
Aus der Abteilung für Allgemeine und Spezielle Pathologie der Ruhr-Universität Bochum
Leiter: Prof. Dr. med. K. Morgenroth ____________________________________
Die Klassifikation der interstitiellen Pneumonien aus pathologisch-anatomischer und klinischer Sicht
Inaugural-Dissertation zur
Erlangung des Doktorgrades der Medizin einer
Hohen Medizinischen Fakultät der Ruhr-Universität Bochum
vorgelegt von Heike Maria Müller
aus Paderborn 2003

II
Dekan: Prof. Dr. med. G. Muhr
Referent: PD Dr. med. D. Theegarten
Koreferent: Prof. Dr. med. Bufe
Tag der mündlichen Prüfung: 22.06.2004

III
Für meine Eltern und ihre Enkel Benedikt Vitus und Johannes Hendrik

IV
Inhaltsverzeichnis Seite 1. Einleitung 1 2. Überblick über die verschiedenen Klassifikationen der interstitiellen Pneumonien 2 2.1 Historische Darstellung 2 2.2 Klassifikation der idiopathischen interstitiellen Pneumonien 4 2.2.1 Klassifikation nach LIEBOW 4 2.2.2 Klassifikation nach KATZENSTEIN 6
2.2.3 Klassifikation der internationalen Arbeitsgruppe der American Thorax Society / European Respiratory Society 10
2.3 Klinische Manifestation 12 2.3.1 IPF 12 2.3.2 DIP 13 2.3.3 RBILD 13 2.3.4 AIP 13 2.3.5 NSIP 14 2.3.6 BOOP 14 2.3 7 LIP 14 3. Material und Methode 16 3.1 Patientengut 16 3.2 Histologische Auswertung 16 3.3 Datenerhebung 17 3.4 Auswertung der Daten 18 4. Ergebnisse 19
4.1 Darstellung des Gesamtkollektivs 19 4.1.1 Allgemeines 19 4.1.2 Histologische Untersuchungen 20 4.1.2.1 Gewöhnliche interstitielle Pneumonie (UIP) 23 4.1.2.2 Respiratorische Bronchiolitis (RB) 28 4.1.2.3 Desquamative interstitielle Pneumonie (DIP) 30 4.1.2.4 Nicht-spezifische interstitielle Pneumonie (NSIP) 31 4.1.2.5 Organisierende Pneumonie (BOOP) 33 4.1.2.6 Lymphozytäre interstitielle Pneumonie (LIP) 35 4.1.3 Anamnese 37 4.1.4 Klinische Untersuchungen 41 4.1.5 Verlauf 46 4.2 Idiopathische pulmonale Fibrose (IPF) 49 4.2.1 Anamnese 49 4.2.2 Klinische Untersuchungen 50 4.2.3 Verlauf 53 4.3 Respiratorische Bronchiolitis mit interstitieller Lungenerkrankung (RBILD) 55 4.3.1 Anamnese 55 4.3.2 Klinische Untersuchungen 56 4.3.3 Verlauf 58

V
4.4 Desquamative interstitielle Pneumonie (DIP) 59 4.4.1 Anamnese 59 4.4.2 Klinische Untersuchungen 59 4.4.3 Verlauf 60 4.5 Nicht-spezifische interstitielle Pneumonie (NSIP) 61 4.5.1 Anamnese 61 4.5.2 Klinische Untersuchungen 62 4.5.3 Verlauf 64 4.6 Bronchiolitis obliterans mit organisierender Pneumonie (BOOP) 66 4.6.1 Anamnese 66 4.6.2 Klinische Untersuchungen 67 4.6.3 Verlauf 69 4.7 Lymphozytäre interstitielle Pneumonie (LIP) 70 4.7.1 Anamnese 70 4.7.2 Klinische Untersuchungen 70 4.7.3 Verlauf 71 4.8 Exogen-allergische Alveolitis (EAA) 72 4.8.1 Anamnese 72 4.8.2 Klinische Untersuchungen 73 4.8.3 Verlauf 75 4.9 Histiozytosis X (HX) 76 4.9.1 Anamnese 76 4.9.2 Klinische Untersuchungen 77 4.9.3 Verlauf 79 4.10 Sekundäre Fälle 80 4.10.1 Anamnese 80 4.10.2 Klinische Untersuchungen 81 4.10.3 Verlauf 82 4.11 Nicht interstitielle Fälle 83 4.11.1 Anamnese 83 4.11.2 Klinische Untersuchungen 83 4.11.3 Verlauf 84 4.12 Kasuistik 85 4.13 Vergleichende Gegenüberstellung des ATS/ERS-Kollektivs 87
4.13.1 Anamnese 87 4.13.2 Klinische Untersuchungen 94 4.13.3 Verlauf 98 4.14 Status in den BAL-Subgruppen 101 5. Diskussion 104 5.1 Gesamtkollektiv 104 5.2 ATS/ERS-Kollektiv 106 5.2.1 Geschlecht- und Altersverteilung 106 5.2.2 Klinik 107 5.2.3 Labor 111 5.2.4 Bronchoalveoläre Lavage 112 5.2.5 Lungenfunktionsanalyse 115 5.2.6 Prognose 117 5.2.7 Histologien 120 5.3 Ausblick 121

VI
6. Zusammenfassung 123 7. Literaturverzeichnis 124 8. Danksagung 138 9. Curriculum Vitae 139

