNPs nach dem Ort
ihres Vorkommens in rSNPs (random SNPs), gSNPs (Gen assoziierte SNPs), cSNPs
(codierende SNPs) und pSNPs(Phänotyp-relevante SNPs) [BGG04].

1 Einleitung
13
1.4.1 Gonosomale STRs
Die meisten STR-Sequenzen sind in nichtkodierenden, hochindividuellen Bereichen der
gesamten DNA lokalisiert und treten dort als Di-, Tri- oder Tetranukeltidrepeats ca. alle
20 bp auf. Sie zählen aufgrund ihrer Struktur zur sogenannten repetitiven DNA. STRs
sind durch sich wiederholende Einheiten (Repeats/Allele) gekennzeichnet. Diese in
STR-Systemen vorkommenden Nukleotidmotive sind zwischen 3 und 7 Basen lang und
durch ihre Vielfältigkeit sehr gut zur genetischen Individualisierung von Personen
geeignet. STR-Sequenzen treten innerhalb genetischer Regionen in Introns,
flankierenden Sequenzen und kodierenden Abschnitten auf. Regionen, in denen diese
Muster erscheinen, sind in Bezug auf die Wiederholungen sehr polymorph. Diese
Wiederholungseinheiten variieren von Individuum zu Individuum und werden demnach
als Längenpolymorphismen beschrieben. Im Laufe der humanen Evolution und unter
Berücksichtigung spezifischer Vererbungsgesetze hat sich eine große Anzahl an
verschiedenen Varianten für jeden STR-Lokus entwickelt. [L13]. Aufgrund der
geringen Allellängen sind STRs ideal zur Amplifikation durch eine
Polymerasekettenreaktion (PCR) mit Hilfe lokusspezifischer Primer geeignet. Durch die
paternale Vererbung der gonosomalen Sequenzvariationen auf dem Y-Chromosom
können Untersuchungen auf Vaterschaft oder Verwandtschaft vorgenommen, sowie
populationsgenetische Fragestellungen betrachtet werden. Warum gerade jene Marker
für diese Analysen verwendet werden, beantwortet der Aufbau des Y-Chromosoms.
Durch die Lokalisation der gonosomalen STRs auf der MSY, befinden sie sich auf dem
rekombinationsfreien Teil des Y-Chromosoms. Sie unterliegen somit keinem crossing-
over und können lediglich durch Mutationen beeinflusst werden. Dies erlaubt eine
genetische Stammbaum- sowie Herkunftsanalyse und die Betrachtung vorväterlicher
Wurzeln.
Nach ersten Untersuchungen dieser polymorphen genetischen Marker durch A. J.
Jeffreys im Jahre 1985 und deren Anwendung als „genetischer Fingerabdruck“ in der
forensischen Spurenanalytik, steigerten die Anerkennung der STR-Systeme zunehmend
[WR03]. Durch weitere Forschungen auf dem Gebiet der STR-Analytik konnten diese
Verfahren optimiert sowie präzisiert werden und ermöglichten den DNA-Nachweis in
alten oder schlecht gelagerter Proben, in denen lediglich degradierte genomische DNA
(aDNA) enthalten war. In Bezug auf populationsgenetische Studien anhand von
Knochenfunden ist eine molekulargenetische Analyse des DNA-Materials meist
schwierig. Um einen hohen Informationsgehalt aus dem degradierten DNA-Material zu
erhalten, bedarf es spezifischer STR-Systeme. Der Fokus liegt hier bei der
Amplifikation von „Mini-STRs“. Diese besitzen eine Amplikonlänge von 70-280 bp
und zählen somit zu den kleineren Vertretern der STR-Systeme. Diese werden
vorzugsweise für die Charakterisierung von aDNA verwendet da diese auch bei
fortgeschrittener DNA-Degradation detektiert werden können.

1 Einleitung
14
Abbildung 4: Mögliche Motivwiederholung eines STR-Systems. Diese schematische Darstellung zeigt die mögliche Motiv-Wiederholung eines STR-Systems. Das Muster besteht hierbei aus der Basenabfolge „CTA“ und wiederholt sich sechsmal. Es kommt hier zur Ausprägung des Allel sechs. Die Nomenklatur der einzelnen STR-Systeme richtet sich nach Ihrem jeweiligen
Genort. Befindet sich ein STR-System in einem solchen nichtkodierenden Bereich,
generiert sich der Name aus der Bezeichnung des jeweiligen Gens und der Nummer des
Introns, in welchem der Marker auftritt. Die Nomenklatur wird am Y-STR-Marker
DYS385 beschrieben:
D: Differenzierung nach Markerart – Es handelt sich hier um einen DNA-Marker
Y: Lokalisation des Markers – Dieser Marker ist auf dem Y-Chromosom lokalisiert
S: Es handelt sich um eine single copy sequence
385: Der Marker stellt den 385 untersuchten Lokus auf dem Y-Chromosom dar
[K05].
Muster
Allel

1 Einleitung
15
1.4.2 Amplifikation gonosomaler STRs
Nach abgeschlossener DNA-Isolation steht dem Weiterarbeiten und der Durchführung
etwaiger Analysen meist nicht ausreichend DNA-Material zur Verfügung. Um die
gewonnene DNA über molekulargenetische Marker ausreichend charakterisieren zu
können, muss eine lokusspezifische Amplifikation mittels Polymerase-Kettenreaktion
(Abbildung 5: ) erfolgen. Durch dieses Verfahren können definierte DNA-Abschnitte
gezielt durch das Anlagern spezifischer Primer und das Erweitern des Template-
Stranges mit freien Nukleotiden durch DNA-Polymerasen vermehrt werden. Umgesetzt
wird dies mit spezifischen Oligonukleotiden, den Primern, welche sowohl forward als
auch reverse auf dem DNA-Strang kurz vor der repetitiven Sequenz hybridisieren. Es
erfolgt so eine eindeutige Definition der Allele an einem STR-Lokus durch
unterschiedliche Anzahlen der Sequenzwiederholungen und der absoluten Länge des
amplifizierten DNA-Fragments in Basenpaaren [R08]. In einem Reaktionszyklus finden
dabei jeweils die Denaturierung des Template-Stranges, das Amplifizieren der beiden
Einzelstränge sowie die Fluoreszenzmarkierung der PCR-Primer statt.
Abbildung 5: Schematische Darstellung einer PCR [mod. B08]. Diese Abbildung veranschaulicht den schematischen Ablauf einer PCR. Zu Beginn kommt es zur Denaturierung der Template-DNA in Einzelstränge und der darauffolgenden Hybridisierung der forward- und reverse-Primer. Mit Hilfe einer sich anlagernden DNA-Polymerase wird der komplementäre DNA-Strang synthetisiert.
Um eine schnellere Analyse von DNA-Proben zu gewährleisten, werden mehrere STRs
gleichzeitig amplifiziert. Diese multiple STR-Analyse basiert auf einer Multiplex-PCR-
Amplifikation welche eine simultane Vervielfältigung sowie Separation und Detektion
der gewünschten DNA-Fragmente erlaubt. Möglich ist dies durch die
Fluoreszenzmarkierung der PCR-Fragmente, was eine Differenzierung von Fragmenten

1 Einleitung
16
mit gleicher Länge ermöglicht [B08, W03]. Es können so hochinformative STR-Profile
erstellt sowie ein Hochdurchsatz an Proben ermöglicht werden. Aufgrund der erhöhten
Anzahl an Primern kann es zur Hybridisierung dieser kommen. Einhergehend müssen
entsprechende Primer designt und im Multiplex aufeinander abgestimmt sein. Hierfür
werden Primer und PCR-Produkte expliziert geprüft, um Wechselwirkungen
untereinander zu reduzieren und als Fehlerquelle auszuschließen. Unter anderem wird
dabei die Konzentration der Primer auf ein Minimum reduziert und die zueinander
komplementären Sequenzabschnitte der verschiedenen Primer und Zielsequenzen
müssen so gering wie möglich gehalten werden [W08, G06]. Das Design der
Oligonukleotide wird in der heutigen Zeit von frei zugänglichen Datenbanken
angeboten, und diverse Software zur Auswertung kann genutzt werden. Das Vorgehen
zur Detektion gonosomaler und autosomaler STRs unterscheidet sich nicht und kann
mittels Kapillarelektrophorese realisiert werden.
Diese Detektionsmethode kann verwendet werden, um neue Polymorphismen zu
scannen und die Allele eines bekannten Polymorphismus in der Zielsequenz zu
bestimmen. Referenzwert dafür stellt das nahezu vollkommen sequenzierte Genom des
Menschen dar, mit der alle anderen Sequenzierungsdaten verglichen werden können.
Häufig verwendete Detektionsmethoden beruhen auf Fluoreszenz, Massenspektrometrie
oder Chemielumineszenz.
1.4.3 Fragmentlängenanalyse mittels Kapillarelektrophorese
Um eine korrekte Detektion der zuvor spezifisch amplifizierten DNA-Fragmente zu
ermöglichen, bedient man sich dem Prinzip der fluorenzbasierten
Fragmentlängenanalyse durch Kapillarelektrophorese, deren schematischer Aufbau in
Abbildung 6 beschrieben wird. Als instrumentelles Analyseverfahren wird die
Kapillarelektrophorese, durch die Kombination einer elektrophoretischen Trennmethode
und der Automatisierbarkeit, vorrangig zu Detektionszwecken verwendet. Die
Auftrennung amplifizierter einzelsträngiger DNA-Fragmente erfolgt in Polyacrylamid-
gelen entsprechend ihrer Länge. Die Wanderungszeit der Fragmente richtet sich dabei
nach der jeweiligen Länge. Die Bestimmung der Allele entsprechend der jeweils
detektierten Fragmentlängen wird mit Hilfe von Längenstandards in jeder Probe
bestimmt und über Allelleitern, welche parallel zu den Proben aufgetragen werden
genau detektiert. Zu den wichtigsten Hauptbestandteilen einer Kapillarelektrophorese
gehören das Hochspannungsnetzteil mit einer Spannung von bis zu 30 kV Stromstärke
sowie ein Detektor. Verantwortlich für die spezifische Auftrennung der DNA-
Fragmente ist eine Glas- oder Quarzkapillare mit einem Innendurchmesser von 50 bis
500 pm und einer Länge von 20 cm bis 100 cm. Diese Kapillare überbrückt zwei
Flüssigkeitsreservoirs, welche mit Pufferlösung befüllt sind. Nach korrektem Einsetzen
der zu detektierenden Proben, findet voll automatisch ein Wechsel der Platinelektrode
aus der Pufferlösung in eine Probe statt. Durch Anlegen einer positiven Spannung wird
die Probe in die Kapillare aufgenommen und im darin befindlichen Polymer (z.B.
Polyacrilamidgel) aufgetrennt. Die Auftrennung erfolgt in Richtung der Anode je nach

1 Einleitung
17
Mobilität der DNA-Fragmente, die sich nach deren jeweiliger Größe richtet [S06]. Nahe
der Anode befindet sich ein auf Laserstrahlung-basierender Detektor. Dieser übermittelt
die Fluoreszenzsignale der vorbei wandernden Fragmente. Mit Hilfe einer Software zur
Auswertung dieser Signale werden Elektropherogramme der jeweiligen Proben erstellt.
Abbildung 6: Schematische Darstellung einer Kapillarelektrophorese [G06]. Abgebildet ist hier der grundlegende Ablauf der fluoreszenzbasierten Detektion von DNA-Fragmenten mittels Kapillarelektrophorese. Der Sequenzer verfügt über zwei Puffergefäße, einen Inlet- und Outlet-Puffer die durch eine Glaskapillare miteinander verbunden sind. Diese Glaskapillare ist mit einem bestimmten Polymer gefüllt durch dieses die DNA-Fragmente laufen. Durch das Anlegen einer positiven Spannung werden die DNA-Proben, welche sich in einem Probentray befinden in die Kapillare injiziert und wandern durch das Gel. Die Wanderungsgeschwindigkeit richtet sich dabei nach der Größe der aufzutrennenden Fragmente. Große Fragmente wandern langsamer durch das Gel als kleinere DNA-Fragmente. Mit Hilfe einer lasergestützten Detektion erfolgt die Fluoreszenzsignalaufnahme und es folgt die Auswertung über eine spezielle Analysesoftware [G06].
Puffer Probe
Kathode
(-) Anode
(+)

1 Einleitung
18
1.5 Formulierung von Haplotypen
Als Y-chromosomaler Haplotyp wird die Gesamtheit von Allelvarianten untersuchter
Y-STRs bezeichnet, welche uniparental über Generationen hinweg in männlicher
Erblinie rekombinationsfrei (unverändert) und gekoppelt weiter vererbt werden [S10].
Das Auftreten von Allelen in der Bevölkerung kann nicht als einheitlich beschrieben
werden. Je nach Bevölkerungsgruppe und Region zeigen sich Unterschiede in der
Allelausprägung. Durch die Haploidie des ChrY wird der Haplotyp klonal entlang der
männlichen Linie vererbt und erlaubt eine simple Bestimmung des allelischen
Zustandes, da nur ein Allel pro Lokus detektiert werden muss [R08]. Genetisch ähnliche
Haplotypen werden in sogenannten Haplogruppen zusammengefasst. Männliche
Individuen, die den gleichen Haplotypen aufweisen, können einer Patrillinie zugeordnet
werden und verfügen über die identische Abstammungslinie. Durch diese Möglichkeit
der Einordnung in Haplogruppen kann, bezogen auf das Vorkommen der jeweiligen
Haplogruppen, eine Verteilung und geographische Abgrenzung von Gebieten erfolgen
sowie Aussagen über Bevölkerungsgruppen getroffen werden. Einsatz finden diese für
die Beantwortung von evolutionsgeschichtlichen und phylogeografischen
Fragestellungen. Durch optimierte Verfahren zur Analyse von aDNA (ancient DNA)
kann mitunter die evolutionäre Vergangenheit früherer Bevölkerungsgruppen dargestellt
sowie Wanderungsprozesse nachvollzogen werden.
Abbildung 7: ChrY –Haplogruppen in Europa [URL-3] . Zu sehen ist hier die wesentliche Verteilung der Y-chromosomalen Haplogruppen in der heutigen Zeit. Deutlich wird hier die regionale Abgrenzung der einzelnen Gruppen.

1 Einleitung
19
Wurzel dieses Stammbaumes bildet ein abgeleiteter, jedoch heute nicht mehr
existierender Haplotyp, der für alle Loki die von Vorfahren geerbten Allelvarianten
aufweist. Dieser Haplotyp aller ChrY wird als deren Most Recent Common Ancestor
(MRCA) bezeichnet und gilt als Vorfahre aller bekannten ChrY [R08]. Der letzte
chromosomale Vorfahre aller ChrY wird als „Y chromosomaler Adam“ definiert und
wird auf ein Alter von 90.000 Jahren detektiert.
In Abbildung 7 wird deutlich, dass sich durch räumliche Trennung und den damit
einhergehenden unterschiedlichen klimatischen und umweltbedingten Unterschieden
die Haplogruppen in ihrer Erscheinung differenzieren. So wurde eine Südost-Nordwest-
Orientierung der Haplogruppen J und R1b festgestellt. Die Haplogruppe R1b
beispielsweise, ist hierbei stark im Westen und kaum im Osten vertreten. Hier kann von
einer geografischen Spezifität gesprochen werden [L09]. In den letzten Jahren wurden
311 variierende ChrY Haplogruppen mit ca. 600 binären Markern gefunden. Um diese
Haplogruppen und ihre phylogeografische Beziehung besser darstellen zu können,
werden jedem monophyletischen Ast, welcher durch einen binären Marker definiert
wird, mit einem Großbuchstaben (derzeit A-T) gekennzeichnet. Verzweigungen
unterhalb der hauptsächlichen Haplogruppen werden mit Zahlen oder Kleinbuchstaben
markiert, die eine feinere Auflösung zulassen (Abbildung 8) [R08].
Abbildung 8: Schematische Darstellung der Haplogruppen-Phylogenie. Abgebildet ist hier der Ursprung der Haplogruppen, entstanden aus dem Y-chromosomalen Adam, sowie die einzelnen monophyletischen Äste mit ihren jeweiligen Verzweigungen, die jeweils eine Haplogruppe ausbilden.

1 Einleitung
20
Diverse phylogeografische Forschungen ergaben, dass der Ursprung der Y-
chromosomalen Stammesgeschichte in Afrika liegt. Bestätigung findet diese Aussage in
den ersten Verzweigungen A und B der Y-Phylogenie, welche mit einigen Ausnahmen
ausschließlich zu afrikanischen Populationen führen. Neben diesen Haplogruppen die
auf eine afrikanische Population zurückzuführen sind, gibt es weitere 3 Hauptgruppen
C, DE und F. Diese Hauptäste C und D sind auf Ostasien bezogen, E deutet auf Afrika,
das südliche Europa und das westliche Asien hin, während die Untergruppen I, J, N und
P der Haplogruppe F auf eine weitere Verbreitung außerhalb Afrikas deuten [Roewer,
2008]. Diese Rekonstruktion demografischer Vorgänge prähistorischer Epochen aus
denen nur wenige archäologische Funde existieren, wird als Phylogeogafie bezeichnet.
Diese leistet einen großen Beitrag zur geografischen Projektion rezenter ChrY
Haplotypen [R08]. Besonders in der forensischen Populationsgenetik können diese
Informationen über die geografische Verteilung der Haplogruppen zur Identifikation der
Herkunft eines männlichen Knochenfundes genutzt werden [L09].