VII
Verzeichnis der Abbildungen und Tabellen
Seite
Abbildung 1: Häufigkeitsverteilung der klinischen Diagnosen
im Gesamtkollektiv 20
Abbildung 2: Histologische Beurteilungssicherheit in Bezug
auf die klinische Diagnose 22
Abbildung 3: UIP; heterogenes Erscheinungsbild 23
Abbildung 4: UIP; fibroblastische Foci 24
Abbildung 5: UIP; fibroblastischer Focus mit typischen Merkmalen 25
Abbildung 6: UIP; honigwabiger Umbau mit bronchiolärer Metaplasie 26
Abbildung 7: UIP; Infiltration des Interstitiums mit Lymphozyten 27
Abbildung 8: RBILD; typische Veränderungen 28
Abbildung 9: RBILD; dicht gedrängt liegende Makrophagen 28
Abbildung 10: RBILD; Pigmentierung der Makrophagen 29
Abbildung 11: RBILD; Makrophagen in der Berliner Blau Färbung 29
Abbildung 12: DIP; uniforme Makrophagenansammlungen 30
Abbildung 13: DIP; intraalveoläres Makrophagenaggregat 30
Abbildung 14: NSIP; uniformer Prozess 31
Abbildung 15: NSIP; fibrotische Form 32
Abbildung 16: NSIP; Schleimstau mit eingelagerten Makrophagen 32
Abbildung 17: BOOP; Granulationsgewebe 33
Abbildung 18: BOOP; Granulationsgewebe 34
Abbildung 19: BOOP; schmetterlingsähnliches Granulationsgewebe 34
Abbildung 20: LIP; Keimzentren 35
Abbildung 21: LIP; Alveolarzellhyperplasie 36
Abbildung 22: Altersverteilung bei den Frauen im Gesamtkollektiv 37
Abbildung 23: Altersverteilung bei den Männern im Gesamtkollektiv 37
Abbildung 24: Verteilung der verschiedenen Grade der Dyspnoe
im Gesamtkollektiv 38
Abbildung 25: Verteilung der Symptome Husten und Auswurf
im Gesamtkollektiv 39
Abbildung 26: Anamnese des Gesamtkollektivs 41
Abbildung 27: Synopsis der klinischen Befunde im Gesamtkollektiv 42
Abbildung 28: BAL-Zelldifferenzierung im Gesamtkollektiv 43

VIII
Abbildung 29: Ergebnisse der Lungenfunktionsanalyse im Gesamtkollektiv 44
Abbildung 30: Prozentsatz jeweils pathologischer Werte bei der
Lungenfunktionsanalyse im Gesamtkollektiv 44
Abbildung 31: Anzahl der Patienten mit pathologischen Werten
in der Blutgasanalyse im Gesamtkollektiv 45
Abbildung 32: Outcome im Gesamtkollektiv 46
Abbildung 33: Verlauf der Lungenfunktionsanalyse über die Zeit
im Gesamtkollektiv 47
Abbildung 34: Verlauf der Blutgasanalyse über die Zeit im Gesamtkollektiv 48
Abbildung 35: Anamnese bei der IPF 50
Abbildung 36: Synopsis der klinischen Befunde bei der IPF 51
Abbildung 37: BAL-Zelldifferenzierung bei der IPF 52
Abbildung 38: Ergebnisse der Lungenfunktionsanalyse bei der IPF 53
Abbildung 39: Outcome bei der IPF 54
Abbildung 40: Anamnese bei der RBILD 56
Abbildung 41: BAL-Zelldifferenzierung bei der RBILD 57
Abbildung 42: Ergebnisse der Lungenfunktionsanalyse bei der RBILD 58
Abbildung 43: Anamnese bei der NSIP 61
Abbildung 44: Synopsis der klinischen Befunde bei der NSIP 62
Abbildung 45: BAL-Zelldifferenzierung bei der NSIP 63
Abbildung 46: Ergebnisse der Lungenfunktionsanalyse bei der NSIP 64
Abbildung 47: Outcome bei der NSIP 65
Abbildung 48: Anamnese bei der BOOP 66
Abbildung 49: Synopsis der klinischen Befunde bei der BOOP 67
Abbildung 50: BAL-Zelldifferenzierung bei der BOOP 68
Abbildung 51: Ergebnisse der Lungenfunktionsanalyse bei der BOOP 68
Abbildung 52: Outcome bei der BOOP 69
Abbildung 53: Anamnese bei der EAA 72
Abbildung 54: Synopsis der klinischen Befunde bei der EAA 73
Abbildung 55: BAL-Zelldifferenzierung bei der EAA 74
Abbildung 56: Outcome bei der EAA 75
Abbildung 57: Anamnese bei der Histiozytosis X 76
Abbildung 58: BAL-Zelldifferenzierung bei der Histiozytosis X 78
Abbildung 59: Outcome bei der Histiozytosis X 79

IX
Abbildung 60: Anamnese bei den sekundären Fällen 80
Abbildung 61: BAL-Zelldifferenzierung bei den sekundären Fällen 81
Abbildung 62: Outcome bei den sekundären Fällen 82
Abbildung 63: Anamnese bei den nicht interstitiellen Fällen 83
Abbildung 64: AIP-ähnlicher Fall, gleichförmige Fibrose 86
Abbildung 65: AIP-ähnlicher Fall, Epithelhyperplasie 86
Abbildung 66: Geschlechtsverteilung im ATS/ERS-Kollektiv 87
Abbildung 67: Altersverteilung im ATS/ERS-Kollektiv 88
Abbildung 68: Raucherstatus im ATS/ERS-Kollektiv 89
Abbildung 69: Husten aufgeschlüsselt im ATS/ERS-Kollektiv 90
Abbildung 70: Dyspnoe aufgeschlüsselt im ATS/ERS-Kollektiv 91
Abbildung 71: Kortison-Gabe im ATS/ERS-Kollektiv 93
Abbildung 72: Trommelschlegelfinger im ATS/ERS-Kollektiv 94
Abbildung 73: Uhrglasnägel im ATS/ERS-Kollektiv 95
Abbildung 74: Zyanose im ATS/ERS-Kollektiv 95
Abbildung 75: inspiratorisches Knisterrasseln im ATS/ERS-Kollektiv 96
Abbildung 76: Ergebnisse der Lungenfunktionsanalyse im ATS/ERS-
Kollektiv 98
Abbildung 77: Outcome im ATS/ERS-Kollektiv 99
Abbildung 78: Kaplan-Meier Analyse im ATS/ERS-Kollektiv 100
Abbildung 79: Ergebnisse der BAL im ATS/ERS-Kollektiv 102
Abbildung 80: BAL-Lymphozyten nach Diagnosen aufgeschlüsselt 102
Abbildung 81: Kaplan-Meier Analyse nach BAL-Lymphozyten-Subgruppen 103
Tabelle 1: Histologische Befunde des Patientenkollektivs
von HAMMAN und RICH 2
Tabelle 2: Klassifikation nach LIEBOW 4
Tabelle 3: Klassifikation nach KATZENSTEIN 7
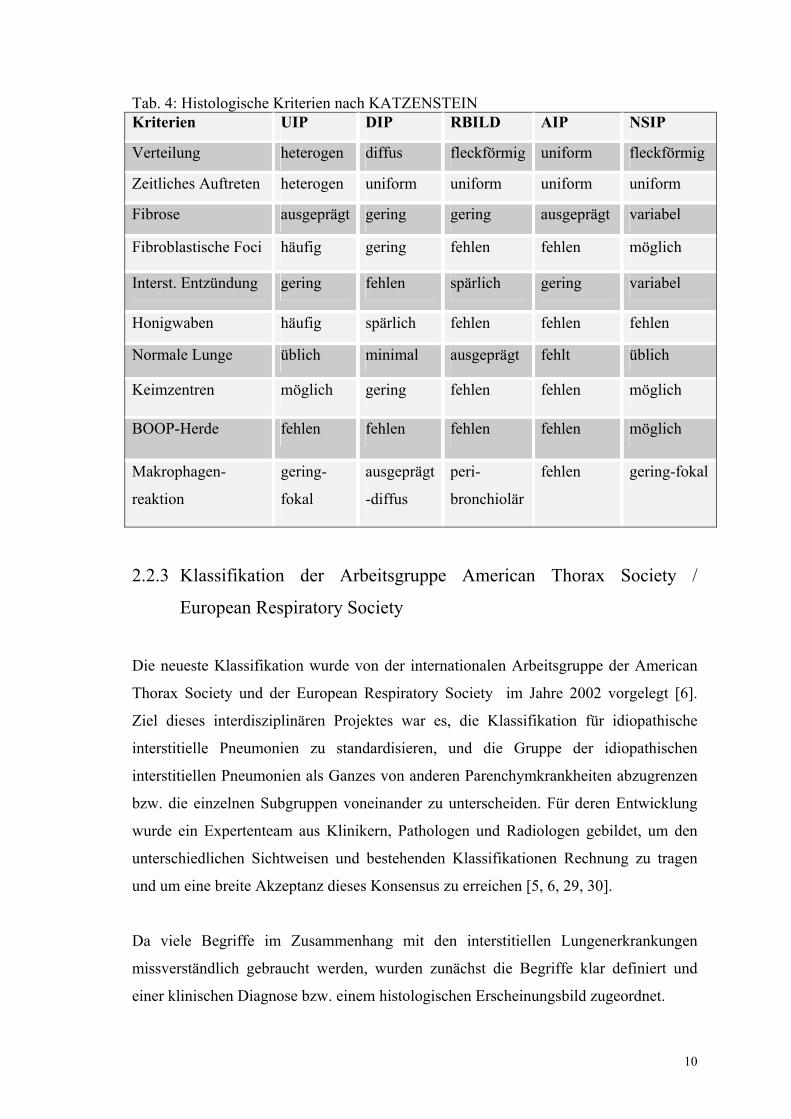
Tabelle 4: Histologische Kriterien nach KATZENSTEIN 10
Tabelle 5: ATS/ERS-Klassifikation: Gegenüberstellung
der histologischen Befunde und der klinischen Diagnose 11
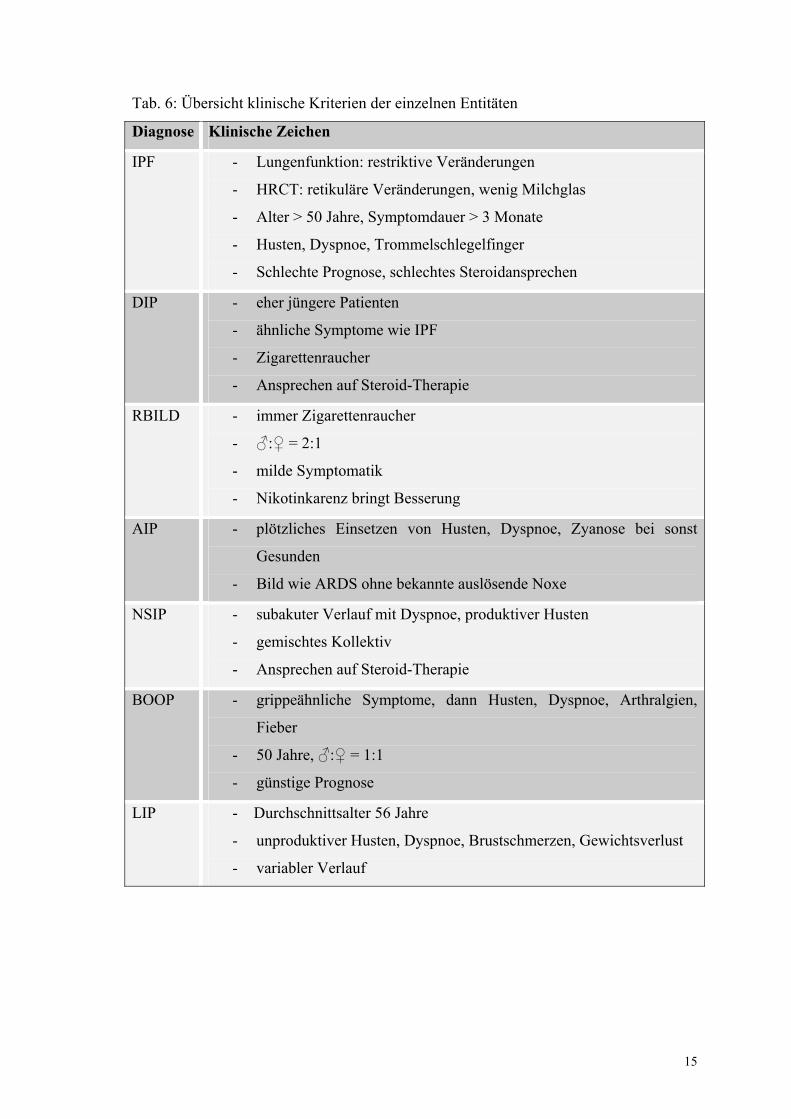
Tabelle 6: Übersicht klinische Kriterien der einzelnen Entitäten 15
Tabelle 7: Gegenüberstellung histologische und klinische Diagnosen 21
Tabelle 8: Anamnestisch bekannte Vorerkrankungen im Gesamtkollektiv 40