1 Einleitung
21
1.7 aDNA-Isolation und Amplifikation
Die Abkürzung aDNA beruht auf dem Begriff ancient DNA und meint damit die
Charakterisierung von altem, vermeintlich degradiertem DNA-Material toter
Lebewesen, unter Verwendung molekulargenetischer Methodiken. In lebenden
Organismen wird die DNA-Struktur unaufhörlich von Reparaturmechanismen
kontrolliert und korrigiert. Nach dem Tod eines Organismus sind diese
Reparaturmechanismen nicht mehr aktiv und körpereigene Autolyseprozesse beginnen,
die DNA zu zerstören [H06]. Der Degradationsprozess des DNA-Materials ist ein
postmortaler, fortschreitender Ablauf der unter Einwirkung und unter Einflussnahme
diverser Umwelteinflüsse beschleunigt oder verlangsamt wird. Vorangegangene
Publikationen zeigen, dass wirkende Umwelteinflüsse größere Auswirkungen auf den
DNA-Erhalt haben als die Dauer der Liegezeit. Es kann somit keine Korrelation
zwischen dem Alter der Probe und dem DNA-Erhalt angenommen werden [BH+99].
Je nach vorherrschenden Umwelteinflüssen und Liegedauer ist in manchen Fällen
lediglich das Knochenmaterial eines Individuums auffindbar. Aufgrund von
Zersetzungsprozessen die eine DNA-Analyse anhand von Gewebe nicht mehr
ermöglichen, kann eine Identifizierung nur noch durch das Zahn- und Knochenmaterial
erfolgen. Doch auch der Erhalt des Skeletts wird durch diverse Einwirkungen gelenkt.
Derartige beeinflussende Faktoren sind Temperatur, Luftfeuchtigkeit, vorherrschender
pH-Wert sowie geochemische Eigenschaften des Bodens und der Grad an mikrobiellem
Befall (Tabelle 1). Bei einer Liegetemperatur um 8 °C kann von langsamen
enzymatischen Abbauprozessen ausgegangen werden und einem guten DNA-Erhalt. Bei
einem pH-Wert im neutralen oder leicht alkalischen Bereich ist ebenso von einem
besseren Erhalt der DNA auszugehen. Eine hohe Luftfeuchte dagegen verursacht den
Angriff des Materials. Insbesondere Zähne werden durch das Angreifen des
Zahnschmelzes über organische Substanzen aus umgebendem Sediment beschädigt.
Weiterhin hat ein hoher Grad an mikrobieller Aktivität durch die Beschleunigung
abbauender Prozesse und einer sukzessiven Auflösung der organischen Matrix negative
Auswirkungen auf den DNA-Erhalt. Neben enzymatischen Abbauprozessen durch
endogene Nukleasen kommt es außerdem durch chemische Reaktionen wie Oxidation
und Hydrolyse zu einem DNA-Zerfall. Oxidative Effekte verursachen Veränderungen
sowie Verluste von Basen wohingegen hydrolytische Prozesse zur Deaminierung von
Basen durch Verlust der Aminogruppe sowie Depurinierung mit einhergehendem
Verlust der Purinbasen Adenin und Guanin oder Depyrimidierung, die zu einer
Destabilisierung des DNA-Moleküls führen können und ein Brechen in einzelne
Fragmente verursachen [H06]. Insbesondere Apurin-Bindungsseiten führen zur
Spaltung von Phosphatverbindungen und so zum Brechen des Zucker-Phosphat-
Rückgrades [I07].
Problematisch sind solche Degradationsmechanismen endogener DNA für die
Amplifikation in einer folgenden PCR. Inhibitoren wie Humin- und Folsäure behindern
die Amplifikation und führen zu unzureichenden DNA-Amplifikaten. Die

1 Einleitung
22
Fragmentverkürzungen und Modifikationen durch Oxidation und Hydrolyse können
außerdem zur Generierung von Mikrosatellit-Artefakten oder zu einem kompletten
Ausfall von erwarteten Fragmenten führen. Um die Koextraktion von PCR
inhibierenden Substanzen so gering wie möglich zu halten und eine fortschreitenden
Degradation des DNA-Materials zu reduzieren, werden etwaige Reinigungsschritte vor
einer molekulargenetischen Untersuchung eingeleitet. Diese sollen die meisten
Inhibitoren entfernen sowie den Ertrag an aDNA optimieren. Problematisch ist dabei
die geringe Menge an zur Verfügung stehendem Material sowie der geringen Menge an
aDNA und den verschiedenen Degradationsstadien. Da sich in degradierten DNA-
Spuren durch Abbauprozesse und enzymatische Spaltung lediglich verkürzte DNA-
Fragmente finden, erschwert dies die Amplifikation langer DNA-Stränge. Kurze
Sequenzlängen der STR-Systeme und die damit verbundenen geringen Amplikonlängen
können hier bei der Amplifikation von historischem DNA-Material hilfreich sein
[WR03]. Seit den Anfängen der aDNA-Isolation aus altem Probenmaterial besteht das
Problem, dass das verbliebene DNA-Material in geringen Mengen sowie in
verschiedenen Degradationsstadien vorliegt. Daher ist es entscheidend, Menge an
verfügbarer DNA unter Verwendung geeigneter enzymatischer Verfahren voll
auszuschöpfen. Mit Hilfe spezifischer PCR Kits, die optimal auf die zu bestimmenden
Positionen definierter STR-Loki abgestimmt sind, lassen sich auch geringe Mengen an
DNA erfolgreich typisieren.
Die Verfahren zur Extraktion und Aufreinigung müssen somit schonend, ohne
aggressive Aufreinigungsmittel und hohe Temperaturen durchgeführt werden [I07]. Das
in dieser Forschungsarbeit verwendete Verfahren wird in den Kapiteln 4.1 bis 4.4
beschrieben.

1 Einleitung
23
Tabelle 1: Übersicht über mögliche Schäden an aDNA [H06]. Dargestellt werden hier die möglichen Einflussfaktoren die einen Zerfall der DNA bewirken können sowie die Form des Schadens und deren Auswirkungen. Weiterhin wird eine mögliche Lösungsstrategie vorgestellt.
Pozess/ Einfluss Schadensform Auswirkungen Lösungsstrategie Mikroorganismen, Nukleasen, Hydrolyse
Strangbrüche Hydrolytische Spaltung der Phosphor-diesterbindungen
Kurze Fragmente Reduzierung der Gesamtmenge
Wahl kurzer und überlappender Amplikons
Oxidation Fragmentierung der Basen bzw. Fragmentierung der Zuckermoleküle durch Hydroxyl-Radikale (-OH) oder Verknüpfung von Purin mit dem Zucker-Phosphatrückgrat
Inhibition der PCR, ggf. Strangabbruch
Oxidation Hydantoine, Beeinträchtigung der Basen durch Auflösung der C-Doppelbindung
Hydrolyse Depurinierung von Adenin und Guanin durch Spaltung der N-glykosidischen Bindungen
Oxidation Guanin > 8-Oxoguanin Transversion G/C� T/A
Mehrere unabhängige PCRs Klonierung und Sequenzierung (Evtl. Einsatz von (UNG; Uracil-N-Glycoslase))
Hydrolyse Deaminierung: von Adenin zu Hypoxantin
Transition A�G
Hydrolyse Deaminierung von Cytosin zu Uracil
Transition C�T
Hydrolyse Deaminierung von 5-Methylcytosin zu Thymin
Transition C�T
Hydrolyse Deaminierung von Guanin zu Xanthin
DNA crosslinks Bindung zwischen DNA-Molekülen oder mit anderen Biomolekülen
sog. Maillard Produkte
Einsatz von N-Phenacyl Thiazoliumbromid (PTB) in Extraktion

1 Einleitung
24
1.6 DNA-Erhalt in historischem Zahnmaterial
Die Identifikation eines Individuums ist eines der wichtigsten
Untersuchungsschwerpunkte in der forensischen Wissenschaft. Die Entdeckung der
Doppel-Helix-Struktur der DNA 1953 durch Watson und Crick erzeugte fundamentale
Veränderungen in nahezu allen Bereichen der Wissenschaft. Diese Entdeckung bildet
die Basis für die Entwicklung von Methoden, die es erlauben, jede Person individuell
auf Grundlage ihrer DNA zu charakterisieren. 1985 entwickelte Jeffreys eine Methodik
zur Detektion hoch variabler DNA-Regionen. Der „genetische Fingerabdruck“ entstand.
Im Laufe der Zeit entwickelten sich zahlreiche Arbeitsweisen zur DNA-Isolation und
somit der Personen-Identifikation aus verschiedenem biologischem Material. Durch
etliche Labortests wurde die DNA-Gewinnung anhand von Knochenmaterial, Haar-
Proben, Blut und anderen Körperflüssigkeiten getestet. Es wurde ersichtlich, dass die
DNA-Extraktion aus praktisch jeder Körper-Probe möglich ist und sich lediglich in der
Qualität und Quantität unterscheidet. Eine wichtige Rolle spielen bei der Kriminologie
und Personen-Identifikation die Zähne. Sie besitzen nicht nur eine hohe Einzigartigkeit
in Hinblick auf ihre Charakterisierung, sondern außerdem eine starke physikalische und
chemische Beständigkeit sowie Langlebigkeit und eine gewisse Resistenz gegenüber
wechselnden Umwelteinflüssen. Zusätzlich werden die dentalen Pulpa-Zellen, welche
die genomische DNA enthalten, durch Zahnschmelz, Dentin und Zement geschützt und
stellen im Hinblick auf historische Skelettfunde eine gute aDNA-Quelle dar. Weiterhin
wird ein natürlicher Schutz der Zähne durch ihre Lage im Kiefer, dem Epithel-und
Bindegewebe sowie Muskel- und Knochengewebe gewährleistet [SG+06]. Durch die
Verankerung der Zahnwurzeln über eine Gomphosis in den Alveolen, sind sie und somit
die enthaltene genomische DNA (gDNA) vor äußeren Umwelteinwirkungen und einem
mikrobiellen Befall geschützt. Die Bedeutung der Zahnmedizin zur Identifikation
humaner Überreste seitens der Forensik wuchs in den letzten Jahren zunehmend. Durch
die DNA-Analyse anhand von Zahnmaterial können auch stark verweste Überreste
sowie Brandopfer oder Knochengräber identifiziert werden. Auch die Verwendung von
Knochenmaterial ist weit verbreitet. Jedoch stellen diese in Bezug auf historische
Skelettfunde oftmals keine ausreichende DNA-Gewinnung in Aussicht. Im Vergleich zu
Zahnmaterial sind Knochen gegenüber Umwelteinflüssen und vorherrschenden
Lagerungsbedingungen durch eine geringere Dichte des Gewebes sowie Porosität
anfälliger. Knochenmaterial verfügt über keinen zusätzlichen Schutz durch Schmelz
oder Zement und wird, abhängig von den Liegebedingungen, schneller von
Mikroorganismen befallen, was eine STR-Analyse mit kommerziell erhältlichen
Multiplex-Kits äußerst erschwert [SS-P+07, BH+99].

1 Einleitung
25
Abbildung 9: Aus einem Kiefer herausgelöste Backenzähne (Molare). Diese Abbildung zeigt zwei Molare, die mit Hilfe mechanischer Einwirkung aus dem Kiefer eines Individuums herausgelöst wurden. Die Zähne werden mit der dazugehörigen Individuennummer und der jeweiligen Position im Kiefer gekennzeichnet.
Bei dem verwendeten Untersuchungsmaterial handelt es sich um Zahnmaterial (Dentes)
(Abbildung 9) der Sekelettfunde. Da aus Erfahrungswerten heraus, im Vergleich zu
Knochenmaterial, eine quantitativ hochwertigere DNA-Menge aus Zahnmaterial
gewonnen werden konnte, wurde in dieser Arbeit ausschließlich auf Zahnmaterial
zurückgegriffen.

1 Einleitung
26
1.8 Populationsgenetische Datenbanken
Um die Ergebnisse der molekulargenetischen Analyse in einen populationsgenetischen
Kontext einordnen zu können, kann dies mit Hilfe von Datenbanken erfolgen. In Bezug
auf die Auswertung Y-chromosomaler Daten können eigens dafür generierte
Datenbanken angewendet werden. Sie können die Auswertung der gewonnenen
Datensätze, unter Angabe definierter Parameter, im Hinblick auf die Zuordnung zu
ethnischen Gruppen übernehmen und die Herkunft der Proben bestimmen.
1.8.1 Y-Search
Die Y-Search (www.ysearch.org/) beschäftig sich vorranging mit Y-chromosomaler
DNA zur Untersuchung von Familienbeziehungen und der Herkunftsbestimmung.
Durch stetig steigenden Dateneingang und Evaluierung dieser kam es Ende 2003 zur
Entwicklung einer web-basierten Datenbank die seither einen freien Zugang für Nutzer
bietet. Unter der Rubrik search for genetic matches wird die Möglichkeit geboten, bis
zu 43 Y-chromosomale Marker einzugeben und diese über selbst gewählte
Parametereinstellungen mit den vorhandenen Daten zu vergleichen. Zu wählende
Parameter sind zum einen: maximal genetische Distanz, die angenommen werden soll
oder Anzahl an Markern, die mit einer genetischen Distanz von 1 vergleichen werden
sollen. Zum anderen kann die Anzahl der zu vergleichenden Marker ausgewählt
werden. Ausgegeben werden ähnliche Haplogruppen die nach steigender genetischer
Distanz geordnet sind sowie die Benutzer-ID desjenigen, der die Daten generiert hat.
Wird die jeweilige ID angewählt, so wird der vom Nutzer hochgeladenen Haplotyp
angezeigt. Unter der Rubrik search by haplogroup ist es möglich, nach Haplogruppen
in verschiedenen Ländern zu suchen. Ausgegeben wird eine Liste von Datenbank-
Nutzern, deren hochgeladene Haplogruppe der gesuchten entspricht. Im Bereich des
research tools können durch die Eingabe einer Benutzer-ID Informationen über Nutzer
herausgefunden werden.
1.8.2 YHRD
Einer der größten populationsgenetischen Datenbanken für Y-chromosomale STR-
Haplotypen ist die YHRD (www.yhrd.org). Diese wurde am Institut für Rechtsmedizin
und forensische Wissenschaften der Charité Berlin in Zusammenarbeit mit mehr als 150
weiteren Instituten aus 45 Ländern erstellt. Die YHRD stellt ein Tool zur forensischen,
anthropologischen und genealogischen Untersuchung allgemein zur Verfügung
[WR13]. Ziel der eingerichteten Datenbank ist es, aufgrund der zu erwartenden
Datenmenge ein Suchprogramm für Haplotypen zu erstellen. Weiterhin sollte es dem
Nutzer möglich sein, zu jedem Zeitpunkt den vollen aktuellen Umfang der Datenbank
abzufragen. Als obligater Datensatz zur Charakterisierung der ChrY wurde hierbei das
international akzeptierte Haplotyp-Format Yh1 unter Einbeziehung des Lokus DYS385
verwendet [R00].
Neben aktuellen Links zu weiteren Datenbanken und Publikationen wird sowohl die
Suche als auch die Analyse von STRs angeboten. Wird eine spezifische Population

1 Einleitung
27
gesucht, so werden die jeweils vertretenen Haplotypen dargestellt. Die Rubrik Analyse
stellt diverse Verfahren bereit, um genetische Distanzen zwischen Populationen zu
untersuchen oder Metapopulationen aus vorliegenden Haplotypen heraus zu filtern.
Unter der Rubrik Forschung können Informationen über aktuelle Untersuchungen
verschiedener Thematiken eingeholt werden.
Weiteres Ziel der DB ist es, Hilfestellung bei der Interpretation von Resultaten aus dem
Vergleich von Beweisproben und bestehenden Referenzdaten vorzunehmen und
Schlussfolgerung daraus zu ziehen.
Da eine starke Substruktur zwischen und innerhalb der Kontinente für Y-Haplotypen
existiert, muss die Datenbank diverse Aspekte von Haplotypen in verschiedenen
geografischen Regionen oder Kontinenten reflektieren. Laut dem Wissenstand im
September 2013 gibt es 115000 9-Lokus Haplotypen (eingeschlossen die über 56000 Y-
Haplotypen) von 851 Probenahmestellen. Diese wurden in 113 Ländern von 237
Instituten und Laboren eingereicht.
Aus geografischer Sicht stammen über 39 % der YHRD Proben aus Europa, 32 % aus
Asien, 4 % aus Afrika und 2 % aus Australien. Außerdem werden kontinuierlich neue
Daten eingereicht und die Datenbank wird ständig evaluiert. Da ständige
Aktualisierungen und Zuwächse den Datenpool erweitern, ist die
Veröffentlichungsanzahl und das Datum der Veröffentlichung ein wichtiger Teil des
„Search Result“-Dokuments.
Populationen, mit geringem oder keinem genetischen Abstand zueinander werden als
Metapopulationen bezeichnet. In dieser Kategorie können alle vordefinierten
Metapopulationen mit den Beschreibungen der jeweiligen Einzugsgebiete, der
geografischen Zerstreuung sowie eine Liste der zugeordneten Populationen angesehen
werden.
Weiterhin ist die Datenbank in der Lage nach einzelnen Allelen und Allel-
Kombinationen zu suchen. Sie unterstützt dabei die am häufigsten genutzten Kits und
Guidelines mit den zugehörigen STRs, wie Minimal, SWGDAM, PowePlex®, YFiler
und Y23. Für diese existieren jeweils verschieden große Datenbanken, die zur
jeweiligen Auswertung als Grundlage dienen. Weiterhin werden die zu den
eingegebenen Haplotypen mögliche Haplogruppen aufgelistet. Die Exaktheit einer
Haplogruppenzuordnung hängt von der zur Verfügung stehenden Referenzdatenmenge
und der eingegebenen Allelanzahl ab. Konnten in einem DNA-Profil nicht alle Allele
detektiert werden, lassen diese Ausfälle in der Auswertung Raum für unspezifische
Haplogruppen.
Weiterhin erlaubt die DB eine SNP-Suche. Die Kategorie Y-SNPs bietet alle
molekularen und phylogenetischen Informationen zu Y-SNP Markern und der
zugeordneten Haplogruppen. Es können dabei Haplogruppen des monophyletischen
Baumes ausgewählt, oder bestimmte Marker betrachtet werden [WR13].

1 Einleitung
28
1.8.3 AMOVA-Analyse
Eine von der YHRD bereitgestellte Analysemethode ist die Analysis of molecular
Variance (AMOVA). Diese Anwendung dient der molekularen Variationsanalyse
zwischen Inter- oder Intra-Populationen, basierend auf molekularen Daten. Mit Hilfe
der AMOVA (www.yhrd.AMOVA.org) ist es möglich, den Umfang an Unterschieden
zwischen zwei oder mehreren Populationen zu bewerten und zu quantifizieren. Mit
Hilfe der molekularen Varianz σ2 wird die durchschnittliche Entfernung eines
Haplotypes eines Indivviduums zu jedem anderen betrachteten Individuum in Form von
1-Schritt-Mutations-Unterschieden beschrieben. Zusätzlich zu einer „gleich“/“ungleich“
Differenzierung bezieht AMOVA das Kriterium „ähnlich“ über die Anzahl vorhandener
1-Mutationsschritte zwischen zwei Haplotypen mit ein. So können genauere Aussagen
über Populationsbeziehungen zueinander auf Basis von Haplotypen getroffen werden.
Die molekulare Varianz σ2 wird wie folgt berechnet [WR13]:
σ� = σ�� + σ�
�
Formel 1: Berechnung der Molekularen Varianz ��. Die molekulare Varianz berechnet sich aus der molekularen Varianz zwischen Populationen σ�
� und der molekularen Varianz zwischen Individuen innerhalb einer Populationσ�
� .
AMOVA unterstützt die Analyse haploider Daten, genannt phi-Statistik. Diese
Berechnungsgrundlage ermöglicht die statistische Bewertung von Unterschieden in der
Varianz zwischen zwei Populationen (ΦST). ΦST beschreibt die genetische Distanz
zwischen zufälligen Haplotypen aus zwei Populationen relativ zu zufälligen Paaren von
Haplotypen, die aus der ganzen Spezies herangezogen wurden. Sie definiert so den
Anteil der molekularen Varianz zwischen Populationen zu der gesamten molekularen
Varianz. ΦST wird wie folgt berechnet [I07]:
Φ�� = σ�
�
σ�
Formel 2: Berechnung der genetischen Distanz ΦST. Die genetische Distanz berechnet sich aus der molekularen Varianz zwischen Populationen σ�
� und der totalen molekularen Varianz σ�.
Weiterhin wird für die Berechnungen der ΦCT -Wert mit einbezogen. Er beschreibt die
Korrelation zwischen der molekularen Diversität von zufälligen Haplotypen innerhalb
einer Gruppe einer Population relativ zu zufälligen Paaren von Haplotypen die aus der
ganzen Spezies herangezogen wurden. Der ΦCT -Wert berechnet sich wie folgt [I07,
WR13]:
Φ � = σ�
σ�
Formel 3: Berechnung der Varianz-Korrelation. Die Korrelation der molekularen Diversitäten berechnet sich aus der Varianz der unterschiedlichen Haplotypen innerhalb einer Population σ� und der Varianz verschiedener Paare von Haplotypen innerhalb einer Artσ�.

1 Einleitung
29
Eine Möglichkeit die molekulare Distanz von Haplotypen zu bestimmen ist die FST
basierte Methode. Der FST -Wert variiert zwischen 0 für Haplotypen die identisch sind,
und 1 für Haplotypen, die keine Gleichheit aufweisen. Er beschreibt das Verhältnis der
durchschnittlichen Anzahl an Unterschieden zwischen Haplotypen innerhalb einer
Population und zufällig gewählten Haplotypen. Der FST –Wert wird wie folgt berechnet
[HW09]:
FST =��
�(�)
Formel 4: Berechnung der molekularen Distanz von Haplotypen. Der FST –Wert berechnet sich aus der durchschnittlichen Anzahl an Unterschieden zwischen Allelpaaren σ� und den zufällig gewählten Allel-unterschieden zwischen Populationen�(�). Je kleiner der Wert, desto ähnlicher sind sich die Populationen.
Eine Berechnungsmöglichkeit für die Distanz zwischen Populationen bietet der RST-
Wert. Die Berechnung des RST -Wertes beruht, im Gegensatz zu dem FST -Wert auf der
Annahme des stufenweisen Mutationsmodells. Dieses Berechnungsmodell spiegelt die
Dynamik von Mikrosatelliten in Bezug auf die Mutation der Allellängen die diese um
eine Wiederholungseinheit verringern oder erweitern können, wieder [I07]. Der RST-
Wert beschreibt die Beziehungen von Allelfrequenzen zwischen Subpopulationen und
ist somit ein Maß für den Grad der Varianz von Allelfrequenzen zwischen zwei
Subpopulationen. Der RST-Wert wird wie folgt berechnet:
��� = �� − ��
��
Formel 5: Berechnung der molekularen Distanz zwischen Populationen. Die Berechnung der molekularen Distanz zwischen Populationen ergibt sich aus der Allel-Varianz in einer totalen Population �� und der Allel-Varianz zwischen Subpopulationen��.
Der RST-Wert kann ebenso durch den �� angegeben werden nachdem die molekulare
Distanz zwischen Allelen bestimmt wurde, basierend auf der Summe der quadratischen
Unterschiede der Repeat-Längen [I07].
Um die Signifikanz der ΦST-Werte beurteilen zu können, wird eine Permutationsanalyse
unter Annahme der Nullhypothese vorgenommen. Dabei werden die Daten des
Datensatzes wahllos kombiniert und diese Ergebnisse mit den „tatsächlich“ in Bezug
auf signifikante Abweichungen verglichen. Das Programm führt hierzu 10.000
Permutationen aus [WR13, I07, HW09].