X
Tabelle 9: BAL-Zelldifferenzierung bei der DIP 60
Tabelle 10: BAL-Zelldifferenzierung bei der LIP 71
Tabelle 11: BAL Daten in den Untergruppen 101
Tabelle 12: Häufigkeit der Subgruppen an Gesamtkollektiven von IIP 104
Tabelle 13: Übersicht der anamnestischen und klinischen Angaben bei der IPF 108
Tabelle 14: Übersicht der anamnestischen und klinischen Angaben
bei der RBILD 109
Tabelle 15: Übersicht der anamnestischen und klinischen Angaben
bei der NSIP 110
Tabelle 16: Übersicht der anamnestischen und klinischen Angaben
bei der BOOP 111
Tabelle 17: Gesamtübersicht der BAL nach Gruppen 114
Tabelle 18: Ergebnisse der Lungenfunktionsanalyse im Gesamtüberblick 116

XI
Verzeichnis der Abkürzungen
ACE Angiotensin converting enzyme
AIP akute interstitielle Pneumonie
AMP alveoläre Makrophagen-Pneumonie
ANOVA analysis of variance
ATS American Thorax Society
BAL bronchoalveoläre Lavage
BE base excess
BIP Bronchiolitis obliterans mit interstitieller Pneumonie
BMI Body-Mass-Index
BOOP Bronchiolitis obliterans mit organisierender Pneumonie
BSG Blutsenkungsgeschwindigkeit
CEA carcinoembryonales Antigen
COP cryptogene organisierende Pneumonie
CT Computertomographie
DAD diffuser Alveolarschaden
DIP desquamative interstitielle Pneumonie
EAA exogen-allergische Alveolitis
ERS European Respiratory Society
EvG Elastica-van-Giesson
FEV1 forciertes expiratorisches Volumen in der 1. Sekunde
FRC funktionelle Residualkapazität
GIP Riesenzellpneumonie
HE Hämatoxylin-Eosin
HIV human immunodeficiency virus
HRCT hochauflösende Computertomographie
HWS/BWS Halswirbelsäule/Brustwirbelsäule
HX Histiozytosis X
IIP idiopathische interstitielle Pneumonie
IPF interstitielle pulmonale Fibrose
IVC inspiratorische Vitalkapazität
LIP lymphozytäre interstitielle Pneumonie
LUFU Lungenfunktionsuntersuchung

XII
n Anzahl
NSIP nicht-spezifische interstitielle Pneumonie
OLB offene Lungenbiopsie
PAS Periodic-Acid-Schiff
pCO2 Kohlendioxidpartialdruck
pO2 Sauerstoffpartialdruck
RAW Atemwegswiderstand
RBILD respiratorische Bronchiolitis mit interstitieller Lungenerkrankung
RLK Ruhrlandklinik Essen
RV Reservevolumen
S Lungensegment
sGaw spezifische Atemleitgeschwindigkeit
Std.abw. Standardabweichung
TBB transbronchiale Biopsie
TLC totale Lungenkapazität
UIP gewöhnliche interstitielle Pneumonie
VC Vitalkapazität

1
1. Einleitung
An der Gesamtheit der Lungenerkrankungen machen die idiopathischen interstitiellen
Lungenerkrankungen nur einen kleinen Teil aus. Die Prävalenz wird auf 80,9 pro
100.000 Männer bzw. 67,2 pro 100.000 Frauen und die Inzidenz auf 31,5 pro 100.000
Männer/Jahr und 26,1 pro 100.000 Frauen/Jahr geschätzt [1]. Die Prävalenz der
idiopathischen pulmonalen Fibrose wird mit 3-6 auf 100.000 Einwohner angegeben [2,
3, 4, 5].
Bei den näheren Untersuchungen der idiopathischen interstitiellen Lungenerkrankungen
hat sich herausgestellt, dass es sich hierbei um eine Gruppe voneinander
unterscheidbarer Erkrankungen mit variablem Therapieansprechen und
unterschiedlicher Prognose handelt. Es wurden bisher eine Vielzahl von Definitionen
verwendet, so dass bis heute uneinheitliche Begriffe benutzt werden. Mit Einführung
der interdisziplinär entwickelten Klassifikation der American Thorax Society /
European Respiratory Society (ATS/ERS) soll eine neue verlässliche Basis für die
Vergleichbarkeit im klinischen Alltag geschaffen werden [6].
Das Ziel dieser Arbeit ist es, ein vorhandenes Kollektiv nach der ATS/ERS-
Klassifikation retrospektiv sowohl histologisch anhand offener Lungenbiopsien als auch
klinisch neu aufzuarbeiten, um den klinischen Nutzen dieser Klassifikation zu
überprüfen und die Ergebnisse in Bezug zur Literatur zu bewerten.

2
2. Überblick über die verschiedenen Klassifikationen der interstitiellen
Pneumonien
2.1 Historische Darstellung
Im Jahre 1872 erfolgte die Erstbeschreibung einer Lungenfibrose durch VON BÜHL.
RINDFLEISCH beschrieb 1897 eine Form der Lungenfibrose als Cirrhosis cystica
pulmonum. SANDOZ veröffentlichte 1907 einen Fall von familiärer idiopathischer
Lungenfibrose, die er als fötale Bronchiektasie bezeichnete. Fünf Jahre später schrieb
VON HANSEMANN über interstitielle Veränderungen der Lunge, für die er den
Namen Lymphangitis reticularis pulmonum einführte [3, 7].
In der englischsprachigen Literatur gelten HAMMAN und RICH als Erstbeschreiber der
idiopathischen Lungenfibrose. Im Zeitraum von 1931 bis 1935 fielen ihnen vier
Patienten mit einer ungewöhnlichen Symptomkonstellation auf. Diese Patienten litten
unter rasch fortschreitender Dyspnoe und Zyanose, die Lungenerkrankung führte im
weiteren Verlauf zu Rechtsherzversagen, wenn die Patienten nicht zuvor an einer
respiratorischen Insuffizienz verstarben [8].
Tab. 1: Histologische Befunde des Patientenkollektivs von HAMMAN und RICH
Histologische Befunde
- Entzündungstyp, der gegen einen bakteriellen Infekt spricht mit Ödemen,
Hämorrhagien und wenigen Leukozyten
- Hypertrophie und Hyperplasie der Pneumozyten
- Nekrosen an alveolärem und bronchiolärem Epithel
- Hyaline Membranen an den Alveolen
- Ausgeprägtes Ödem und Fibrinablagerungen innerhalb der Alveolarwände
- Extensive, diffuse und progressive interstitielle Proliferation von Bindegewebe
- Eosinophile Granulozyten innerhalb des Interstitiums
- Abwesenheit von Bakterien

3
Aufgrund der histologischen Befunde bezeichneten HAMMAN und RICH dieses
Krankheitsbild als akute diffuse interstitielle Lungenfibrose (AIP) [8]. Später wurde es
auch als Hamman-Rich-Syndrom bekannt. Es stellt die akute Verlaufsform der
idiopathischen interstitiellen Fibrosen dar, weitaus häufiger sind jedoch die chronisch
verlaufenden Formen [9].
LIEBOW befürwortete den Begriff interstitielle Pneumonie. Er war der Ansicht, dass
das Wort Alveolitis die morphologischen Veränderungen nicht ausreichend beschreibt,
da auch das extraalveoläre Gewebe verändert ist. Er unternahm 1974 als erster den
Versuch, eine Klassifikation anhand histologischer Kriterien zu erstellen [9].
Die unterschiedliche Gewichtung einzelner Aspekte der interstitiellen Pneumonien hat
dazu geführt, dass von den Autoren eine Vielzahl von Synonymen benutzt wurden [10].
CEGLA bezeichnete die idiopathische Lungenfibrose beispielsweise als idiopathisch
fibrosierende Alveolitis [11], SCADDING als diffus fibrosierende Alveolitis [12],
STACK als kryptogenetisch fibrosierende Alveolitis [13] und CRYSTAL als
idiopathische interstitielle Lungenfibrose [14].
Eine Bronchiolitis obliterans mit organisierender Pneumonie (BOOP) wurde erstmals
1901 von LANGE als „eigenthümliche Erkrankung der kleinen Bronchien und
Bronchiolen“ beschrieben. 1985 schlug EPLER den Begriff BOOP für ein
eigenständiges idiopathisches Krankheitsbild vor, das mit einer relativ guten Prognose
und einem guten Ansprechen auf Steroidtherapie verbunden ist [15].
KITAICHI hat 1990 52 Fälle mit idiopathischer interstitieller Fibrose untersucht, von
denen 2 in keine bisher bekannte Kategorie passten und somit als „unklassifizierbare
interstitielle Pneumonie“ beschrieben wurden. Diese wurden als neue Entität der nicht-
spezifischen interstitiellen Pneumonie NSIP erstmals von ihm eingeführt [16].
Anfang der 90er Jahre gab es mehrere neue Vorschläge, die interstitiellen Fibrosen
anhand der Pathogenese einzuteilen: GABBRIELLI führte eine Klassifikation ein, die
„echte“ Fibrosen (reparative, kongestive und reaktive Fibrosen) und „falsche“ Fibrosen
(nicht-systematische und systematische Fibrosen) unterscheidet [17]. HOGG schlägt
dagegen vor, zwischen entzündlichen und neoplastischen Formen zuunterscheiden [18].

4
1997 stellte dann KATZENSTEIN ihre Klassifikation vor [19]. Die aktuelle Einteilung
stammt von der ATS/ERS Arbeitsgruppe, die im Jahr 2002 veröffentlicht wurde [6].
2.2 Klassifikation der interstitiellen Pneumonien
2.2.1 Klassifikation nach LIEBOW
Die idiopathischen interstitiellen Lungenfibrosen wurden bisher üblicherweise nach der
Klassifikation von LIEBOW aus dem Jahr 1974 eingeteilt [9]. LIEBOW unterschied 5
Typen:
Tab. 2: Klassifikation nach LIEBOW
Typen
1. Gewöhnliche interstitielle Pneumonie (UIP = usual interstitial pneumonia) 2. Bronchiolitis obliterans mit klassischer interstitieller Pneumonie (BIP =
bronchiolitis obliterans with classical interstitial pneumonia)
3. Desquamative interstitielle Pneumonie (DIP = desquamative interstitial pneumonia)
4. Lymphozytäre interstitielle Pneumonie (LIP = lymphoid interstitial pneumonia)
5. Interstitielle Riesenzellpneumonie (GIP = giant cell interstitial pneumonia)
LIEBOW legte besonderen Wert darauf, dass seine Einteilung lediglich auf
histologischen Kriterien beruht und die unterschiedlichen Gruppen verschiedene Typen
von Gewebereaktionen sind, woraus keine sicheren Schlüsse auf die Pathogenese
gezogen werden können. Dennoch unterscheiden sich die Gruppen bezüglich der Klinik
und ihrem Ansprechen auf eine Cortison-Therapie [9].
Gewöhnliche interstitielle Pneumonie (UIP = usual interstitial pneumonia)
LIEBOW beschrieb die UIP als diffusen Alveolarschaden (DAD = diffuse alveolar
damage). Als Prototyp galt hierbei das histologische Muster, wie es bei beatmeten
Patienten mit hoher Sauerstoffzufuhr auftritt. Dabei kommt es schnell zu einem
ausgedehnten interstitiellen Ödem mit mononukleärer Infiltration, einer starken
Ausbildung von hyalinen Membranen, einer Ansammlung von azidophilem,
proteinreichem Material in den Alveolen, nach drei Tagen zu einer fibroblastischen
Proliferation und nach acht Tagen zu einer Epithelhyperplasie. Erst im Endstadium