2 Zielstellung
30
2 Zielstellung
Ziel dieser Studie ist es, unter Verwendung forensischer DNA-Analysemethoden auf
populationsgenetische Fragestellungen einzugehen. Mit Hilfe molekulargenetischer
Analysen von Skelettmaterial des historischen Gräberfeldes Görzig, soll eine
Einordnung in den populationsgenetischen Kontext erfolgen. Die Untersuchung von 23
molekulargenetischen Markern (Y-STRs) des Y-Chromosoms soll Aufschluss über eine
populationsspezifische Haplogruppenverteilung liefern. So kann ein Vergleich der
resultierenden Haplogruppen zur heutigen Haplogruppenverteilung gezogen- sowie
eventuelle verwandtschaftliche Verhältnisse und Veränderungen innerhalb der
Population dargelegt werden. Nach einer osteologisch morphologischen Begutachtung
des Skelettmaterials und der Geschlechts- und Sterbealtersabschätzung der einzelnen
Individuen, zu denen der Schädel (Cranium), der jeweilige Zahnstatus und die unteren
Extremitäten (Membrum inferius) herangezogen wurden, erfolgen nun
molekulargenetische Untersuchungen. Hierzu wurde vorrangig das Zahnmaterial
(Dentes) aufgearbeitet und chromosomale sowie extrachromosomale DNA extrahiert.
Da im Rahmen dieser Forschungsarbeit die Analyse männlicher Individuen im
Vordergrund steht, soll eine Geschlechtsbestimmung der DNA-Proben erfolgen. Die
maskulin getesteten Individuen werden im weiteren Verlauf mittels PowerPlex® Y23
Kit auf Y-chromosomale STRs untersucht.
Die erhaltenen Y-STR-Daten sollen die Grundlage für die Bestimmung jeweiliger
Haplotypen bilden. Hierzu wird die Y chromosome Haplotype Reference Database
(YHRD; http://www.yhrd.org/) herangezogen. Die resultierenden Haplotypen sollen
anschließend Haplogruppen zugeordnet werden. Diese Resultate sind fundamental für
die Darstellung der Haplogruppenverteilung innerhalb des Grabungsfeldes. Um
eventuelle Ähnlichkeiten zwischen Populationen bewerten zu können, sollen die
Ergebnisse durch eine AMOVA-Analyse mit Daten der YHRD verglichen, und mit
Hilfe eines biostatistischen Programms ausgewertet werden. Das Tool wurde eigens für
die Auswertung der erhaltenen Y-chromosomalen Daten geschrieben und ist speziell
auf die Erfordernisse der Auswertung abgestimmt. Mit Hilfe dieses Tools ist eine
Betrachtung der Allelfrequenz sowie Haplotyp- und Gen-Diversität möglich. Weiterhin
wird die Haplotypfrequenz sowie Discrimination-Capacity angegeben. Im Rahmen
dieser Forschungsarbeit sollen 82 Zahnproben aufgearbeitet werden. Es erfolgt eine
DNA-Isolation sowie die Analyse autosomaler und Y-chromosomaler STR-Systeme.

3 Material
31
3 Material
3.1 Geräte und Software
Die folgende Übersicht enthält alle benötigten Geräte, Materialien und Software für die
mechanische Aufbereitung des Probenmaterials, der DNA-Isolation sowie der STR-
Analyse.
Gerät Bezeichnung Hersteller
Feinwaage CP224 S Sartorius, Hamburg
Filtermembranen MFTM- Membrane Filters, 0,05 mm V MWP Milipore
Filterpapier MN615 Macherey-Nagel, Düren
Kapillarelektrophorese-System
ABI PrismTM 310 Genetic Analyzer
Life Technologie (Applied Biosystems, Weiterstadt)
Kapillarelektrophorese-System
ABI PrismTM 3130 Genetic Analyzer
Life Technologie (Applied Biosystems, Weiterstadt)
Kugelmühle MM200 Retsch, Düsseldorf
Rotor Rotator 2-1175 neoLab, Heidelberg
Rundschale Milliporerundschalen ø47 mm Milipore
Handschleifer Dremel Multipro 395 Dremel® Europe, NL
Thermocycler GeneAmp® PCR-System 9700
Life Technologie (Applied Biosystems, Weiterstadt)
Tischzentrifuge Centrifuge 5424 Eppendorf, Hamburg
Trockenschrank BE 500 Memmert, Schwabach
UV-Schrank DNA/RNA UV-Cleaner UVC/F-M-AR
Kisker, Steinfurt
Zentrifuge Universal 320 Hettrich, Weiterstadt
Analysesoftware Firma
GenMapper® ID v3.2 (Analysesoftware ABI 310)
Life Technologies (Applied Biosystems, Weiterstadt)
GenMapper® ID-X 1.0.1 (Analysesoftware ABI 3130)
Life Technologies (Applied Biosystems, Weiterstadt)

3 Material
32
Data collection Software Firma
Data collection Software ABI 310 Life Technologie (Applied Biosystems, Weiterstadt)
Data collection Software ABI 3130 Life Technologie (Applied Biosystems, Weiterstadt)

3 Material
33
3.2 Chemikalien und Verbrauchslösungen
Nachfolgend sind alle benötigten Chemikalien gelistet, die während der entsprechenden
Arbeitsschritte verwendet wurden.
Dekalzifizierung, Lyse und Proteinase K- Verdau
Chemikalie Bezeichnung Hersteller Bestandteile
All-tissue DNA-Kit GEN-IAL®
DNA-Exitus PlusTM
A 7089, 1000 AppliChem, Darmstadt
Ethanol EtOH J.T. Baker absolut (100%)
Ethylendiamin-tetraessigsäure (EDTA)
Triplex® III (Ethylendinitrilotetraessigsäure, Dinatriumsalz- Dihydrat)
MERCK
Proteinase K
EC 3.4.21.14, 100 mg Lyophilisat, 30 m Anson U/mg, c = 20 mg/ml
MERCK
DNA-Aufreinigung mittels Phenol-Chloroform/Isoamylakohol
Chemikalie Bezeichnung Hersteller Bestandteile
Chloroform/Isoamylalkohol ((v/v) 24:1)
ROTH
Ethanol EtOH J.T. Baker
Natriumchlorid (NaCl) NaCl (5M) Kmf optiChem
Phenol ROTH
TE-Puffer Buffer AE (Elutionbuffer)
QIAGEN 10 mM Tris-HCl, 0.5 mM EDTA (pH 9,0)
TN-Puffer 10 mM Tris-HCl (pH 7,6), 10 mM NaCl

3 Material
34
Quantifizierung genomischer DNA (gDNA)
Chemikalie Bezeichnung Hersteller
Quantifiler® Human DNA Quantification Kit
Life Technologie (Applied Biosystems, Weiterstadt)
Amplifikation und Detektion der STR-Systeme
Chemikalie Bezeichnung Hersteller
Hi-DiTM Formamid Life Technologie (Applied Biosystems, Weiterstadt)
PowerPlex® S5 System Promega
PowerPlex® Y23 System Promega

4 Methoden
35
4 Methoden
Nachfolgend werden die Methoden zur Zahnaufbereitung, DNA-Isolation sowie der
molekulargenetischen Analysen mittels STR-Systemen aufgeführt und näher erläutert.
4.1 Aufarbeitung des Zahnmaterials
Da sich in früheren Studien eine DNA-Isolation aus Zahnmaterial bezüglich der DNA-
Qualität und –Quantität als effektiv erwies, erfolgte die DNA-Extraktion aus
verbliebenen Zahnproben.
Vorzugsweise wurden als Probenmaterial für die DNA-Extraktion die hinteren
Backenzähne (Molaren) aus Ober-und Unterkiefer verwendet. Aufgrund des größeren
Volumens im Vergleich zu den Premolaren, Caninen und Incisoren steigt die
Wahrscheinlichkeit auf eine höhere DNA-Ausbeute durch einen größeren Gehalt an
dentalen Pulpa-Zellen in denen sich unter anderem die gDNA befindet [SS-P+07]. Die
entfernten Zähne wurden durch Abschleifen von anheftendem Schmutz und Leimresten
befreit und die Zahnkrone entfernt. Dieser Vorgang der mechanischen Aufreinigung ist
fundamental für die Weiterverarbeitung, da durch den Abschleifungsvorgang die
Kontaminationsgefahr durch anheftende Fremd-DNA verringert wurde. Anschließend
wurden die präparierten Zähne mit 99 %igem Ethanol benetzt und mit kurzwelligem
UV-Licht behandelt. Dieser Dekontaminationsvorgang dient der Fragmentierung
anhaftender Fremd-DNA, um das Amplifikationsergebnis nicht zu beeinträchtigt. Mit
Hilfe einer Kugelmühle wurden die aufbereiteten Zähne zu feinstem Zahnmehl
zerkleinert. Die flächenmäßige Vergrößerung des Zahnmaterials bietet eine größere
Wirkungsoberfläche für die darauf folgende Dekalzifizierung. Es wurde jeweils ein
Zahn pro Individuum bearbeitet. Eine Mahlsequenz erstreckte sich über 30 sek mit
anschließender Pause (30 sek). Dies sollte den Grad der Erhitzung so gering wie
möglich halten, da sonst eine zusätzliche DNA-Schädigung entstehen würde. Nach
jedem Mahldurchgang wurden alle genutzten Materialien gründlich gewaschen und
Dekontaminationsschritte durchgeführt, um dem Risiko einer DNA-Verschmutzung mit
einer anderen Probe zu entgehen.

4 Methoden
36
4.2 Dekalzifizierung, Zelllyse und Proteinase K-Verdau
Die Basis für eine sensitive STR-Analytik bildet die Präparation hochreiner DNA aus
kleinsten Zellmengen. Daher ist es von größter Wichtigkeit im Umgang mit dem zu
bearbeitenden Zahnmaterial in voller Schutzkleindung, sowie Haar- und Mundschutz zu
hantieren, um die Kontaminierung mit Fremd-DNA so minimal wie möglich zu halten.
Grundlegende Schritte der DNA-Präparation aus Zahnmaterial ist die Dekalzifizierung
sowie die Zell- und Kernlyse.
Nach abgeschlossener Zerkleinerung der Zahnproben erfolgt die Isolation der DNA
unter Verwendung des All-tissue DNA-Kit der Firma GEN-IAL. Das gewonnene
Zahnmehl wurde zu Beginn einer Dekalzifizierung unterzogen. Hierbei handelt es sich
um das Herauslösen des Minerals Kalzium aus der Probe. Umgesetzt wird dieser
Vorgang durch den Einsatz von Ethylendiamintetraessigsäure (EDTA). EDTA ist ein
starker Komplexbildner, welcher Chelatkomplexe mit zweiwertigen Kationen eingehen
kann. Hierzu wurde 0,5 M EDTA mit einem eingestellten pH-Wert von 7,5 verwendet.
Zu Beginn des Dekalzifizierungsvorgangs wurden die Proben mit 25 mL EDTA versetzt
und über Nacht in einem Rotor bei 4 °C inkubiert. Am folgenden Tag wurden die
Proben für 10 min bei 4000 rpm zentrifugiert. Der entstandene Überstand wurde
verworfen. Die erneute Zugabe von 25 mL EDTA sowie eine anschließende Inkubation,
dreimal über Nacht und zweimal über Tag, erfolgten insgesamt fünfmal. Nach der
Dekalzifizierung wurden die Proben abschließend zentrifugiert und der Überstand
verworfen. Anschließend erfolgte eine Waschung des Zahnmehls. Hierzu wurden die
Proben mit 30 mL dest. Wasser versetzt, mit dem Vortexer gemischt und für 10 min bei
4000 rpm zentrifugiert. Der Überstand wurde verworfen. Dieser Vorgang wurde
dreimal wiederholt. Für die Zelllyse wurden dem Pellet 1 mL Lyse 1-Puffer, 100 mL
Lyse 2-Puffer, 50 µL Proteinase K sowie 25 µL Dithiothreitol- Lösung (DTT) (1 M)
zugegeben und über Nacht inkubiert. Lyse-Puffer und Proteinase K dienen in diesem
Schritt dem Zellaufschluss und der Denaturierung der Zellbestandteile. DTT ist ein
stark reduzierendes Reagenz und reduziert, unterstützend zur Proteinase-K, die
Disulfidbrücken der Proteine. Die Lösung wurde im Schüttelbad bei 56 °C über Nacht
inkubiert. Anschließend wurden 750 µL Lyse3-Puffer hinzugefügt und 20-30 sek leicht
gemischt um alle Inhibitoren und Störfaktoren der nachfolgenden PCR sowie Proteine
durch Präzipitation restlos zu entfernen [URL-4]. Die verwendeten Lyse-Puffer sowie
das Enzym Proteinase K sind Bestandteile des GEN-IAL Kits. Von den entstandenen
Lysaten wurden jeweils 1 mL in Reaktionsgefäße (2 mL) überführt. Das Proben wurde
bei -20 °C gelagert.

4 Methoden
37
4.3 DNA-Aufreinigung mittels Phenol-Chloroform/Isoamylalkohol
Um auch hier zur Minimierung bzw. Verminderung von Kontamination der DNA-
Proben während des Routineprozesses beizutragen, sowohl auch zum Selbstschutz
mussten Sicherheitsvorkehrungen getroffen werden.
Zur Beseitigung der noch in den Lysaten befindlichen Inhibitoren wird eine sogenannte
PCI-Aufreinigung durchgeführt. Effizienz und Sensitivität einer solchen
Extraktionsmethode sind dabei ausschlaggebend. Die Phenol-
Chloroform/Isoamylalkohol-Aufreinigung ist ein solches Verfahren zur Aufreinigung
von Nukleinsäuren aus wässriger Lösung. Nachteil dieser Methodik ist die Toxizität der
eingesetzten Chemikalien, der hohe Zeitaufwand sowie eine hohe geforderte Präzision.
Wird ein Arbeitsschritt nicht ordnungsgemäß und exakt ausgeführt, kann es zu
Verunreinigungen und möglichen PCR-Inhibitoren in den DNA-Isolaten kommen.
Trotz dieser Tatsache hat sich diese Extraktionsmethode durch ihre Effektivität und
Sensitivität der Aufreinigung forensischer Proben, speziell der DNA-Extraktion aus
historischem Knochen- und Zahnmaterial, etabliert. Die Ausbeute an Gesamt-DNA ist
hoch und zudem qualitativ hochwertig [KNP05].
Die im Tiefkühlschrank gelagerten Lysate wurden mit 1 mL Phenol versetzt, kräftig
durchmischt und anschließend für 15 min im Rotor vermengt und weiterhin für 15 min
bei 3000 rpm zentrifugiert. Phenol agiert hierbei als Phasentrenner von organischen
Bestandteilen und gelösten Nukleinsäuren. Nach der Zentrifugation haben sich zwei
Phasen ausgebildet. Die erste der entstandenen Phasen, in der sich hydrophile
Nukleinsäuren befinden, wurde in ein neues Reaktionsgefäß überführt und mit 0,5 mL
Phenol und 0,5 mL Chloroform/Isoamylalkohol versetzt. Zwischen der ersten und
zweiten Phase bildete sich eine sogenannte Interphase aus. In ihr lagern sich
denaturierte Proteine ab. Diese und die zweite, lipophile organische Phase, in welcher
sich Zellreste und anorganische Bestandteile absetzen, wurden verworfen. Nach dem
Versetzen mit Chloroform/Isoamylalkohol ((v/v) 24:1) wurden die Proben kräftig
durchmischt und anschließend für 15 min bei 3000 rpm zentrifugiert. Der Überstand
wurde in ein neues Reaktionsgefäß überführt. Im folgenden Schritt wurden die Proben
mit 1 mL Chloroform/Isoamylalkohol, kräftig durchmischt und für 15 min bei 3000 rpm
zentrifugiert. Chloroform dient zum Auswaschen von Phenolresten und dem entfernt
restliche lipophile organische Bestandteile aus der wässrigen Phase. Isoamylalkohol
dient zur Verhinderung von Schaumbildung und ist nicht zwingen von Nöten. Der
entstandene Überstand wurde in ein neues Reaktionsgefäß überführt. Für die
anschließende Präzipitation der DNA wurde den Proben 1 mL Ethanol (100%) sowie 40
µL NaCl (5 M) zugegeben und diese 1 h bei -20 °C inkubiert. Ethanol bewirkt das
Lockern der Hydrathülle um die DNA-Moleküle und verändert so deren Löslichkeit.
Durch Zugabe von NaCl wird die Kationenzahl in der Lösung erhöht, was ebenfalls die
Löslichkeit herabsetzt. Die Kationen des NaCl binden durch die niedrige
Dielektrizitätskonstante des Ethanols effektiv an die negativen Phosphatgruppen der
DNA und führen so zu einer Präzipitation. Die Temperatur von -20 °C unterstützt
diesen Vorgang. Nach der vorgesehenen Inkubationszeit erfolgte für 15 min eine

4 Methoden
38
Zentrifugation bei 14000 rpm. Anschließend wurde das Ethanol ab dekantiert und
überschüssiges Ethanol wurde vorsichtig abpipettiert. Das entstandene Pellet wurde zur
Trocknung 1 h bei 45 °C im Trockenschrank gelagert. Im Folgenden wurden die
getrockneten Pellets mit 50 µL TE-Puffer resuspendiert und über Nacht bei 45 °C im
Trockenschrank gelagert. Der TE-Puffer agiert dabei als Stabilisator der DNA und
verlangsamt Abbauprozesse durch Degradation [URL-5]. Zur Entsalzung der DNA-
Isolate wurde abschließend mit einer dafür vorgesehenen Milipore-Membran gearbeitet.
Dazu wurde eine Milipore-Rundschale zur Hälfte mit TN-Puffer befüllt und eine
Milipore-Membran mit der glatten Oberfläche nach oben aufgelegt. Bei dieser handelt
es sich um ein semipermeable Membran die in Kombination mit TN-Puffer das in den
Proben enthaltene Natrium herausfiltern. Vor dem Auftragen der DNA-Isolate auf die
Membran werden diese kurzzeitig für 30 sek bei 1000 rpm zentrifugiert. Im Anschluss
erfolgte das tropfenweise Auftragen zu je 25 µL (eine Probe pro Membran und maximal
sechs Tropfen) und eine Inkubation bei Raumtemperatur für 1 h bis 1,5 h. Nach dieser
Zeit wurden die Tropfen wieder aufgenommen und in ein 1,5 mL Reaktionsgefäß
überführt. Die Stellen an denen sich die Tropfen befanden wurden mit 5 µL Aqua dest.
nachgespült und ebenfalls in dasselbe Reaktionsgefäß überführt. Bis zur nachfolgenden
Quantifizierung wurden die DNA-Isolate bei -20 °C gelagert.