5
treten dann Honigwaben und Verlust der Lungengerüststruktur auf. Der Anteil der
schleichend verlaufenden Fälle ohne erkennbare Ätiologie wurde von LIEBOW mit 30-
40% beziffert [9].
Bronchiolitis obliterans mit klassischer interstitieller Pneumonie (BIP =
bronchiolitis obliterans with classical interstitial pneumonia)
Die BIP ist durch ein fibrinöses intrabronchioläres Exsudat gekennzeichnet, in dem
auch große einkernige Makrophagen vorkommen. Hinzu kommt eine Infiltration der
interalveolären Septen durch kleine mononukleäre Zellen, sowie auch durch
Plasmazellen und polymorphkernigen Leukozyten. Das fibrinöse intrabronchioläre
Exsudat wird organisiert und durch Granulationsgewebe ersetzt [9].
Desquamative interstitielle Pneumonie (DIP = desquamative interstitial
pneumonia)
Die Erstbeschreibung einer DIP erfolgte bereits 1965. LIEBOW fand bei 18 Fällen eine
Histologie, die sich klar von der anderer interstitieller Pneumonien unterschied und
auch klinisch unterschieden sich die Fälle von den anderen idiopathischen interstitiellen
Fibrosen. Das Kennzeichen der DIP ist die uniforme und gleichmäßig diffuse
Ansammlung von großen mononukleären Zellen in den Alveolarräumen. Diese Zellen
liegen dicht gedrängt, jedoch kann jede Zelle einzeln abgegrenzt werden. Das
Zytoplasma enthält feine gelb-braune Granula, und lässt sich im Allgemeinen nicht mit
Eisen anfärben. LIEBOW interpretierte diese mononukleären Zellen als granuläre
Pneumozyten, die durch Desquamation von den Alveolardeckzellen hervorgehen [9,
20]. Die These von Liebow, dass es sich bei den intraalveolären Zellen um
desquamierte Pneumozyten Typ II handelt, wurde häufig diskutiert und gilt heute als
widerlegt [21]. Dennoch findet man bei der DIP häufig auch proliferierte Pneumozyten.
Wenn die Läsion nur schwach ausgeprägt ist, sind die Veränderungen peribronchiolär
angeordnet. Die interstitielle Fibrose ist zumeist nur mäßig ausgeprägt. Sie enthält ein
unterschiedlich starkes inflammatorisches Infiltrat aus Lymphozyten, teilweise liegen
die Lymphozyten in Gruppen beieinander und bilden stellenweise Keimzentren. Nicht
selten findet man eine Proliferation der glatten Muskelzellen. Erst in sehr
fortgeschrittenen Fällen stößt man bei der DIP auch auf honigwabigen Umbau, im
Gegensatz zur UIP fehlen aber die fibroblastischen Foci [9, 20].

6
Lymphozytäre interstitielle Pneumonie (LIP = lymphoid interstitial pneumonia)
Die LIP ist insbesondere durch ausgedehnte lymphozytäre Infiltrationen der Lunge
gekennzeichnet. Das lymphozytäre Infiltrat besteht aus Lymphozyten, Plasmazellen
und Riesenzellen. Die LIP ist in ca. einem Drittel der Fälle mit einem Sjögren-Syndrom
assoziiert, häufig finden sich erhöhte Immunglobuline [9].
Interstitielle Riesenzellpneumonie (GIP = giant cell interstitial pneumonia)
Bei der Riesenzellpneumonie (GIP) liegt ein interstitielles Infiltrat ähnlich dem der UIP
vor, jedoch finden sich zahlreiche große, bizarr geformte mehrkernige Zellen. Teilweise
können diese Riesenzellen eine ganze Alveole ausfüllen. Histomorphologisch erinnert
die GIP an eine Masernpneumonie, kann aber durch die Größe der Zellen, einer
ausgeprägte Phagozytose, dem Fehlen von Einschlusskörperchen sowie der streng
intraalveolären Lage der Riesenzellen unterschieden werden [9].
2.2.2 Klassifikation nach KATZENSTEIN
Die Liebow-Klassifikation wurde 1993 durch KATZENSTEIN modifiziert [19]. Es
sollte eine Klassifikation der idiopathischen interstitiellen Pneumonien geschaffen
werden, die für den Kliniker und den Pathologen gleichermaßen praktikabel ist und
gleichzeitig der Begriffsverwirrung entgegenwirken soll. Diese unterscheidet sich in
folgenden Punkten von der Liebow-Klassifikation:
Sowohl die BIP, die LIP als auch die GIP wurden aus der Klassifikation
herausgenommen.
Die BIP wird heute als Bronchiolitis obliterans mit organisierender Pneumonie (BOOP)
bezeichnet. Hierbei handelt es sich um ein eher primär bronchioläres Geschehen,
welches auf die Alveolarräume übergreift. Sie entspricht also weniger einem
interstitiellen Prozess [19, 22, 23].
Die Entitäten GIP und LIP erscheinen nicht mehr, weil sie nicht mehr als idiopathisch
gelten konnten [19, 22]. Der Riesenzellpneumonie wird als Ursache eine
Hartmetallpneumokoniose zugeschrieben [24]. Die LIP wird eher den