4 Methoden
39
4.4 DNA-Quantifizierung
Für die Bestimmung der DNA-Quantität der DNA-Isolate werden mit Hilfe einer
Quantifizierungsmethode die jeweiligen Proben analysiert. Die verwendete
Quantifizierungsmethode beruht auf einer Real-time-PCR unter Verwendung von
Hydrolysesonden (TaqMan®-Sonden). Die resultierenden Ergebnisse geben Aufschluss
über die DNA-Konzentration. Durchgeführt wurde diese Methodik nach Anleitung des
Quantifilers® Human DNA Quantification Kit der Firma Applied Biosystems. Die
Detektion erfolgte durch das firmengleiche 7500 Fast Real-Time PCR System. Die
Auswertung der generierten Daten wurde mit systemeigener SDA-Software realisiert.
Die Quantifizierung wurde unter folgenden Reaktionsbedingungen durchgeführt
(Tabelle 2). Tabelle 2: Reaktionsansatz zur DNA-Quantifizierung. Die Reaktion wurde unter Standardbedingungen durchgeführt.
Reaktionskomponenten Volumina Quantifiler Primer-Mix 10,5 µL Quantifiler PCR-Reaktions-Mix 12,5 µL Probe/Standard/IPC 2 µL
Die Methode der quantitativen Real-Time-PCR (qrtPCR) basiert auf einer durch
spezifische Primer-definierte enzymatische in vitro Replikation. Durch sich
wiederholende Replikationszyklen wird eine annähernd exponentielle Amplifikation der
Zielsequenz erreicht. Es kommt anschließend zur Messung der Produktakkumulation in
der exponentiellen Phase in Echtzeit. Umgesetzt wird diese Methodik durch
Hydrolysesonden (TaqMan®-Sonden). Diese Oligonukleotidsonden sind mit
Fluorochromen versehen. Eine solche fluorogene Sonde ist am 5´-Terminus mit einem
Reporter (Fluoreszenzfarbstoff) und am 3´-Terminus mit einem Quencher markiert.
Dieser Quencher unterbindet die Fluoreszenz des Reporters. Die Unterdrückung der
Fluoreszenz des Reporter-Farbstoffs wird durch einen Fluoreszenz-Einergietransfer
(FET) nach Förster unterbunden. Zu Beginn der Amplifikation liegen dementsprechend
die Hydrolysesonden im fluoreszenzunterdrückten Zustand vor. Um die Quantifizierung
gewünschter DNA-Abschnitte zu gewährleisten werden targentspezifische forward und
reverse Primer verwendet, welche den Bereich der Hybridisierung flankieren. Während
des Amplifikationsvorgangs hybridisiert die TaqMan®-Sonde sequenzspezifisch zur
komplementären Template-DNA. Die zur Quantifizierung genutzte Taq-Polymerase
stößt während der Primer-Hybridisierung auf die Fluoreszenz-Sonde und verdrängt
diese. Durch die 5´-3´-Exonukleaseaktivität der DNA-Polymerase wird die Fluoreszenz-
Sonde hydrolysiert. Die Hydrolyse verursacht das Abspalten des Reporters (Abbildung
10) [W03].

4 Methoden
40
Abbildung 10: Schematische Darstellung der Extensionsphase einer TaqMan® PCR [S06]. 1. Spezifische Anlagerung der TaqMan®-Sonde und eines PCR-Primers; 2. Primer-Extension und Sondenhydrolyse durch 5´-3´-Exonukleaseaktivität der DNA-Polymerase; 3. TaqMan®-Sonde wird vollständig hydrolysiert, Reporter und Quencher werden getrennt, PCR-Produkt wird synthetisiert; 4. Reportersignal wächst in Abhängigkeit der freigesetzten Reporter-Moleküle.
Aufgrund dieser räumlichen Trennung des Reporters vom Quencher fluoresziert der
Reporter [W03, S06]. Die Fluoreszenzintesität des Reporters ist proportional zur DNA-
Konzentration. Je stärker das zu detektierende Signal ist, desto mehr DNA ist in der
Probe enthalten.
1
2
3
4

4 Methoden
41
4.5 Autosomale STR-Analyse mittels PowerPlex® S5 Kit
Nach abgeschlossener Quantifizierung der DNA-Proben wurden diese anschließend
mittels PCR amplifiziert und typisiert. Die Amplifikation der DNA-Isolate erfolgte
durch das Mini STR PowerPlex® S5 Kit der Firma Promega. Ziel dieser Analyse war
eine Differenzierung zwischen femininen und maskulinen Individuen mit Hilfe des
Amelogenin-Gens. Dual konnte das Kit auf seine Effizienz der Betrachtung von aDNA-
Proben hin charakterisiert werden. Durch das Kit ist eine simultane Amplifikation der
STR-Marker D18S51, D8S1179, TH01, FGA und dem geschlechtsspezifischen Marker
Amelogenin möglich.
Die Amplifikation der DNA-Isolate splittet sich in zwei Arbeitsprozesse. Der Prä- und
Post-Amplifikation. Für die Prä-Amplifikation ist die Anfertigung eines Mastermix von
Vorteil der je Probe 5 μl PowerPlex ® S5 5x MasterMix und 2,5 μl PowerPlex® S5 10x
PrimerPairMix enthält (Tabelle 3). Das Volumen der einzusetzenden Template-DNA
variierte ja nach ermittelter Konzentration. Die einzusetzende DNA-Menge wurde auf
0,15 ng festgelegt und die eingesetzten Template-DNA auf maximal 150 pg minimiert. Tabelle 3: Reaktionsansatz des PowerPlex® S5 Kits für die Amplifikation. Die Menge der Template-DNA wurde den einzelnen Quantifizierungsergebnissen angepasst.
PCR Amplifikationskomponenten Volumina Amplifikationswasser Auf Reaktionsvolumen von 25 µl
auffüllen PowerPlex® S5 Master Mix 5 µl PowerPlex® S5 Primer Paar Mix 2,5 µl Template-DNA (0,15 ng) 2 – 17,5 µl Reaktionsvolumen 25 µl Um ausreichend auswertbare Profile der stark degradierter aDNA zu erzielen, variierte
die Zyklenzahl der PCR-Reaktion, je nach Probe, zwischen 32 und 34 Zyklen (Tabelle
4). Erfahrungswerte zeigten, dass bei einer Erhöhung der Zyklenzahl von degradierten
Spuren die Wahrscheinlichkeit eines auswertbaren Ergebnisses steigt, bzw. es zu
qualitativ hochwertigeren Amplifikationsergebnissen kommen kann. Bei stark
degradiertem DNA-Material kann jedoch eine Erhöhung der Zyklenzahl durch zu starke
Fragmentierung kein positives Ergebnis erzielen. Die Primerbindungsstellen sind
hierbei so stark modifiziert, dass eine komplette Bindung zum Teil nicht mehr möglich
ist. Durch die Degradation entstandene Artefakte werden bei einer Erhöhung der
Zyklenzahl ebenfalls stark angereichert und einer Verbesserung des Profils wird nicht
erreicht [M09].

4 Methoden
42
Tabelle 4: Amplifikationsprotokoll PowerPlex® S5. Je nach Quantifizierungsergebnis der DNA wurde die Zyklenzahl auf 32 bzw. 34 Zyklen angepasst, um ausreichend auswertbare Profile der einzelnen Systeme zu erzielen.
Amplifikationsprotokoll 96 °C für 2 min 94 °C für 30 sec 60 °C für 2 min 72 °C für 90 sec 32 – 34 Zyklen 60 °C für 45 min 4 °C abkühlen
Nach Abschluss der PCR wurden die Proben auf 4 °C herunter gekühlt und bis zur Post-
Amplifikation bei -20 °C gelagert. Im zweiten Arbeitsprozess folgt die Post-Amplifikation. Hierfür wurde ein weiterer Mastermix aus Formamid, und Standard angefertigt. Die Nachfolgende Tabelle 5 zeigt die notwendigen Komponenten sowie deren Volumina. Tabelle 5: Reaktionsansatz der Post-Amplifikation des PowerPlex® S5 Kits. Die Tabelle zeigt die einzelnen Komponenten der Post-Amplifikation. Die Template-DNA wird mit Hilfe von Formamid unter Wärmeeinwirkung denaturiert und für die folgende Kapillarelektrophorese vorbereitet.
Komponente Volumina Hi-DiTM Formamid 12 μl ILS600 Standard 0,5 µl Template-DNA/ Allelleiter 1 µl
Die Amplifikate wurden durch den Sequenzer ABI PRISM® 310 der Firma Applied
Biosystems mittels Kapillarelektrophorese detektiert. Anschließend erfolgte eine
Auswertung der Ergebnisse mit Hilfe der GenMapper® ID Software v3.2. Die
Amplifikate wurden bei -20 °C gelagert.
4.6 Haplotypisierung mittels PowerPlex® Y23 Kit
Im weiteren Verlauf wurde der Y-chromosomale Haplotyp der männlichen Individuen,
durch die Analyse von 23 spezifischen STR-Systemen, bestimmt. Die dafür genutzten
Y-STRs wurden mit Hilfe des PowerPlex® Y23 Kit der Firma Promega amplifiziert.
Dieses Multiplexkit ermöglicht eine simultane Amplifikation von 23 STR-Markern:
DYS576, DYS389I, DYS448, DYS389II, DYS19, DYS391, DYS481, DYS549,
DYS533, DYS438, DYS437, DYS570, DYS635, DYS390, DYS439, DYS392,
DYS643, DYS393, DYS458, DYS385a/b, DYS456 und Y-GATA-H4.

4 Methoden
43
Die Amplifikation wird auch hier in Prä- und Post-Amplifikation unterteilt. Für die Prä-
Amplifikation wird pro Probe ein Mastermix aus 5 µL PowerPlex® Y23 5x MasterMix
und 2,5 µL PowerPlex® Y23 10x PrimerPairMix angefertigt (Tabelle 6). Das Volumen
der einzusetzenden Template-DNA variierte ja nach ermittelter Konzentration. Die
einzusetzende DNA-Menge wurde auf 0,15 ng festgelegt. Die Menge der
einzusetzenden Template-DNA wurde auf maximal 150 pg minimiert. Tabelle 6: Reaktionsansatz des PowerPlex® Y23 Kits für die Amplifikation. Die Menge der Template-DNA wurde den vorangegangenen Quantifizierungsergebnissen angepasst.
PCR Amplifikationskomponenten Volumina Amplifikationswasser Auf Reaktionsvolumen von 25 µl
auffüllen PowerPlex® Y23 Master Mix 5 µl PowerPlex® Y23 Primer Pair Mix 2,5 µl Template-DNA (0,15 ng) 2 – 17,5 µl Reaktionsvolumen 25 µl Tabelle 7: Amplifikationsprotokoll PowerPlex® Y23. Je nach Quantifizierungsergebnis der DNA wurde die Zyklenzahl auf 32 bzw. 34 Zyklen angepasst um ausreichend auswertbare Profile der einzelnen Systeme zu erzielen.
Amplifikationsprotokoll 96 °C für 2 min 94 °C für 30 sec 60 °C für 2 min 72 °C für 90 sec 32 – 34 Zyklen 60 °C für 45 min 4 °C abkühlen
Nach Abschluss der PCR wurden die Proben auf 4 °C herunter gekühlt und bis zur Post-
Amplifikation bei -20 °C gelagert.
Bei der anschließenden Post-Amplifikation wurde ein MasterMix aus Formamid und
einem Längenstandard angefertigt. Die notwendigen Komponenten mit dazugehörigen
Volumina kann der nachfolgenden Tabelle 8entnommen werden.

4 Methoden
44
Tabelle 8: Reaktionsansatz der Post-Amplifikation des Power-Plex® Y23 Kit. Die Tabelle zeigt die einzelnen Komponenten der Post-Amplifikation. Die Template-DNA wird mit Hilfe von Formamid unter Wärmeeinwirkung denaturiert und für die folgende Kapillarelektrophorese vorbereitet.
Komponente Volumina Hi-DiTM Formamid 12 μl ILS500 Standard 0,5 µl Template-DNA/ Allelleiter 1 µl
Die zu amplifizierenden Fragmente wurden durch den Sequenzer ABI PRISM® 3130
detektiert. Anschließend erfolgte eine Auswertung der Ergebnisse mittels der
GenMapper® ID-X 1.0.1 Software. Die amplifizierten Proben werden bei -20 °C
gelagert.

4 Methoden
45
4.7 Genealogische Charakterisierung des Datensatzes
Für die Auswertung genealogischer Daten existieren verschiedene biostatistische
Methoden und Strategien die eine Einordnung in den populationsgenetischen Kontext
sowie einen Intra- und Interpopulationsvergleich zulassen. Die genetischen Variationen
innerhalb der vorliegenden Population können durch die Aufstellung einer
Allelverteilung sowie der Betrachtung der einzelnen Haplotypen und der Berechnung
der Gen- und Haplotyp-Diversität untersucht werden. Im Nachfolgenden sind die in
dieser Forschungsarbeit betrachteten Gesichtspunkte zur Charakterisierung der
vorliegenden Population und der Evaluierung neuer Daten beschrieben. Für die Analyse
der Allelverteilung, der Haplotyp- und Gen-Diversität sowie der Discrimination-
Capacity, die ein Maß für die genetische Vielfalt innerhalb einer Population darstellen,
wurde ein eigens dafür entwickeltes Programm verwendet. Bereitgestellt und entworfen
wurde dieses Programm von dem Bioinformatik-Promovend M. Sc. Herrn Florian
Heinke der Hochschule Mittweida. Die dafür genutzten Berechnungsgrundlagen werden
im Folgenden beschrieben [RK+96, CHB13, I07].
4.7.1 Haplogruppenverteilung
Mit Hilfe der durch Y-chromosomale STRs generierten Haplotypen lässt sich die
Zuordnung der Individuen zu ethnische Gruppen ermöglichen. Eine erste
Differenzierung zwischen Populationen nach Region und Herkunft ist somit umsetzbar.
Um aus den Haplotypen die entsprechenden Haplogruppen herausfiltern zu können,
bedarf es der Nutzung entsprechender Datenbanken.
4.7.2 Allelverteilung
Um den erhaltenen Datensatz genau charakterisieren zu können werden diverse Daten
aus den Ergebnissen herausgefiltert. Die Allelverteilung und –frequenz spielt dabei eine
große Rolle. Auf Grund von Formeln die dem Programm zu Grunde liegen konnten die
Allelfrequenzen für jeden Lokus berechnet werden. Um eine Verteilung aufzustellen
werden die einzelnen Loki bezüglich ihrer auftretenden Allele untersucht und die
Summe der jeweils auftretenden Allele gebildet. Anschließend kann die Allelfrequenz
berechnet werden. Sie ist ein Maß für die relative Häufigkeit eines Allels pro Lokus.
Diese ergibt sich aus folgender Formel [I07]:
��� =���
�
Formel 6: Berechnung der Allelfrequenz. Die Formel für die Berechnung der Allelfrequenz ��� ergibt sich aus der Gesamtzahl ��� der Allele ! des Lokus " und der Anzahl der Individuen N in der Population.

4 Methoden
46
4.7.3 Haplotyp-Diversität, Gen-Diversität und Discrimination-Capacity
Die Haplotyp-Diversität ist ein Maß für die Einzigartigkeit eines Haplotyps in einer
Population. Sie beschreibt somit den Anteil an unterschiedlichen Haplotypen die
innerhalb einer untersuchten Population auftreten. Je mehr identische Haplotypen in
einer Population entdeckt werden, desto kleiner wird der Diversitätswert. Die Haplotyp-
Diversität wird nach folgender Formel berechnet[CH+13]:
�# = ∗ (1 − ∑ '��
� )�
− 1
Formel 7: Berechnung der Haplotyp-Diversität. Grundlage der Berechnung bildet die Frequenz ' des Haplotyps ! in der Population j und die Gesamtzahl der Haplotypen N.
Die Haplotypfrequenz ergibt sich dabei aus der Formel für die Berechnung der
Allelfrequenz.
Die Gen-Diversität wird pro Lokus berechnet und ist ein Maß für die genetische
Vielfalt eines Lokus. Sie richtet sich nach Allelausprägung und –frequenz. Sie wird wie
folgt berechnet [CH+13]:
(# = ∗ (1 − ∑ )�
�)�
− 1
Formel 8: Berechnung der Gen-Diversität. Für die Berechnung der Gen-Diversität wird die Gesamtzahl N der Allele und die Frequenz pi des Allels i einbezogen.
Die sogenannte Discrimitation-Capacity gibt an, wie vielfältig die auftretenden
Haplotypen in Bezug auf die Anzahl der betrachteten Individuen sind. Sie berechnet
sich aus dem Verhältnis der vorkommenden Haplotypen zu der Gesamtanzahl an
betrachteten Individuen. Je mehr einzigartige Haplotypen vertreten sind, desto höher ist
der Diskriminierungswert [CH+13].
#* = #�,
Formel 9: Berechnung der Discrimination-Capacity. Grundlage für die Berechnung der Discrimination-Capacity sind die Anzahl vorkommenden Haplotypen #-. sowie die Gesamtanzahl / an betrachteten Individuen.

5 Ergebnisse
47
5 Ergebnisse
5.1 Mechanische Aufarbeitung des Zahnmaterials und DNA-Isolation
Nach abgeschlossener Bestandsaufnahme des Knochenmaterials durch vorangegangene
Forschungsarbeiten ist von einer Probenanzahl von 82 auszugehen. Sofern vorhanden,
wurde das Zahnmaterial aus dem Ober- und Unterkiefer der Individuen entfernt. In
einigen Fällen kam es vor, dass einem Individuum (Benennung durch
Individuennummern aus vorangegangenen anthropologischen Studien) mehrere Kiefer
und lose Zähne zugeteilt wurden sowie loses Zahnmaterial sichergestellt wurde. Diese
losen Zähne bekamen jeweils eine eigene Individuennummer und wurden als ein
Individuum angesehen und aufgeführt. Es ist somit möglich, dass Zahnmaterial aus
vermeintlich unterschiedlichen Individuen ein und demselben Individuum zugeordnet
werden kann. Anhand der erstellten DNA-Profile konnte die Zusammengehörigkeit von
Ober- und Unterkieferproben eines Individuums bestätigt, und eine eventuelle
Zusammengehörigkeit einiger Proben angenommen werden.
Der allgemeine Zustand der Zähne kann als verwertbar beschrieben werden. Die
Zahnproben wurden mechanisch aufgereinigt und zu Zahnmehl zerkleinert. Je nach
aufbereiteter Lysatmenge ergaben sich nach anschließender PC/I-Aufreinigung die
einzelnen DNA-Isolat-Volumina. Diese lagen im Bereich von 180 µL bis 240 µL.
5.2 DNA-Quantifizierung
Die Quantifizierung der DNA-Isolate erfolgte unter Verwendung des Quantifilers®
Human DNA Quantification Kit der Firma Applied Biosystems. Die Detektion wurde
durch das firmengleiche 7500 Fast Real-Time PCR System durchgeführt. Die
Quantifizierungsergebnisse der einzelnen Proben zeigten dabei deutliche
Schwankungen zwischen 0,000 ng/µL und 0,15 ng/µL. Einige Proben wiesen eine
unbestimmte DNA-Konzentration auf.
5.3 Geschlechtsbestimmung anhand autosomaler STR-Analysen mittels
PowerPlex® S5 Kit
Die unter Verwendung des PowerPlex® S5 Kit durchgeführte Bestimmung von vier
autosomalen STR-Systemen sowie dem geschlechtsspezifischen Gen Amelogenin
diente in erster Linie der genotypischen Geschlechtsbestimmung der einzelnen
Individuen. Durch eine Analyse der erhaltenen Ergebnisse mittels GenMapper® ID
Software v3.2 konnte eine Differenzierung der erhaltenen DNA-Profile in feminin und
maskulin vorgenommen werden. Wurde in einem STR-System nur ein Peak detektiert,
erfolgte bei ausreichender Peak-Höhe die Einstufung im Sinne eines homozygoten
Ergebnisses. Je nach Degradationsstatus der einzelnen Proben entstanden qualitativ
unterschiedliche Profile. Bei einigen DNA-Profilen kam es zu Farbdurchschlägen oder
zu sogenannten Artefakten, die durch Strand-Slippage entstehen. Diese nicht
zuordenbaren Peaks erschweren die korrekte Interpretation der Profile und wurden

5 Ergebnisse
48
durch Verdünnung des DNA-Isolats einer erneuten Post-Amplifikation unterzogen. Die
Analyse von DNA-Isolaten niedriger Konzentration lieferten häufig unzureichende
Profile in denen durch Allelic Drop-outs keine Aussage zum Geschlecht getroffen
werden konnte. Weiterhin trat das Phänomen auf, dass in einigen Proben der
Amelogenin-Markers (AMEL) nicht bestimmt werden konnte. In einigen Fällen wurde
außerdem ein abweichendes Ergebnis zu den morphologischen Resultaten erzielt. In
nahezu allen DNA-Profilen ließ sich ein Abfall der RFU bei steigender Fragmentlänge
feststellen. Zurückzuführen ist dies auf den DNA-Degradationsgrad durch lange
Lagerungszeit des Skelettmaterials im Boden und den ausgesetzten
Witterungsbedingungen. Das folgende DNA-Profil einer analysierten Probe konnte
erfolgreich als maskulin identifiziert werden. Es ist deutlich ein X- und ein Y-Allel zu
erkennen. Weiterhin konnten in allen autosomale STR-Systemen heterozygote Allele
detektiert werden. Aufgetretene Stutter-Peaks konnten nach näherer Betrachtung sowie
einem folgenden Ausschlussverfahren ignoriert werden.
Abbildung 11: Elektropherogramme (PowerPlex® S5). Dieses Elektropherogramm zeigt deutliche heterozygote Allele in jedem STR-System. Anhand des hier detektierten Amelogenin-Markers kann die Probe als eindeutig männlich identifiziert werden. Mit einem „?“ gekennzeichnete Peaks konnten anhand der Software keinem Allel zugeordnet werden da sie nicht auf der Allelleiter liegen. Da sie in ihren Fragmentlängen identisch mit den im System FGA aufgetretenen Allelen sind, handelt es sich um Farbdurchschläge. Sie sind nicht aussagekräftig und somit unrelevant. Der mit einem „?“ gekennzeichnete Peak mit einer Fragmentlänge von 109,85 bp ist ebenfalls nichtig, da es sich hier um einen weiteren Farbdurchschlag des Allels 9,3 im System TH01 handelt.
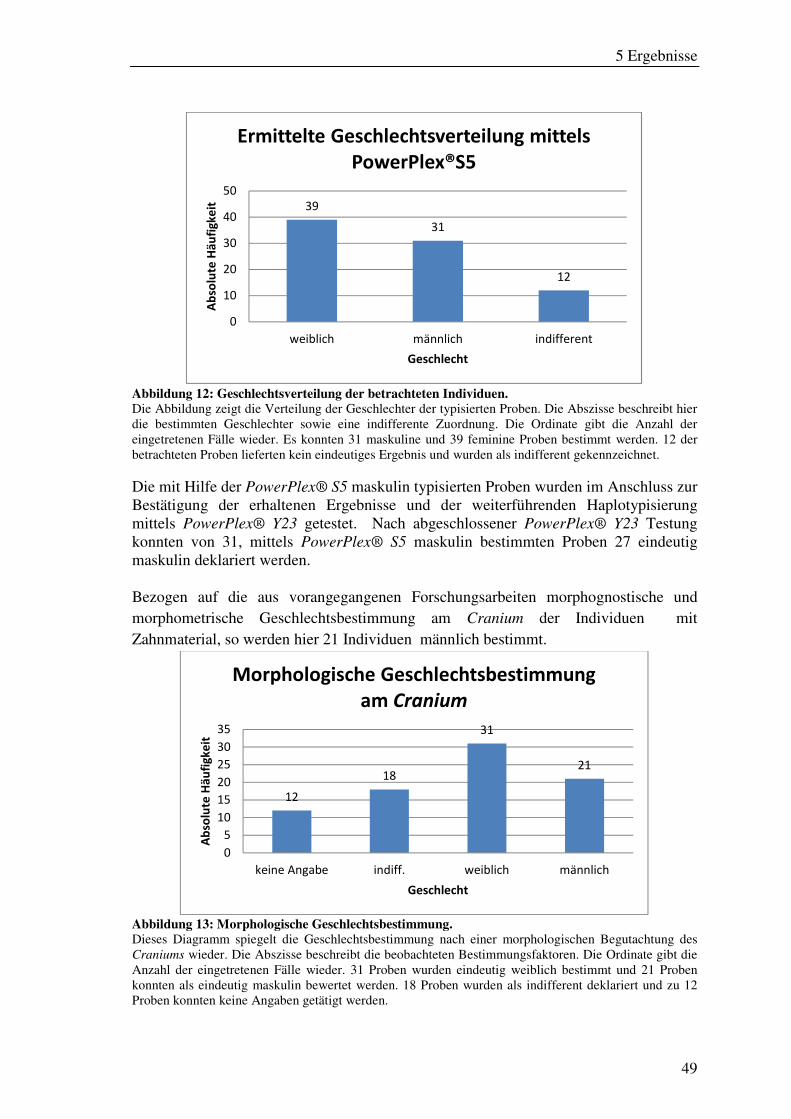
5 Ergebnisse
49
Abbildung 12: Geschlechtsverteilung der betrachteten Individuen. Die Abbildung zeigt die Verteilung der Geschlechter der typisierten Proben. Die Abszisse beschreibt hier die bestimmten Geschlechter sowie eine indifferente Zuordnung. Die Ordinate gibt die Anzahl der eingetretenen Fälle wieder. Es konnten 31 maskuline und 39 feminine Proben bestimmt werden. 12 der betrachteten Proben lieferten kein eindeutiges Ergebnis und wurden als indifferent gekennzeichnet.
Die mit Hilfe der PowerPlex® S5 maskulin typisierten Proben wurden im Anschluss zur Bestätigung der erhaltenen Ergebnisse und der weiterführenden Haplotypisierung mittels PowerPlex® Y23 getestet. Nach abgeschlossener PowerPlex® Y23 Testung konnten von 31, mittels PowerPlex® S5 maskulin bestimmten Proben 27 eindeutig maskulin deklariert werden. Bezogen auf die aus vorangegangenen Forschungsarbeiten morphognostische und
morphometrische Geschlechtsbestimmung am Cranium der Individuen mit
Zahnmaterial, so werden hier 21 Individuen männlich bestimmt.
Abbildung 13: Morphologische Geschlechtsbestimmung. Dieses Diagramm spiegelt die Geschlechtsbestimmung nach einer morphologischen Begutachtung des Craniums wieder. Die Abszisse beschreibt die beobachteten Bestimmungsfaktoren. Die Ordinate gibt die Anzahl der eingetretenen Fälle wieder. 31 Proben wurden eindeutig weiblich bestimmt und 21 Proben konnten als eindeutig maskulin bewertet werden. 18 Proben wurden als indifferent deklariert und zu 12 Proben konnten keine Angaben getätigt werden.
39
31
12
0
10
20
30
40
50
weiblich männlich indifferent
Ab
solu
te H
äu
fig
ke
it
Geschlecht
Ermittelte Geschlechtsverteilung mittels
PowerPlex®S5
12
18
31
21
0
5
10
15
20
25
30
35
keine Angabe indiff. weiblich männlich
Ab
solu
te H
äu
fig
ke
it
Geschlecht
Morphologische Geschlechtsbestimmung
am Cranium

5 Ergebnisse
50
Abbildung 14: Vergleich der molekulargenetischen und morphologischen Geschlechtsbestimmung. Die Abbildung veranschaulicht die mit Hilfe der beiden Methoden bestimmte Geschlechtsverteilung. Die dunkelblau hinterlegten Balken bezeichnen die mit Hilfe der molekulargenetischen PowerPlex® S5 Analyse durchgeführte Genotypisierung. Die hellblau unterlegten Balken veranschaulichen die morphologische Geschlechtsbestimmung. Die Abszisse spiegelt die einzelnen Bestimmungsfaktoren wieder. Die Ordinate beschreibt die Anzahl der eingetretenen Fälle. Es wird deutlich, dass weder bei der maskulinen, noch bei der femininen Geschlechtsbestimmung eine Übereinstimmung vorliegt. Die weibliche Geschlechtsbestimmung durch beide Methoden weist eine Differenz von acht auf. Die männliche Betrachtung zeigt einen Unterschied von elf.
Werden die Ergebnisse der morphologischen Geschlechtsbestimmung mit denen der
DNA-Typisierung verglichen, so ergeben sich Unterschiede. Eine Übereinstimmung der
Geschlechtsbestimmung wiesen 21 feminin bestimmte und 14 maskulin deklarierte
Proben auf. Elf Proben wurden durch die molekulargenetische und morphologische
Bestimmung differente Geschlechter zugewiesen. Anhand fehlender Vergleichswerte
durch unzureichende molekulargenetische DNA-Typisierung sowie unmögliche
morphologische Begutachtung konnte eine Zuordnung der restlichen 36 Proben nicht
erfolgen.
5.4 Haplotypisierung mittels PowerPlex® Y23 Kit
Nach abgeschlossener Geschlechtsbestimmung erfolgte die Haplotypisierung der
maskulin getesteten Individuen. Mittels PowerPlex® Y23 Kit wurden 23 gonosomale
STR-Systeme amplifiziert. Die ermittelten STR-Profile eines DNA-Isolats werden als
Haplotyp bezeichnet. Die eruierten Haplotypen der einzelnen Individuen tragen zur
geografischen Eingrenzung der Herkunft bei und geben Aufschluss über eine mögliche
Migration. Der Allelstatus auf den jeweiligen STR-Loci ist im Gegensatz zu den
PowerPlex® S5-Pofilen homozygot, d.h. es wird nur ein Allel pro STR-System
ausgeprägt. Liegt in dem jeweiligen STR-System jedoch eine Mutation in Form einer
Duplikation vor, werden zwei Allele ausgebildet. Wie bereits im vorangegangenen
Kapitel (Autosomale STR-Analyse mittels PowerPlex® S5 Kit) beschrieben, hängt die
39
31 31
20
8
19
3
11
0
5
10
15
20
25
30
35
40
45
weiblich männlich indifferent keine Angabe
Ab
solu
te H
äu
fig
ke
it
Geschlecht
Vergleich der molekulargenetischen und
morphologischen Gelschlechtsbestimmung

5 Ergebnisse
51
Qualität der Profile von der jeweiligen DNA-Konzentration sowie dem Grad der
Degradation ab.
Abbildung 15: Elektropherogrammausschnitt eines mit PowerPlex® Y23 amplifizierten DNA-Isolats. Anhand dieses Profilausschnitts eines Unterkiefers lässt sich der Degradationsprozess sehr gut verdeutlichen. Ab einer Fragmentlänge von 140 bp nimmt die Signalstärke der einzelnen Systeme zunehmend ab. Mit zunehmenden Fragmetlängen konnten Ausfälle in den jeweiligen STR-Systemen beobachtet werden. Diese Allelic Drop-outs können in Folge der auftretenden DNA-Degradation entstanden sein.
DNA-Isolate denen mittels Genotypisierung kein eindeutiges Geschlecht zugeordnet
werden konnte, wurden anhand einer Y-SNP-Testung auf ihr Geschlecht hin bestimmt.
Weiterhin wurden Proben, die unzureichende Ergebnisse der Haplotypisierung lieferten,
mit Hilfe der Y-SNPs erneut typisiert. Für solche Zwecke werden ancestry SNPs
verwendet. Diese „Vorfahren-informativen-Marker“ sind definierte Sätze von
Einzelbasen-Polymorphismen zur Differenzierung von DNA-Sequenzen. Diese
unterscheiden sich im Wesentlichen in ihrer Frequenz zwischen einzelnen Populationen
aus verschiedenen geografischen Regionen der Welt. Unter Verwendung von
Multiplex-Assays wurde dieses 52 Y-SNP Panel im Rahmen einer laufenden
Graduierungsarbeit etabliert und dient parallel zu den Y-STRs zur
Herkunftsbestimmung.
Nach einem Abgleich der Profile untereinander ergaben sich Parallelen die auf eine
Zusammengehörigkeit schließen lassen. Proben aus einem Ober- und Unterkiefer
wurden zu einem Individuum zusammengefasst. Aus den 82 typisierten Proben wird so
eine Individuen Anzahl von 55 angenommen. Daraus ergaben sich 22 maskulin
typisierte Individuen. Eine Zusammenfassung zu diesen war für die darauffolgende
Haplogruppenbestimmung innerhalb der betrachteten Population notwendig.
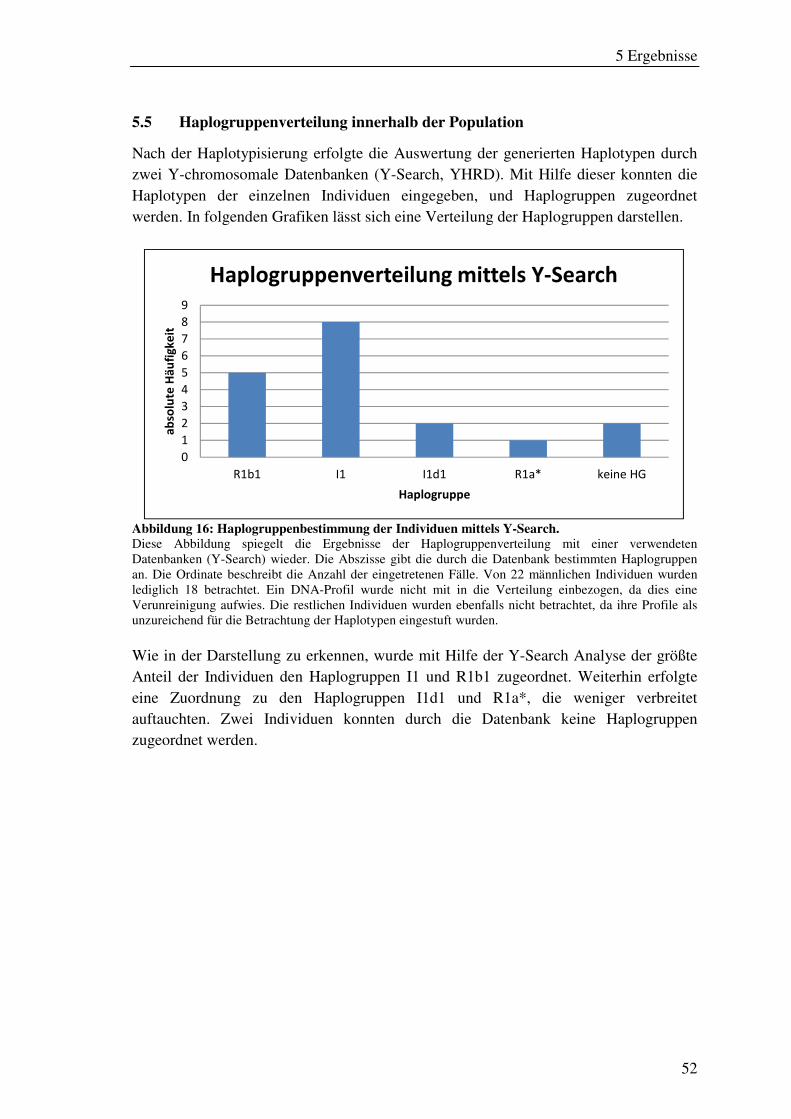
5 Ergebnisse
52
5.5 Haplogruppenverteilung innerhalb der Population
Nach der Haplotypisierung erfolgte die Auswertung der generierten Haplotypen durch
zwei Y-chromosomale Datenbanken (Y-Search, YHRD). Mit Hilfe dieser konnten die
Haplotypen der einzelnen Individuen eingegeben, und Haplogruppen zugeordnet
werden. In folgenden Grafiken lässt sich eine Verteilung der Haplogruppen darstellen.
Abbildung 16: Haplogruppenbestimmung der Individuen mittels Y-Search. Diese Abbildung spiegelt die Ergebnisse der Haplogruppenverteilung mit einer verwendeten Datenbanken (Y-Search) wieder. Die Abszisse gibt die durch die Datenbank bestimmten Haplogruppen an. Die Ordinate beschreibt die Anzahl der eingetretenen Fälle. Von 22 männlichen Individuen wurden lediglich 18 betrachtet. Ein DNA-Profil wurde nicht mit in die Verteilung einbezogen, da dies eine Verunreinigung aufwies. Die restlichen Individuen wurden ebenfalls nicht betrachtet, da ihre Profile als unzureichend für die Betrachtung der Haplotypen eingestuft wurden.
Wie in der Darstellung zu erkennen, wurde mit Hilfe der Y-Search Analyse der größte
Anteil der Individuen den Haplogruppen I1 und R1b1 zugeordnet. Weiterhin erfolgte
eine Zuordnung zu den Haplogruppen I1d1 und R1a*, die weniger verbreitet
auftauchten. Zwei Individuen konnten durch die Datenbank keine Haplogruppen
zugeordnet werden.
0
1
2
3
4
5
6
7
8
9
R1b1 I1 I1d1 R1a* keine HG
ab
solu
te H
äu
fig
ke
it
Haplogruppe
Haplogruppenverteilung mittels Y-Search

5 Ergebnisse
53
Abbildung 17: Haplogruppenbestimmung der Individuen mittels Y-Search. Diese Abbildung spiegelt die Ergebnisse der Haplogruppenverteilung mit der zweiten verwendeten Datenbanken (YHRD) wieder. Die Abszisse gibt die durch die Datenbank bestimmten Haplogruppen an. Die Ordinate beschreibt die Anzahl der eingetretenen Fälle. Von 22 männlichen Individuen wurden lediglich 18 betrachtet. Ein DNA-Profil wurde nicht mit in die Verteilung einbezogen, da dies eine Verunreinigung aufwies. Die restlichen Individuen wurden ebenfalls nicht betrachtet, da ihre Profile als unzureichend für die Betrachtung der Haplotypen eingestuft wurden.
Anhand der eingegebenen Haplotypen erfolgte eine gröbere bzw. oberflächlichere Einteilung durch die YHRD. Es konnte lediglich eine Zuordnung in die zwei Haupt-Haplogruppen R und I erfolgen. Durch die ermittelten Haplogruppen konnte eine geografische Eingrenzung erfolgen. Da
die Haplogruppen R1b1 und I1d1 Untergruppen der Haupt-Haplogruppen R1b und I1
sind, werden hier ausschließlich diese Haplogruppen betrachtet. Wie in Abbildung 18
zu erkennen, ist die Haplogruppe R1b vorwiegend in West Europa, der Atlantischen
Küste sowie Teilen Russlands zu finden. Die Haplogruppe I1 erstreckt sich über die
Gebiete Nordeuropas wie Skandinavien und Finnland. Die Haplogruppe R1a ist
hauptsächlich in Polen und der Ukraine zu finden sowie in Teilen der Slowakei.
Ganzheitlich betrachtet kann davon ausgegangen werden, dass diese Haplogruppen
vorwiegend im europäischen Raum zu Hause sind bzw. dort ihren Ursprung haben und
sich über angrenzende Gebiete durch Migration ausbreiteten [URL-6]. Die Verteilung
der Haplogruppe R1b verlief in nördliche und südliche, als auch in östliche Richtung bis
hin in den asiatischen Raum. Die Haplogruppe R1a mit ihrer Quelle in Osteuropa sowie
der Ukraine verbreitete sich im mittel- und nordeuropäischen Raum sowie in West- und
Mittelasien. Vom Ursprung der Haplogruppe I1 verbreitete sich diese in ganz Europa
sowie in angrenzenden asiatischen Gebieten.
5
1
12
0
2
4
6
8
10
12
14
I R keine HG
ab
solu
te H
äu
fig
ke
it
Haplogruppen
Haplogruppenverteilung mittels YHRD
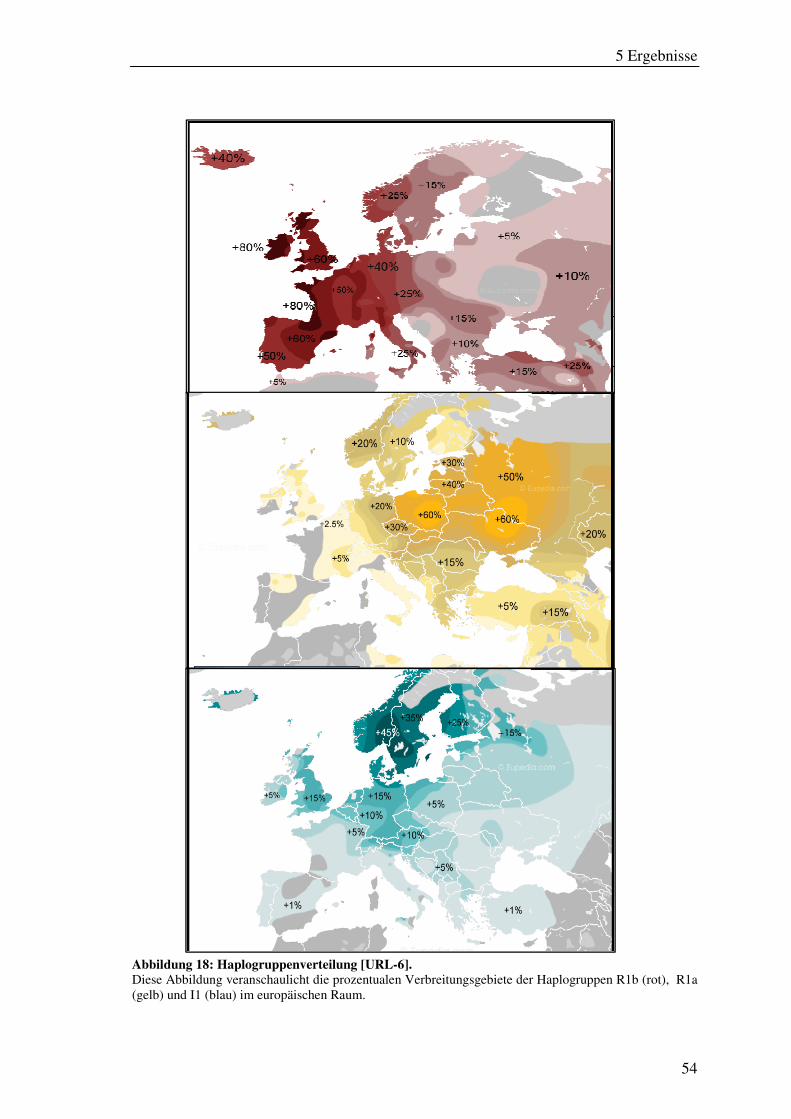
5 Ergebnisse
54
Abbildung 18: Haplogruppenverteilung [URL-6]. Diese Abbildung veranschaulicht die prozentualen Verbreitungsgebiete der Haplogruppen R1b (rot), R1a (gelb) und I1 (blau) im europäischen Raum.