7
lymphoproliferativen Erkrankungen zugerechnet und als lymphoide Läsion der Lunge
bezeichnet. Sie ist häufig mit Immunschwächen assoziiert [19, 22].
Hinzugenommen wurden hingegen zwei Entitäten, nämlich die respiratorische
Bronchiolitis mit interstitieller Lungenerkrankung (RBILD = respiratory bronchiolitis
with interstitial lung disease), die vermutlich in Zusammenhang mit der DIP steht, und
die neue Entität der nicht-spezifischen interstitiellen Pneumonie (NSIP = non-specific
interstitial pneumonia), unter der die Fälle zusammengefasst werden, die nicht die
Kriterien für eine der anderen spezifischen Erkrankungen erfüllen. Außerdem wurde die
akute interstitielle Pneumonie (AIP = acute interstitial pneumonia) von der Gruppe der
gewöhnlichen interstitiellen Pneumonien abgegrenzt um deutlich zu machen, dass der
Verlauf der einen Gruppe hochakut und der der anderen chronisch-schleichend ist.
Nach KATZENSTEIN werden somit die folgenden 5 Entitäten unterschieden.
Tab. 3: Klassifikation nach KATZENSTEIN Entitäten
- Gewöhnliche interstitielle Pneumonie (UIP = usual interstitial pneumonia)
- Desquamative interstitielle Pneumonie (DIP = desquamative interstitial pneumonia)
- Respiratorische Bronchiolitis mit interstitieller Lungenerkrankung (RBILD =
respiratory bronchiolitis with interstitial lung disease)
- akute interstitielle Pneumonie (AIP = acute interstitial pneumonia)
- nicht-spezifische interstitielle Pneumonie (NSIP = non-specific interstitial pneumonia)
Gewöhnliche interstitielle Pneumonie (UIP = usual interstitial pneumonia)
Im Unterschied zur Liebow-Klassifikation sind hier nur UIP-Fälle zulässig, die einen
schleichenden, chronischen Verlauf haben. Daher treten auch ausschließlich zeitlich
fortgeschrittene Formen auf.
Das histologische Bild der UIP zeichnet sich vor allem durch ein heterogenes, nicht-
uniformes Bild aus. Die Läsionen scheinen unterschiedlich alt zu sein. Als Orte akuter
Lungenschädigung gelten die fibroblastischen Foci. Das sind lamellenartige
Fibroblasten-Aggregate, die häufig mit hyperplastischem Epithel überzogen sind. Sie
enthalten nur wenig Kollagen und sind im Lichtmikroskop schon bei geringer
Vergrößerung gut vom umliegenden Gewebe zu unterscheiden. Eine zeitlich ältere

8
Läsion stellt die interstitielle Fibrose dar, die durch eosinophile Kollagenablagerung
gekennzeichnet ist. Das inflammatorische Infiltrat besteht hauptsächlich aus
Lymphozyten, die teils als Keimzentren vorliegen, Plasmazellen, Neutrophile,
Eosinophile sowie Mastzellen. Ein weiteres wichtiges Charakteristikum ist der
honigwabige Umbau, wobei die erweiterten, umstrukturierten Alveolen oft mit Mucus
ausgefüllt sind. Darüber hinaus liegt gelegentlich auch eine Proliferation glatter
Muskelzellen nahe der Alveolen, der respiratorischen Bronchioli bzw. der Gefäße oder
eine Akkumulation von Makrophagen innerhalb der Alveolen ähnlich wie bei der DIP
vor. Diese Veränderungen kommen bunt gemischt nebeneinander vor und wechseln
sich mit gesundem Lungengewebe ab [19, 22, 25].
Desquamative interstitielle Pneumonie (DIP = desquamative interstitial
pneumonia)
In der Gruppe der DIP wurden von LIEBOW nur idiopathische Fälle beschrieben, so
dass die DIP unverändert von KATZENSTEIN übernommen wurde. Es wird lediglich
herausgestellt, dass die Läsion an allen Stellen offensichtlich gleich alt ist, und in
keinem Fall fibroblastische Foci auftreten [22].
Respiratorische Bronchiolitis mit inerstitieller Lungenerkrankung (RBILD =
respiratory bronchiolitis with interstitial lung disease)
Eine Neuerung in der Katzenstein-Klassifikation ist die Erweiterung um die RBILD.
Hierbei handelt es sich um ein ähnliches Erscheinungsbild wie bei der DIP. Im
Unterschied dazu findet sich hier jedoch ein fleckförmiger Prozess, der sich vorwiegend
im peribronchiolärem Parenchym abspielt. Die Markophagenanhäufungen liegen fest
akkumuliert in den Alveolen und respiratorischen Bronchioli. Die Wände sind nur in
geringem Ausmaß verdickt und enthalten nur ein spärlich entzündliches Infiltrat.
Außerhalb der fleckförmigen Läsionen findet sich normales Lungengewebe. Die
respiratorische Bronchiolitis wurde zuerst bei Autopsien beschrieben, allerdings haben
diese Veränderungen keine klinischen Auswirkungen gehabt [19, 22, 26]. Es wird
diskutiert, ob die RBILD ein eigenständiges Krankheitsbild oder lediglich ein frühes
Stadium der DIP darstellt [27]. Im Allgemeinen - wie auch in dieser Arbeit - werden die
DIP und die RBILD zusammen betrachtet, zuvor jedoch getrennt dargestellt. Die
RBILD kommt definitionsgemäß nur bei Rauchern vor [6].