5 Ergebnisse
55
5.6 Auswertung der genealogischen Daten
Im nachfolgenden werden die Ergebnisse der biostatistischen Auswertung und die
Ergebnisse der verwendeten Datenbank (YHRD) dargelegt.
5.6.1 Allelverteilung
Für die Charakterisierung des Datensatzes wurden die Allelverteilungen der jeweiligen
Loki bestimmt. Angegeben wird hier die Anzahl eines auftretenden Allels pro Lokus.
Für das STR-System DYS576 wird die Allelverteilung beispielhaft dargestellt. Die
Allelverteilungen der gesamten Loki werden im Anhang (Abbildung Anhang 1-3)
dargestellt.
Abbildung 19: Säulendiagramm der Allelverteilung für den Lokus DYS576. Abgebildet ist ein Auszug der mit Hilfe des PowePlex® Y23 Kits erhaltenen Allelverteilungen. Betrachtet wird hier das STR-System DYS576. Die Abszisse stellt die aufgetretenen Allele sowie die ausgefallenen Allele (n.a.) dar. Die Ordinate enthält die jeweiligen Allelanzahlen der in diesem System aufgetretenen Allele.
Die Allelfrequenz beschreibt die relative Häufigkeit des Auftretens eines Allels auf einem Lokus. Folgend wird beispielhaft die Allelfrequenz des Lokus DYS570 dargestellt. In Tabelle 9: Übersicht der ermittelten Allelfrequenzen aller Loci findet sich eine Übersicht der berechneten Allelfrequenzen für jeden Lokus. Die Allelfrequenzen der gesamten Loci werden im Anhang (Abbildung Anhang 4-6) dargestellt.
Abbildung 20: Säulendiagramm der Allelfrequenz des Lokus DYS570. Abgebildet ist hier ein Auszug der ermittelten Allelfrequenzen. Berechnet wurden diese mit Hilfe des bereitgestellten Programms. Betrachtet wird hier das STR-System DYS570. Die Abszisse gibt die aufgetretenen Allele sowie die ausgefallenen Allele (n.a.) wieder. Die Ordinate enthält die berechneten Frequenzen der in diesem System aufgetretenen Allele.
0
2
4
6
8
10 12 14 16 18 20 22 24 26 28 9
Ab
s. H
äu
fig
ke
it
Allele
Allelverteilung pro Lokus
DYS576
0
0,1
0,2
0,3
10 13 16 19 22 25 28 n.a.
All
elf
req
ue
nz
Allel
DYS570

5 Ergebnisse
56
Tabelle 9: Übersicht der ermittelten Allelfrequenzen aller Loci. Zu sehen ist hier die Gesamtheit der ermittelten Allelfrequenzen. Die erste Spalte beinhaltet die betrachteten Loki. Die erste Zeile stellt alle aufgetretenen Allele sowie die Anzahl der in diesem System ausgebliebenen Allele (n.a.) dar.
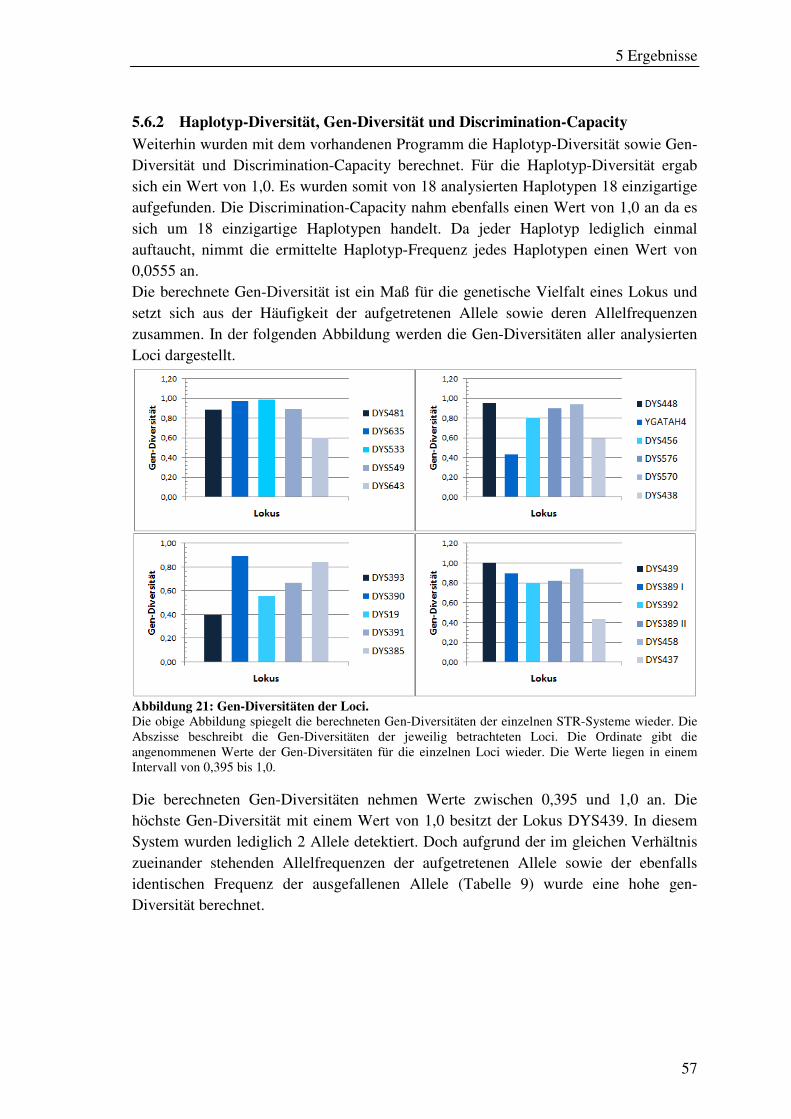
5 Ergebnisse
57
5.6.2 Haplotyp-Diversität, Gen-Diversität und Discrimination-Capacity
Weiterhin wurden mit dem vorhandenen Programm die Haplotyp-Diversität sowie Gen-
Diversität und Discrimination-Capacity berechnet. Für die Haplotyp-Diversität ergab
sich ein Wert von 1,0. Es wurden somit von 18 analysierten Haplotypen 18 einzigartige
aufgefunden. Die Discrimination-Capacity nahm ebenfalls einen Wert von 1,0 an da es
sich um 18 einzigartige Haplotypen handelt. Da jeder Haplotyp lediglich einmal
auftaucht, nimmt die ermittelte Haplotyp-Frequenz jedes Haplotypen einen Wert von
0,0555 an.
Die berechnete Gen-Diversität ist ein Maß für die genetische Vielfalt eines Lokus und
setzt sich aus der Häufigkeit der aufgetretenen Allele sowie deren Allelfrequenzen
zusammen. In der folgenden Abbildung werden die Gen-Diversitäten aller analysierten
Loci dargestellt.
Abbildung 21: Gen-Diversitäten der Loci. Die obige Abbildung spiegelt die berechneten Gen-Diversitäten der einzelnen STR-Systeme wieder. Die Abszisse beschreibt die Gen-Diversitäten der jeweilig betrachteten Loci. Die Ordinate gibt die angenommenen Werte der Gen-Diversitäten für die einzelnen Loci wieder. Die Werte liegen in einem Intervall von 0,395 bis 1,0.
Die berechneten Gen-Diversitäten nehmen Werte zwischen 0,395 und 1,0 an. Die
höchste Gen-Diversität mit einem Wert von 1,0 besitzt der Lokus DYS439. In diesem
System wurden lediglich 2 Allele detektiert. Doch aufgrund der im gleichen Verhältnis
zueinander stehenden Allelfrequenzen der aufgetretenen Allele sowie der ebenfalls
identischen Frequenz der ausgefallenen Allele (Tabelle 9) wurde eine hohe gen-
Diversität berechnet.

6 Diskussion
58
6 Diskussion
6.1 STR-Analysen zur Geschlechtsbestimmung und Haplotypisierung
Um Aussagen hinsichtlich des Geschlechts der Individuen treffen zu können, fand eine
DNA-Typisierung mit Hilfe vier autosomaler STR-Systeme sowie dem
geschlechtsspezifischen Marker Amelogenin statt. Anschließend wurde die Analyse
gonosomaler STR-Systeme zur Haplotypisierung der maskulinen Individuen
durchgeführt. Erschwert wurde die Auswertung der Profile durch das Auftreten von
Artefakten und Verunreinigungen. Einige DNA-Profile wiesen nicht eindeutig
durchgängige STR-Merkmale auf, die aufgrund von Allelic Drop-outs hervorgerufen
worden. Allelic Drop-outs bezeichnen eine Ausprägung von Artefakten und verursachen
nicht reproduzierbare STR-Merkmale. Hervorgerufen wird dieser Effekt
möglicherweise durch eine zu starke Degradation der aDNA. Diese erschwert zudem
die Detektion zunehmender Fragmentlängen. Weiterhin ist ein stetiger Abfall der RFU
(Decay Curve) mit steigender Fragmentlänge zu beobachten, was auf die Degradation
der aDNA zurückzuführen ist. Aufgrund zu starker Fragmentierung in Folge der
Degradation werden die Primer-Bindestellen zerstört und eine Hybridisierung der
Primer an das Template ist nicht mehr möglich. STR-Systeme ab einer Länge von 140
bp konnten so nur erschwert bis gar nicht detektiert werden. Eine weitere Ursache für
das Ausbleiben von Signalen können Punktmutationen auf den Bindestellen der Primer
verursachen. Durch die punktuelle Veränderung können die Primer nicht mehr an der
Zielsequenz hybridisieren und es kommt zu einem Signalausfall. Eine weitere Form
von Artefakten sind Stutter-peaks. Diese Signale treten meist vor oder nach einem
Hauptpeak aufgrund ihrer geringen Höhe gut von einem Hauptpeak zu unterscheiden.
Ursache dieser Signalausprägung ist das sogenannte Strand-Slippage welches durch das
Verrutschen der DNA-Polymerase während der Replikation verursacht wird und zu
einer Schlaufenbildung des neusynthetisierten Stranges führt. In der folgenden
Reparatur wird der überschüssige Strang jedoch nicht herausgeschnitten, sondern der
Matritzenstrang erweitert. Es entstehen so Stutter-peaks die entweder um eine
Wiederholungseinheit kürzer oder länger als der Hauptpeak sind.
Weitere Probleme entstanden durch den geschlechtsspezifischen Marker Amelogenin.
Bei diesem geschlechtsspezifischen Amelogenin-System handelt es sich nicht um ein
STR-System sondern um die Sequenz des Gens, welches für das Protein Amelogenin
codiert das zur Zahnschmelzbildung beiträgt. Dieses Gen liegt auf beiden
Geschlechtschromosomen (AMELX, AMELY) in unterschiedlicher Ausprägung vor, und
kann somit zur Geschlechtsdifferenzierung genutzt werden. Verwendete
sequenzspezifische forward und reverse Primer amplifizieren für das X-Chromosom
eine Sequenz des Intron 1, welche eine Länge von 106 bp aufweist. Das Amplikon des
Y-Chromosoms ist mit einer Länge von 112 bp um sechs Basenpaare länger. Ein
weiblicher Genotyp prägt somit einen Peak aus, während bei dem männlichen Genotyp

6 Diskussion
59
zwei Peaks erwartet werden. Werden nach abgeschlossener Genotypisierung die
Elektropherogramme betrachtet, so erfolgt die Zuordnung eines DNA-Profils zu einem
femininen Genotyp ausschließlich über den fehlenden Y-Peak und einen homozygot
ausgeprägten X-Peak. Die Zuordnung zu einem maskulinen Genotyp geschieht nach der
dualen Detektion eines X- und Y-Peaks. Einigen Profilen konnten aufgrund eines
Ausfalls dieses Markers und durch Artefakt-Anhäufungen auf dem Amelogenin-Lokus
kein Geschlecht zugeordnet werden. Eine mögliche Ursache eines Ausfalls des
Amelogenin-Systems könnte durch eine Deletion im AMELY hervorgerufen werden
oder es entstehen Allelic Drop-outs durch zu starke aDNA-Degradation oder
Mutationen an den Primer-Bindungsstellen. Weiterhin könnten zu hohe DNA-
Konzentrationen ein Rauschen und Anhäufungen von Stutter-Peaks im Profil erzeugen.
Hier wäre eine Verdünnung der Post-Amplifikation mit anschließender Typisierung
denkbar. Eine weitere Möglichkeit Interpretationsfehler zu umgehen, wäre die
Sequenzierung des spezifischen Amelogenin-Lokus.
Profile, bei denen aufgrund der DNA-Typisierung ein männlicher Genotyp nur vermutet
werden konnte, wurden durch spezifische Y-chromosomale Marker mittels PowerPlex®
Y23 Kit getestet. In den meisten Fällen konnten Y-chromosomale STR-Systeme
nachgewiesen, und der maskuline Genotyp bestätigt werden. Trotz der Anfälligkeit auf
Fehlinterpretation dieses Systems bietet der Amelogenin-Marker eine schnelle und
einfache Methode der Geschlechtsbestimmung. Erhaltene Ergebnisse sollten, in Bezug
auf das Alter der Proben und der fortschreitenden DNA-Degradation jedoch immer mit
einer gewissen Vorsicht interpretiert werden sowie weiteren molekulargenetischen
Untersuchungen unterzogen werden, um eine Fehlbestimmung zu umgehen.
Weiterhin traten Allel-Duplikationen auf einigen STR-Systemen auf. Da es bei der Y-
chromosomalen Haplotypisierung zu einer Ausprägung homozygoter Allele kommt, ist
von einer Veränderung durch Mutation auszugehen. Dem System DYS385 konnten in
14 von 18 haplotypisierten Profilen Duplikationen nachgewiesen werden. Grund für
diese hohe Anzahl an Duplikationen ist jedoch die Besonderheit, dass das System
DYS385 ein Multi-Kopie-Lokus ist, welche typischer Weise zwei Kopien ausbilden. Er
wird in DYS385a und DYS385b unterschieden. Hier liegt kein Mutationsereignis vor.
Weiterhin konnten in sechs von 18 getesteten Profilen Duplikationen detektiert werden
die aufgrund von Mutation entstanden. Insgesamt wurden 13 Duplikationen in den
STR-Systemen DYS390, DYS391, DYS448, DYS456, DYS576, DYS570, DYS481,
DYS635 und DYS549 festgestellt werden. Das auftretende Phänomen der Duplikation
entsteht durch unbalancierte Aberration. Dabei kommt es zu einer Verdopplung eines
Genabschnitts durch das illegitime crossing-over von homologen Chromosomen oder
Schwesterchromatiden während der, in der Spermatogenese ablaufenden, Meiose. In
einem Haplotypisierungsprofil traten in sechs STR-Loki Duplikationen auf. Aufgrund
der Annahme, dass eine Mutation nur aller 1000 Generationen pro Lokus auftritt, kann
von einem verunreinigten DNA-Isolat ausgegangen werden [DK+07].
Anhand der gemessenen RFU der Duplikationen, welche sich in einem Verhältnis von
1:2 ausprägten, wird zwischen einem Haupt- und Nebenverursacher differenziert.

6 Diskussion
60
Artefakte können hier ausgeschlossen werden, da alle Peaks eine RFU größer 50
aufwiesen und somit als echt angesehen werden können. Da keine Y23-Profile der
männlichen Bearbeiter erstellt wurden, konnte kein Profil-Abgleich und somit keine
Identifizierung des Haupt- und Nebenverursachers erfolgen. Zur Klärung dieses
Sachverhalts wäre eine nochmalige DNA-Isolation der Probe mit anschließender
Typisierung denkbar. Eine weitere Möglichkeit bestände durch die Haplotypisierung
der männlichen Bearbeitungspersonen und einem Abgleich des verunreinigten DNA-
Profils.
Für die Genotypisierung der aDNA-Isolate kam das PowerPlex® S5 Kit zum Einsatz. In
Anbetracht der Gesamtzahl der im Kit enthaltenen STR-Systeme zu den detektierten,
konnte das PowerPlex® S5 Kit im Vergleich zu anderen Multiplex-Kits überzeugen
[HR+12]. Weiterhin schnitt das Kit bei der durchschnittlichen Detektion der STR-
Systeme und Amelogenin gut ab und befindet sich im Mittelfeld. Die in Tests
analysierte und relativ hohe Amplifikations-Rate von über 50 % ist im Vergleich zu
anderen Multiplex-Kits, welche mit einer größeren Anzahl an STR-Markern arbeiten,
jedoch weniger aussagekräftig. Trotzdem ist das S5 Kit nützlich und empfehlenswert in
Form einer Screening-PCR, um die DNA-Qualität kostengünstig zu überprüfen
[HR+12]. Für die Haplotypisierung wurde das PowerPlex® Y23 Kit verwendet. Es
enthält neben den 17, in den meisten kommerziell erhältlichen Kits sechs weitere stark
aussagekräftige STR-Marker. Diese sechs weiteren Marker besitzen eine höhere Gen-
Diversität und erlauben eine bessere Unterscheidung nicht verwandter männlicher
Individuen. Darüber hinaus sind zwei schnell mutierende Loki enthalten, die eine
Differenzierung verwandter Individuen ermöglichen [TE+2012]. Dieses
Amplifikationskit erweist sich als eine robuste und sensitive Methode zur Y-STR-
Detektion und erlaubt auch mit geringen Mengen an aDNA eine zuverlässige
Amplifikation die eine hohe Resistenz gegenüber PCR-Inhibitoren aufweist. Den
generierten STR-Profile der verwendeten PowerPlex® S5 und Y23 Kits konnten, bis
auf wenige Ausnahmen, balancierte und verwertbare Peaks entnommen werden.
6.3 Vergleich der molekulargenetischen und morphologischen
Geschlechtsbestimmung
Anhand des Vergleichs der molekulargenetischen und morphologischen
Geschlechtsbestimmung lassen sich, wie in Abbildung 14 veranschaulicht, einige
Unterschiede feststellen. Es fand von insgesamt 55 Individuen lediglich eine
Übereinstimmung des Geschlechts bei 14 femininen und 10 maskulinen bestimmten
Individuen statt. In einigen Fällen konnte sowohl auf molekulargenetischer als auch
morphologischer Ebene keine genaue Aussage getroffen werden. Ursache der
unzureichenden Genotypisierung kann auf einen zu hohen Degradationsgrad der aDNA
sowie vorliegende PCR-Inhibitoren und zu starke Artefaktbildung zurückzuführen sein.
Eine wiederholte Analyse mittels PowerPlex® S5 Kit wäre hier ratsam. Zur

6 Diskussion
61
Genotypisierung abweichende morphologische Ergebnisse können auf einem schlechten
Erhalt der Knochen und einer zu starken Fragmentierung des Skelettmaterials basieren.
Abweichungen in der Bestimmung könnten anhand einer wiederholten Aufnahme der
Geschlechtsmerkmale überprüft und gegebenenfalls verbessert werden.
6.2 Haplogruppenverteilung innerhalb der Population
Mit Hilfe der verwendeten Datenbanken (YHRD, Y-Search) konnte eine
Charakterisierung des Datensatzes bezüglich der Herkunft vorgenommen werden. Dabei
wird ersichtlich, dass sich die Ergebnisse der Haplogruppen-Zuordnung sich
voneinander unterscheiden. Verursacht wird dies durch die unterschiedlichen Daten und
Parametern, die den Datenbanken zu Grunde liegen. Betrachtet man die Y-Search, so ist
diese von ihrem Datenumfang kleiner als die YHRD und ihre Daten werden durch
verschiedene Nutzer generiert und evaluiert. Eine Haplogruppen-Zuordnung erfolgt hier
durch die Angabe der gewünschten Gen-Diversität zu vergleichender Haplotypen was
eine spezifischere Suche zulässt. Jedoch hängt der Sucherfolg von den eingespeisten
Haplotypen hab. Eine individuelle Parametereinstellung ist bei Benutzung der YHRD
nicht möglich. Datenbankintern erfolgt hier ein Abgleich des eingegebenen Haplotyps
mit in der Datenbank hinterlegten Haplogruppen. Aufgrund der häufigen Allel-Ausfälle
konnte der Großteil der eingegebenen Haplotypen nicht bestimmt werden da zu wenig
Allele für einen Vergleich zur Verfügung standen. Ein weiterer Punkt könnte die
fehlende Permutation von aufgetretenen Allel-Duplikationen sein, die in der
Auswertung durch die YHRD nicht beachtet wurde. Diese Tatsache kann eine mögliche
Ursache für die grobe Haplogruppen-Einordnung darstellen. Eine erneute
Haplogruppenbestimmung mit vorheriger Permutation wäre denkbar und von Vorteil
für die evtl. Bestätigung der Ergebnisse und einer genaueren Zuordnung. Diese starken
Unterschiede zwischen den verwendeten Datenbanken können so auf die differenten zu
Grunde liegenden Datensätze zurückzuführen sein sowie auf die Tatsache, dass es sich
um eine historische Population handelt. Die populationsgenetische Betrachtung
historischer Knochenfunde und deren Einordnung in den populationsgenetischen
Kontext stehen noch am Anfang. So liegen momentan noch keine ausreichenden
Referenzdaten zur genauen Herkunftsbestimmung vor und es kann nur eine grobe
Einordnung erfolgen. Aufgrund der hohen Allelic Drop-out-Anzahlen in nahezu allen
DNA-Profilen, ist die genaue Haplogruppen-Einordnung weiterhin fraglich. Da die
Datenbanken für die Haplotyp-Betrachtung nur die detektierten Allele mit einbeziehen,
lassen die ausgefallenen Allele Raum für Fehlbestimmungen. Eine erneute DNA-
Isolation mit anschließender Haplotypisierung wäre aufgrund des hohen
Bearbeitungsaufwandes und dem begrenzt zur Verfügung stehenden DNA-Material nur
bedingt denkbar.
Aufgrund von Kenntnissen der Verbreitung der betrachteten Haplogruppen (Abbildung
18) die ausnahmslos das Gebiet Deutschland mit einbeziehen, kann von der Korrektheit
der erhaltenen Haplogruppen-Ergebnisse ausgegangen werden. Wie in Abbildung 22 zu
erkennen ist, treffen die Haplogruppen R1a, R1b und I1 während ihrer

6 Diskussion
62
Migrationsbewegung im mitteleuropäischen Raum aufeinander. Es ist somit nicht
abwegig, diese Haplogruppen des geborgenen Knochengrabes bei Görzig vorzufinden.
Abbildung 22: Schematische Darstellung der Haplogruppen-Migration [W08]. Zu sehen ist hier die schematische Darstellung der Migrationswege der einzelnen Haplogruppen aus dem
Ursprungsland Afrika. Grau hinterlegte Haplogruppen spiegeln die genetisch weiter entwickelten
Haplogruppen wieder. Farbig hinterlegt sind die ungefähren geografischen Verbreitungsgebiete der vier
Haupt-Populationen. Die roten, gelben, blauen und orangenen Pfeile veranschaulichen die
Wanderungsbewegungen der Populationen während der Rekolonisation Nordeuropas in den letzten
10.000 bis 16.000 Jahre. Die grünen Pfeile deuten auf die Besiedlung des Balkans und der
Mittelmeerküste durch frühzeitige Bauern in den letzten 10.000 Jahren hin. Die beigefügten Jahreszahlen
zu jeder Haplogruppe beschreiben die vergangenen Jahrtausende.
Vergleicht man die heutige Verteilung der Haplogruppen in Abbildung 7 (Kapitel 1.5
Formulierung von Haplotypen) mit dem Ursprung und der angenommenen Verbreitung
der Haplogruppen (Abbildung 22), können Parallelen gezogen werden. Mit dem Fall
des Römischen Reiches vor rund 2000 Jahren begann die Wanderung der Teutonen
nach Italien und der Zusammenschluss der römischen und germanischen Zivilisation,
welcher den Ursprung der westlichen europäischen Population markiert und die
Haplogruppe R1b in diesen Regionen verankerte. Abweichend zu früheren
Ausbreitungsgebieten der Haplogruppen, dominiert die Haplogruppe R1b in dem Gebiet
in und um Deutschland herum. Wie in Abbildung 7 zu erkennen, finden sich jedoch
immer noch Regionen in diesem Gebiet, die durch die Migrationsbewegung die
Haplogruppen R1a und I1 ausprägen. Im Gegensatz dazu erfuhr der Westen Europas ein
Ausbleiben der Migration nomadischer Populationen was die Beständigkeit der

6 Diskussion
63
Haplogruppe R1a in Westeuropa erklärt. Im Laufe der populationsgenetischen
Entwicklung kam es so durch Migrationsereignisse wie Besiedlung, Kolonisation und
diversen Einwanderungswellen zur Vermischung des Genpools der
Ursprungspopulationen mit ausländischen Einwanderern. Dies führte zur Ausprägung
weiterer Haplogruppen die sich in den jeweiligen Gebieten etablierten. So sind
unteranderem Einwanderunswellen aus dem asiatischen Raum sowie Teilen des
Schwarzen Meeres nachweisbar [R00]. Es ist demnach anzunehmen, dass in der Zeit
des Umbruch und der Einwanderung vor ca. 2000 Jahren mit der Besiedlung von
Ländern um Europa der Grundstein für die jetzige Haplogruppenverteilung gelegt
wurde. Eine erkennbare Abgrenzung der heutigen Haplogruppen-Gebiete voneinander
prägte sich zu einen durch die vorgegebenen Siedlungsgebiete der Haplogruppen sowie
herrschende klimatische Unterschiede, geografische Gegebenheiten wie Gebirgen oder
entstandenen Landesgrenzen aus.
6.4 Aussagekraft der biostatistischen Ergebnisse
Die Ergebnisse der biostatistischen Auswertung bieten eine weitere Grundlage für die
Forschung auf dem Gebiet der Populationsgenetik. Anhand der genealogischen
Auswertung kann eine erste Datenevaluierung erfolgen und eine Einordnung in den
populationsgenetischen Hintergrund vorgenommen werden. Aufgrund eines hohen
Allel-Ausfalls durch Allelic Drop-outs und deren Einbeziehung in die biostatistische
Auswertung ist die Aussagekraft der erhaltenen Ergebnisse unstrittig. Um
aussagekräftigere Ergebnisse zu erzielen wäre eine erneute Haplotypisierung mit
anschließender biostatistischer Betrachtung notwendig.
Für weitere Betrachtungen des Datensatzes im Hinblick auf Vergleiche zwischen Inter-
und Intra-Populationsdaten kann eine molekulare Varianzanalyse durchgeführt werden.
Mit Hilfe des AMOVA-Tools kann der Umfang an Unterschieden zwischen zwei oder
mehreren Populationsdaten bewerten und quantifiziert werden. Anhand der molekularen
Varianz σ2 kann die durchschnittliche Entfernung eines Haplotypes eines Indivviduums
zu jedem anderen betrachtet und die Nähe von Populationen zueinander in einem MDS-
Plot grafisch dargestellt werden. So können genauere Aussagen über
Populationsbeziehungen zueinander auf Basis von Haplotypen getroffen werden.

7 Zusammenfassung und Ausblick
64
7 Zusammenfassung und Ausblick
Aggregierend aus den Ergebnissen der autosomalen und Y-chromosomalen STR-
Analysen kann abschließend gesagt werden, dass die Charakterisierung der Individuen
des Gräberfeldes Görzig hinsichtlich des Geschlechts und der Haplotypisierung sowie
Haplogruppen-Verteilung der maskulinen Individuen erfolgreich war. Die Kombination
aus exakt durchgeführten Arbeitsmethoden der Aufarbeitung und DNA-Isolation sowie
der anschließenden molekulargenetischen Untersuchung lieferten aussagekräftige
Ergebnisse die eine Einordnung in den populationsgenetischen Hintergrund zuließen.
Hilfestellung bei diesen genografischen Fragestellungen bildeten die dafür generierten
Datenbanken, welche einen Vergleich mit hinterlegten Daten ermöglichte. Die erlangten
Resultate tragen zum Fortschritt populationsgenetischer Fragestellungen bei und dienen
der Datenerweiterung- und evaluierung historischer Populationen.
Fortführend ist die Klärung verwandtschaftlicher Beziehungen innerhalb der
untersuchten Population denkbar. Weiterhin könnte die bisher erfolgreich
durchgeführte DNA-Typisierung zum Ausschluss von Fehlerquellen wiederholt, und
mit Hilfe neuer Prä- und Post-Amplifikate eine Verbesserung der
Haplotypisierungsergebnisse erzielt werden. Außerdem könnten in Bezug auf die
Qualität und –Quantität extrahierter DNA Vergleiche zwischen Zahn-und
Knochenmaterial erfolgen. Ein weiterer Gesichtspunkt fortwährender Arbeiten könnte
die Durchführung einer AMOVA-Analyse sein sowie die zusätzliche Testung von
Verwandtschaftsgraden mit Hilfe X-chromosomaler STRs sowie die Haplotypisierung
auf maternaler Linie sein.
Für weitere erfolgversprechende genomweite Assoziationen bleibt die
Charakterisierung von Referenzpopulationen unabdingbar [R08]. Wünschenswert wären
zudem die vollständige Etablierung der Haplotypisierungstechnik sowie die
systematische Evaluierung und Anpassung der molekulargenetischen Methoden sowie
deren internationale Harmonisierung [R10]. Das bis heute erlangte Wissen über die
Entstehung und Charakterisierung von Haplotypen und ihre Anwendung auf
historisches sowie rezentes Knochenmaterial ist vorerst der Grundbaustein für weitere
Forschungen auf dem Gebiet der Populationsgenetik. Auch sind die Analysen auf dem
Gebiet der Rekonstruktion von Episoden der Humangenetik noch am Anfang. Durch die
extreme Stratifizierung der Y-chromosomalen Haplotypen werden Y-STR-Systeme
immer häufiger zur Bestimmung der ethnografischen und geografischen Herkunft eines
männlichen Knochenfundes angewendet.

Literaturverzeichnis
65
Literaturverzeichnis
[KNP05] Köchl, S.; Niederstätter, H.; Parson, W. (2005): DNA Extraction and
Quantitation of Forensic Samples Using the Phenol-Chloroform Method
and Real-Time PCR Methods. Molecular Biology, Forensic DNA Typing Protocols 297
[S06] Schild, T. A. (2006); Applied Biosystems GmbH; Einführung in die
Real-Time TaqMan® PCR-Technologie
[W03] Wilhelm, J. (2003); Entwicklung real-time-PCR-basierter Methoden für
die moderne DNA-Analytik, Justus-Liebig-Universität Gießen [K05] Krause, D. (2005); Eine weltweite Datenbank für Allelfrequenzen
autosomaler STR-Systeme, Universitätsklinikum Münster Institut für Rechtsmedizin
[BH+99] Burger, J., Hummel, S., Herrmann, B., Henke, W. (1999); DNA
preservation: A microsatellite-DNA study on ancient skeletal remains, Electrophoresis 1999
[P89] Pääbo, S. (1989); Ancient DNA: Extraction, characterization, molecular
cloning, and enzymatic amplification, Proc. Natl. Acad. Sci. USA [RH07] Rohland, N., Hofreiter, M. (2007); Ancient DNA extraction from bones
and teeth, Max Planck Institute for Evolutionary Anthropology [I07] Immel, U. -D. (2007); Ausgewählte Aspekte und Anwendungen Y-Short
Tandem Repeats (Y-STRs), Martin-Luther-Universität Halle-Wittenberg [H06] Haak, W. (2006); Populationsgenetik der ersten Bauern Mitteleuropas
Eine aDNA-Studie an eolithischem Skelettmaterial, Johannes Gutenberg Universität Mainz
[B00] Bemmann, J. (2000); „Mitteldeutschland in der jüngeren Römischen
Kaiserzeit und Völkerwanderungszeit – eine von den Körperbestattungen
ausgehende Studie (2000) “
[K11] Kleinecke, J. (2011);„Das Gräberfeld Görzig, Kr. Köthen - eine
archäologische und anthropologische Untersuchung (2011)“
[PSH08] PSH Pop’- & Evol’biol (2008); Populationsgenetik: Gene und ihre
Frequenzen, Kap. 3: Gene und ihre Frequenzen
[M00] ]Müller, S. (2000); Populationsgenetische Untersuchungen der
Tetranukleotid Tandem Repeat Polymorphismen D11S488, D18S51,

Literaturverzeichnis
66
D19S246, HUMFIBRA und HUMVWFA31/A, Johann Wolfgang Goethe-Universität Frankfurt am Main
[SS-P+07] Ricardo Henrique Alves da SILV1, Arsenio SALES-PERES, Rogério Nogueira de OLIVEIRA, Fernando Toledo de OLIVEIRA, Sílvia Helena de Carvalho SALES-PERES2 (2007); USE OF DNA TECHNOLOGY IN
FORENSIC DENTISTRY; Jornal of Applied Oral Science, Vol.: 15, No.: 3
[SG+06] Marı´a Lourdes Sampietro, M. Thomas P. Gilbert, Oscar Lao, David Caramelli, Martina Lari, Jaume Bertranpetit, Carles Lalueza-Foxk (2006); Tracking down Human Contamination in Ancient Human Teeth; Molecular Biology and Evolution, Vol.: 23, No.: 9
[HS+98] Hidding, M., Schmitt, C., Broicher, C., Staak M. (1998); Zwei Beispiele
zur Anwendbarkeit Y-chromosomaler DNA-Polymorphismen in der
forensischen Spurenanalytik, Institut für Rechtsmedizin, Universität zu Köln. Springer-Verlag 1998
[L13] Labudde, D. (2013); Bioinformatik und Forensik, Skript 2013 [WR03] Wiegand, P., Rolf, B. (2003) Analyse biologischer Spuren, Rechtsmedizin Springer-Verlag 2003 [R08] Roewer, L. (2008) Populationsgenetik des Y-Chromosoms, medizinische
Genetik Springer Verlag [L09] Lünnemann, P. A. (2009); Analyse binärer Y-chromosomaler
Polymorphismen in unterschiedlichen Populationen [K05] Kaiser, C. (2005); Untersuchungen zur Degradation von DNA in
Knochen und deren Anwendbarkeit für die Liegezeitbestimmung von
Skelettfunden
[B08] Balogh, M. K. (2008); Forensisch relevante SNP-Genotypisierung
mittels elektronischer Microarray-Technologie
[BGG04] Bundesgesundheitsbl - Gesundheitsforsch - Gesundheitsschutz (2004):
Genetische Polymorphismen (Sequenzvariationen) von Fremdstoff-
metabolisierenden Enzymen und ihre Bedeutung in der Umweltmedizin Springer Medizin Verlag
[H12] Harthun, M. (2012) Forensische DNA-Analysen – Einarbeitung in
autosomale und gonosomale STR-Analysen, Projektarbeit [W08] Weigand, N. (2008): Multiplex Polymerasekettenreaktion mit
fluoreszenzoptischer Produktdetektion durch den Einsatz von Primern
mit „primerintegrierten Reportersequenzen“

Literaturverzeichnis
67
[R08] Reuss, E. (2008); Anwendungen der PCR in der forensischen DNA-
Analyse, ZENTRUM DER RECHTSMEDIZIN, JOHANN WOLFGANG GOETHE-UNIVERSITÄT FRANKFURT
[G06] Graf, M. (2006); Analytik von Proteinen in der Kapillarelektrophorese,
Fakultät für Lebenswissenschaften der Technischen Universität Carolo-Wilhelmina zu Braunschweig
[WR13] Willuweit, S., Roewer, L. (2013); The Y-Chromosome Haplotype
Reference Database – Directions for Use –, Institute of Legal Medicine and Forensic Sciences, Charité – Universitätsmedizin Berlin
[HW09] Holsinger, K.E., Weir, B.S. (2009); Genetics in geographically
structured populations: defining, estimating and interpreting FST, Fundamental ConCepts in GenetiCs, Vol.: 10
[M09] Müller, K.V. (2009); Optimierung der DNA-Analyse für degradierte
biologische Spure, Medizinische Fakultät der Universität Ulm [RK+96] Roewer, L., Kayser, M., Dieltjes, P., Nagy, M., Bakker, E., Krawczak,
M., de Knijff , P. (1996); Analysis of molecular variance (AMOVA) of Y-
chromosome-specific microsatellites in two closely related human
populations, Hum Mol Genet 5:1029–1033 [CH+13] Coble, M., Hill, C., Butler, J.M. (2013); Haplotype data for 23 Y-
chromosome markers in four U.S. population groups, Forensic Science International: Genetics 7 (2013) e66–e68
[DK+07] Decker, A.E., Kline, M.C., Redman, J.W., Reid, T.M., Butler, J.M.
(2007); Analysis of mutations in father–son pairs with 17 Y-STR loci, Forensic Science International: Genetics Vol.: 2
[HR+12] Harder, M., Renneberg, R., Meyer, P., Ben Krause-Kyoral, B., von
Wurmb-Schwark (2012), N.; STR-typing of ancient skeletal remains:
which multiplex-PCR kit is the best?, FORENSIC SCIENCE Croat Med Nr.: 53: 416-422
[TE+2012] Thompson, J.M., Ewing, M.M., Frank W.E., Pogemiller, J.J., Craig A.
Nolde, C.A., Koehler, D.J., Shaffer, A.M., Rabbach D.R., Fulmer, P.M., Cynthia J. Sprecher, C.J., Storts, D.R. (2012); Developmental validation
of the PowerPlex1 Y23 System: A single multiplex Y-STR analysis system
for casework and database samples, Forensic Science International: Genetics
[BL+14] Balitzki, B., Laberke, P.J., Jegge, L., Kübler, E. (2014);
Geschlechtsbestimmung mit dem Amelogeninsystem Kasuistische

Literaturverzeichnis
68
Betrachtung möglicher Fehlerquellen, Rechtsmedizin Springer-Verlag Berlin Heidelberg
[F00] Foernzler, D. (2000); SNPs – kleine Unterschiede mit großer Wirkung,
BioWorld, Juni 2000 [R00] Roewer, L. (2000); Die Haplotypisierung des Y-Chromosoms –
Grundlagen und Anwendungen einer neuen molekulargenetischen
Identifizierungsmethode, Medizinische Fakultät der Humboldt-Universität zu Berlin (Charité)
[W08] Wiik, K. (2008); WhereDid European Men Come From?, Journal of
Genetic Genealogy Internetquellen [SH07] Schulte, T. ; Hummes, S. (2007): Der genetische Fingerabdruck aus
historischen Skeletten DNA-Analysen zur Verwandtschaftsfeststellung
der Individuen der Wittekindsburg; 50-55 URL: http://www.gefao.de/bilder/publikation/AiO04-PDF/AiO4-06.pdf
[URL-1] (05.02.2014), gutefrage.net GmbH; Markus Wölflick: Chromosomensatz des Menschen URL: http://upload.wikimedia.org/wikipedia/commons/2/21/DNA_human_male _chromosomes.gif [URL-2] (06.07.2014) niceWeb: Y-Chromosom URL: http://bio3400.nicerweb.com/Locked/media/ch07/Y_chromosome.html [URL-3] (12.10.2014), igenia xenDach; Haplogruppen URL: http://www.igenea.com/forum/threads/y-dna-haplogruppen-karten.26/ [URL-4] (19.05.2014) Chemgapedia Wiley Information Services GmbH: EDTA-
Komplexe; Proteinase K, Lyse-Puffer URL: http://www.chemgapedia.de [URL-5] (01.07.2014) Open WetWare, Etienne Robillard: TE-Buffer URL: http://openwetware.org/wiki/TE_buffer [URL-6] (23.06.2012) Eupedia, eupedia.com: Y-chromosomal Haplogroups URL: http://www.eupedia.com/europe/origins_haplogroups_europe.shtml [URL-7] (11.06.2014) Heinz-Ulrich Hartung : Deutschland URL: http://www.up-up-and-away.info/html/deutschland.html [URL-8] (11.06.2014) GoogleMaps: Görzig URL: https://www.google.de/maps/place/G%C3%B6rzig/@51.6716328,11.9988272 ,9174m/data=!3m1!1e3!4m2!3m1!1s0x47a67228a9d3cf93:0xa01e49465c655762
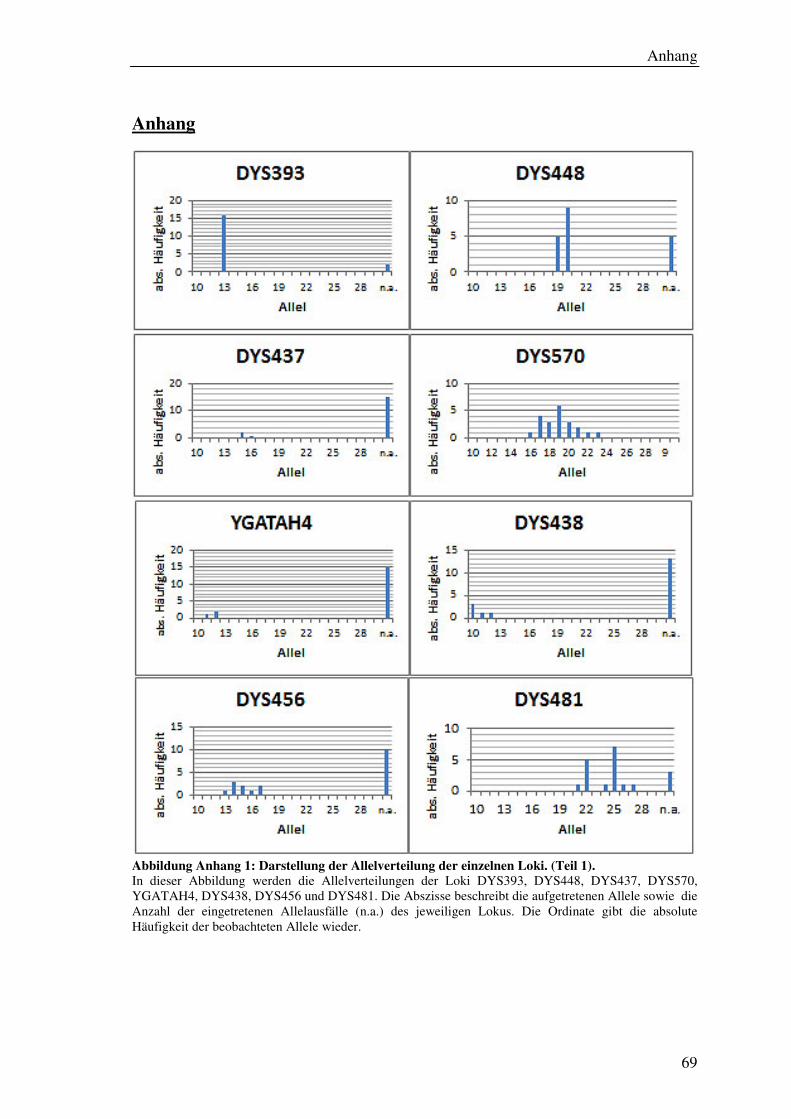
Anhang
69
Anhang
Abbildung Anhang 1: Darstellung der Allelverteilung der einzelnen Loki. (Teil 1). In dieser Abbildung werden die Allelverteilungen der Loki DYS393, DYS448, DYS437, DYS570, YGATAH4, DYS438, DYS456 und DYS481. Die Abszisse beschreibt die aufgetretenen Allele sowie die Anzahl der eingetretenen Allelausfälle (n.a.) des jeweiligen Lokus. Die Ordinate gibt die absolute Häufigkeit der beobachteten Allele wieder.
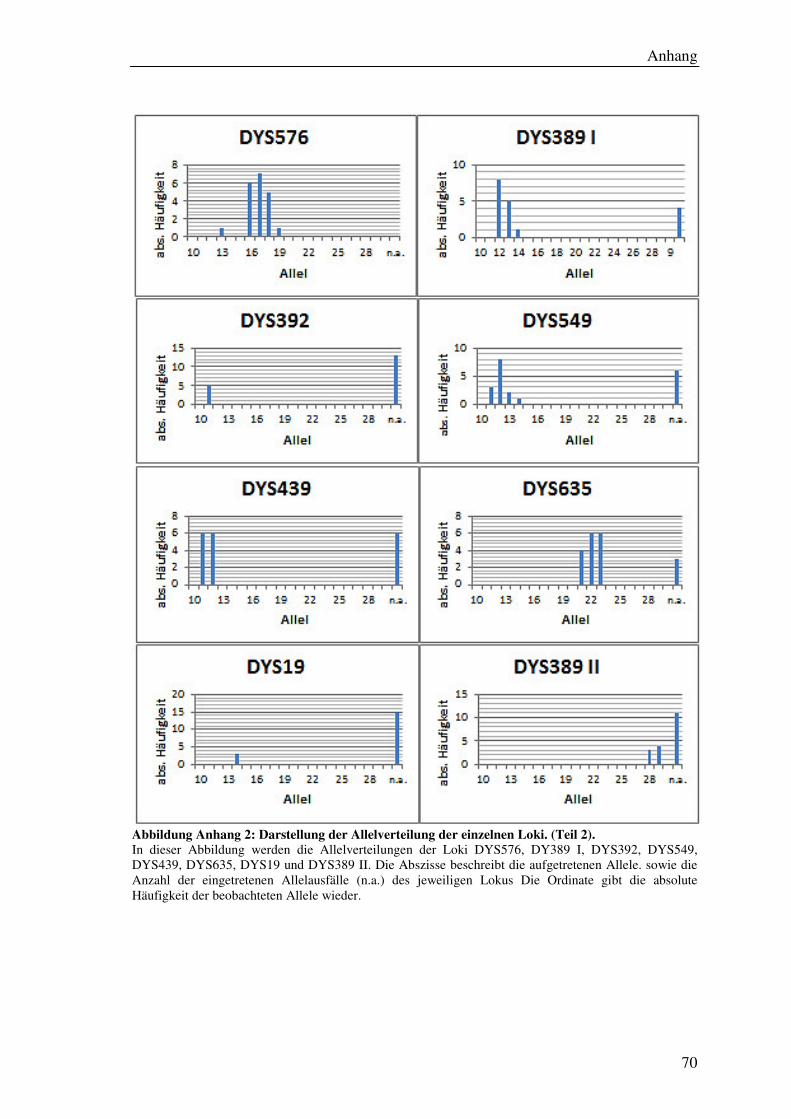
Anhang
70
Abbildung Anhang 2: Darstellung der Allelverteilung der einzelnen Loki. (Teil 2). In dieser Abbildung werden die Allelverteilungen der Loki DYS576, DY389 I, DYS392, DYS549, DYS439, DYS635, DYS19 und DYS389 II. Die Abszisse beschreibt die aufgetretenen Allele. sowie die Anzahl der eingetretenen Allelausfälle (n.a.) des jeweiligen Lokus Die Ordinate gibt die absolute Häufigkeit der beobachteten Allele wieder.
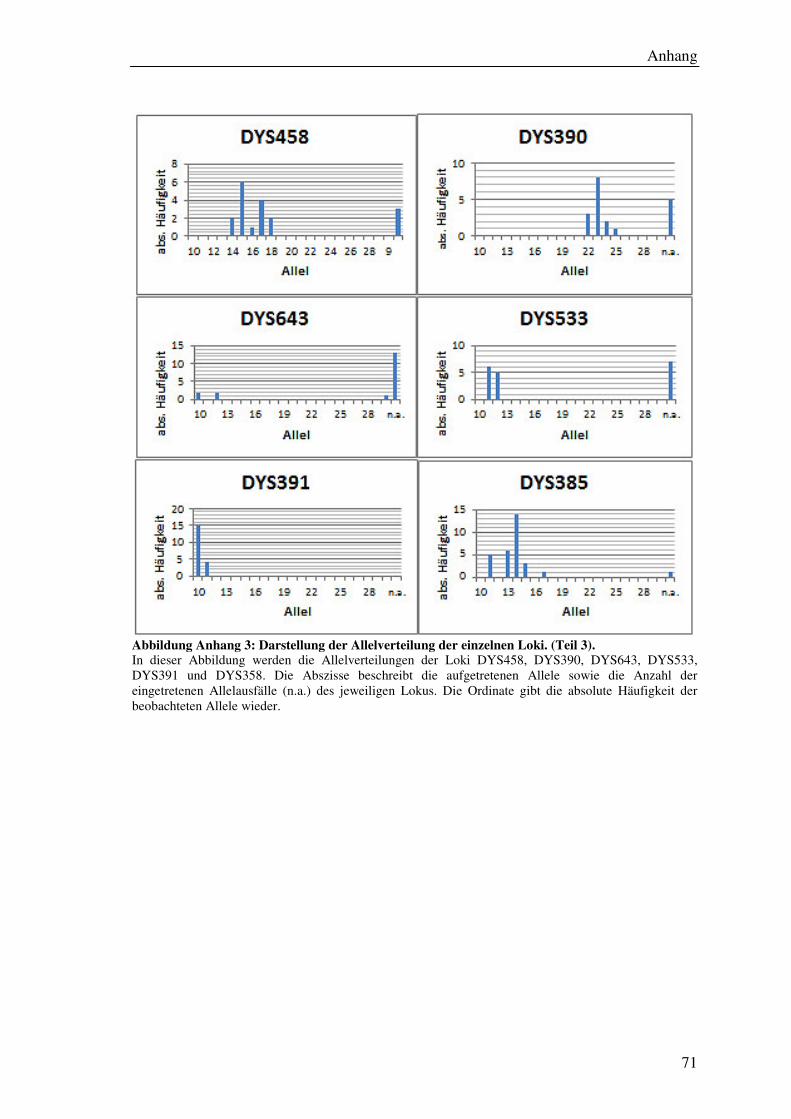
Anhang
71
Abbildung Anhang 3: Darstellung der Allelverteilung der einzelnen Loki. (Teil 3). In dieser Abbildung werden die Allelverteilungen der Loki DYS458, DYS390, DYS643, DYS533, DYS391 und DYS358. Die Abszisse beschreibt die aufgetretenen Allele sowie die Anzahl der eingetretenen Allelausfälle (n.a.) des jeweiligen Lokus. Die Ordinate gibt die absolute Häufigkeit der beobachteten Allele wieder.

Anhang
72
Abbildung Anhang 4: Darstellung der Allelfrequenzen der einzelnen Loki. (Teil 1). Diese Abbildung spiegelt die Allelfrequenzen der einzelnen Loki DYS393, DYS390, DYS439, DYS392, DYS389 II, DYS448, DYS458 und DYS437. Die Abszisse beschreibt die aufgetretenen Allele sowie die Anzahl der eingetretenen Allelausfälle (n.a.) des jeweiligen Lokus. Die Ordinate gibt die berechnete Allelfrequenz der aufgetretenen Allele im jeweiligen System wieder.
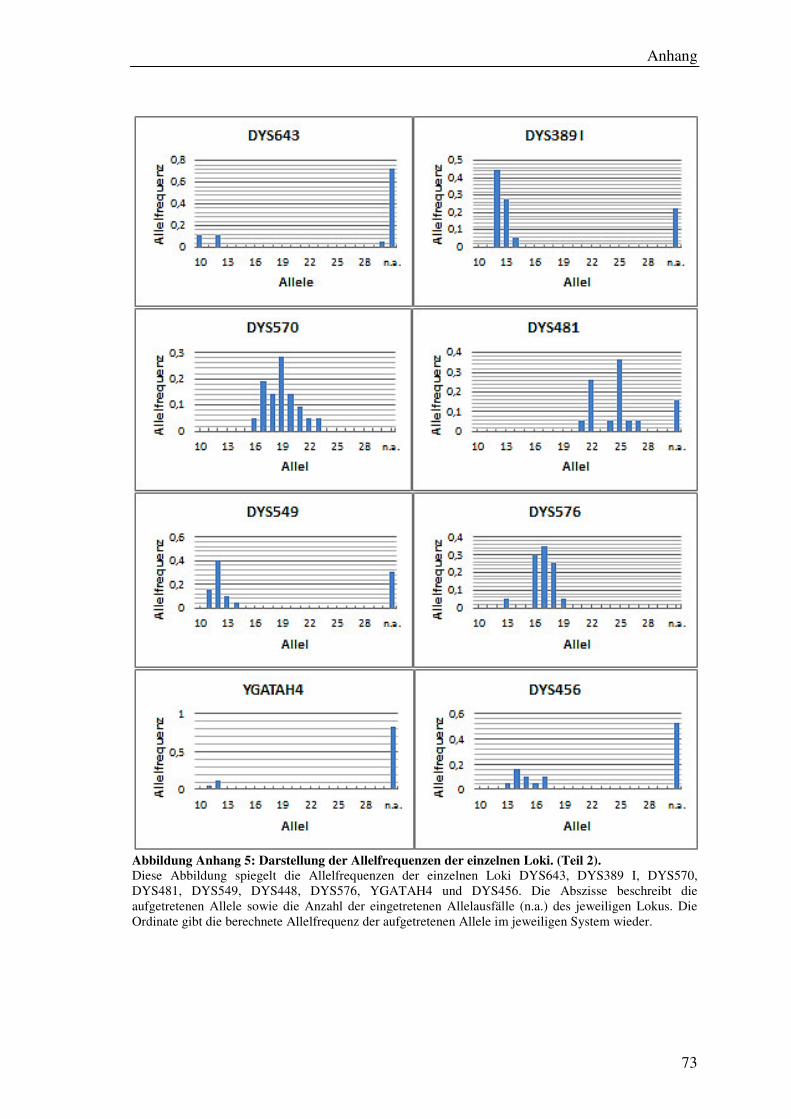
Anhang
73
Abbildung Anhang 5: Darstellung der Allelfrequenzen der einzelnen Loki. (Teil 2). Diese Abbildung spiegelt die Allelfrequenzen der einzelnen Loki DYS643, DYS389 I, DYS570, DYS481, DYS549, DYS448, DYS576, YGATAH4 und DYS456. Die Abszisse beschreibt die aufgetretenen Allele sowie die Anzahl der eingetretenen Allelausfälle (n.a.) des jeweiligen Lokus. Die Ordinate gibt die berechnete Allelfrequenz der aufgetretenen Allele im jeweiligen System wieder.
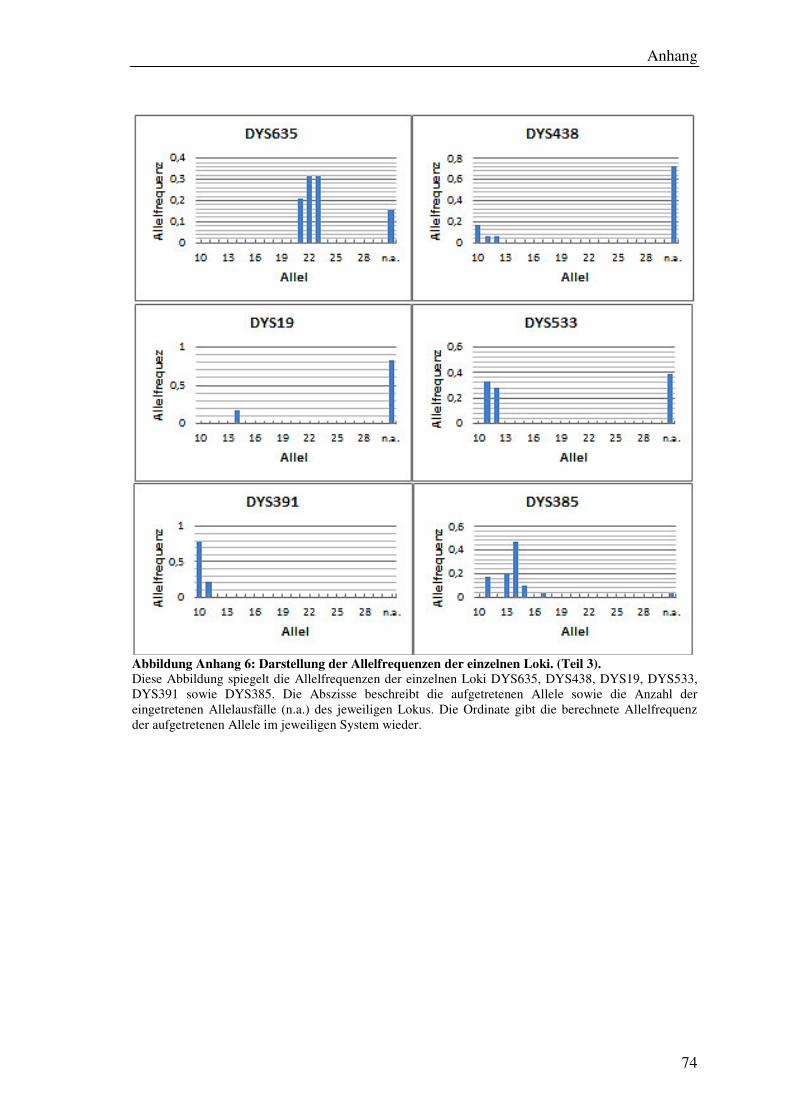
Anhang
74
Abbildung Anhang 6: Darstellung der Allelfrequenzen der einzelnen Loki. (Teil 3). Diese Abbildung spiegelt die Allelfrequenzen der einzelnen Loki DYS635, DYS438, DYS19, DYS533, DYS391 sowie DYS385. Die Abszisse beschreibt die aufgetretenen Allele sowie die Anzahl der eingetretenen Allelausfälle (n.a.) des jeweiligen Lokus. Die Ordinate gibt die berechnete Allelfrequenz der aufgetretenen Allele im jeweiligen System wieder.

Selbstständigkeitserklärung
75
Selbstständigkeitserklärung
Hiermit erkläre ich, dass ich die vorliegende Arbeit selbstständig und nur unter Verwendung der angegebenen Literatur und Hilfsmittel angefertigt habe.
Stellen, die wörtlich oder sinngemäß aus Quellen entnommen wurden, sind als solche kenntlich gemacht.
Diese Arbeit wurde in gleicher oder ähnlicher Form noch keiner anderen Prüfungsbehörde vorgelegt.
Mittweida, den 25.08.2014
Nadine Friedewald