9
Akute interstitielle Pneumonie (AIP = acute interstitial pneumonia)
Bei der AIP findet sich eine ausgeprägte interstitielle Fibroblastenproliferation, die zu
einer starken Wandverdickung führt und die Alveolarräume unterschiedlich groß und
teils schlitzartig erscheinen lässt. Es ist nur wenig Kollagen zu sehen, dafür findet man
ein unterschiedlich starkes chronisch-entzündliches Infiltrat. Die AIP präsentiert sich
auffallend gleichförmig, alle Veränderungen scheinen gleich alt zu sein. Häufig
kommen auch Zell-Atypien und Mitosen im hyperplastischen Alveolarepithel vor.
Gelegentlich treten hyaline Membranen auf. Sie entspricht dem histologischen Bild des
diffusen Alveolarschadens (DAD) [8, 23, 28].
Nicht-spezifische interstitielle Pneumonie (NSIP = non-specific interstitial
pneumonia)
Der Begriff der NSIP wurde insbesondere für solche Fälle eingeführt, bei denen nicht
alle Kriterien für eine UIP erfüllt waren. Wie der Name NSIP schon sagt, fehlen hier die
spezifischen Veränderungen, die für eine UIP, DIP oder AIP sprechen würden.
Die NSIP ist durch ein fleckförmiges, in sich aber homogenes Bild gekennzeichnet. Die
Läsionen scheinen alle gleich alt zu sein und liegen bevorzugt peribronchiolär. Das
interstitielle Infiltrat besteht zumeist aus Lymphozyten mit unterschiedlich starker
Beimengung von Plasmazellen mit nur wenig Fibrose, was als zellulärer Typ bezeichnet
wird. Beim fibrotischen Typ überwiegt die Fibrose, das entzündliche Infiltrat ist
dagegen nur gering ausgeprägt. Die Alveolen sind oft mit prominenten
Alveolarephithelzellen vom Typ II ausgekleidet.
Darüber hinaus können bei der NSIP auch in sehr geringem Umfang Merkmale der
anderen Gruppen, wie beispielsweise eine intraalveoläre Makrophagenreaktion,
einzelne fibroblastische Foci oder auch vereinzelt BOOP-Herde auftreten [19, 22, 25,
26].
10
Tab. 4: Histologische Kriterien nach KATZENSTEIN Kriterien UIP DIP RBILD AIP NSIP
Verteilung heterogen diffus fleckförmig uniform fleckförmig
Zeitliches Auftreten heterogen uniform uniform uniform uniform
Fibrose ausgeprägt gering gering ausgeprägt variabel
Fibroblastische Foci häufig gering fehlen fehlen möglich
Interst. Entzündung gering fehlen spärlich gering variabel
Honigwaben häufig spärlich fehlen fehlen fehlen
Normale Lunge üblich minimal ausgeprägt fehlt üblich
Keimzentren möglich gering fehlen fehlen möglich
BOOP-Herde fehlen fehlen fehlen fehlen möglich
Makrophagen-
reaktion
gering-
fokal
ausgeprägt
-diffus
peri-
bronchiolär
fehlen gering-fokal
2.2.3 Klassifikation der Arbeitsgruppe American Thorax Society /
European Respiratory Society
Die neueste Klassifikation wurde von der internationalen Arbeitsgruppe der American
Thorax Society und der European Respiratory Society im Jahre 2002 vorgelegt [6].
Ziel dieses interdisziplinären Projektes war es, die Klassifikation für idiopathische
interstitielle Pneumonien zu standardisieren, und die Gruppe der idiopathischen
interstitiellen Pneumonien als Ganzes von anderen Parenchymkrankheiten abzugrenzen
bzw. die einzelnen Subgruppen voneinander zu unterscheiden. Für deren Entwicklung
wurde ein Expertenteam aus Klinikern, Pathologen und Radiologen gebildet, um den
unterschiedlichen Sichtweisen und bestehenden Klassifikationen Rechnung zu tragen
und um eine breite Akzeptanz dieses Konsensus zu erreichen [5, 6, 29, 30].
Da viele Begriffe im Zusammenhang mit den interstitiellen Lungenerkrankungen
missverständlich gebraucht werden, wurden zunächst die Begriffe klar definiert und
einer klinischen Diagnose bzw. einem histologischen Erscheinungsbild zugeordnet.

11
Folgende klinische Entitäten sind in der Klassifikation enthalten: idiopathische
pulmonale Fibrose (IPF), desquamative interstitielle Pneumonie (DIP), respiratorische
Bronchiolitis mit interstitieller Lungenerkrankung (RBILD), cryptogene organisierende
Pneumonie (COP)/ Synonym: Bronchiolitis obliterans mit organisierender Pneumonie
(BOOP), akute interstitielle Pneumonie (AIP), nicht-spezifische interstitielle Pneumonie
(NSIP) und lymphozytäre interstitielle Pneumonie (LIP). Tabelle 5 zeigt die
entsprechenden histologischen Erscheinungsbilder [6].
Tab. 5: ATS/ERS-Klassifikation: Gegenüberstellung der histologischen Befunde und
der klinischen Diagnose
Histologisches Erscheinungsbild Klinische Diagnose
Gewöhnliche interstitielle Pneumonie Idiopathische pulmonale Fibrose
Desquamative interstitielle Pneumonie Desquamative interstitielle Pneumonie
Respiratorische Bronchiolitis Respiratorische Bronchiolitis mit
interstitieller Lungenerkrankung
Organisierende Pneumonie Cryptogene organisierende Pneumonie
Diffuser Alveolarschaden Akute interstitielle Pneumonie
Nicht-spezifische interstitielle Pneumonie Nicht-spezifische interstitielle Pneumonie
Lymphozytäre interstitielle Pneumonie Lymphozytäre interstitielle Pneumonie
Von der ATS/ERS wurde für die desquamative interstitielle Pneumonie (DIP) zunächst
der Begriff der alveolären Makrophagen-Pneumonie (AMP) bevorzugt. Obwohl die
intraalveoläre Zellansammlung vorwiegend aus Makrophagen besteht und nicht, wie
von Liebow angenommen, aus desquamierten epithelialen Zellen, konnte sich der
Begriff der AMP jedoch nicht durchsetzten [6, 20, 21, 31].
Für den im deutschsprachigen Raum gängigen Begriff BOOP wird von der ATS/ERS
Arbeitsgruppe der Begriff cryptogene organisierende Pneumonie bevorzugt, da der
Name BOOP zu Verwirrungen mit Erkrankungen der Atemwege führen könnte. In die
Klassifikation wurde die COP aufgenommen, weil sie mit anderen Formen der
idiopathischen intersitiellen Pneumonien verwechselt werden kann [6].
Als Differentialdiagnosen zur COP kommen neben der NSIP, DIP und UIP auch der
diffuse Alveolarschaden (DAD = diffuse alveolar damage) in Frage. Dieser ist durch ein
diffuses, uniformes Bild mit ausgeprägtem Wandödem und hyalinen Membranen

12
gekennzeichnet, das dem der AIP entspricht. Jedoch liegt dem DAD eine bekannte
Ursache zugrunde, während die AIP idiopathisch ist [6, 23, 32].
Die NSIP wird zunehmend als eine eigenständige Entität anerkannt, und nicht mehr nur
als Sammeltopf für alle Fälle angesehen, die nur schwer klassifizierbar sind. Im
Gegenteil schafft die ATS/ERS-Klassifikation für die Rarität, dass sich ein Fall
überhaupt nicht zuordnen lässt, die diagnostische Hintertür der unklassifizierbaren
idiopathischen Pneumonie [6].
Die LIP, die von KATZENSTEIN mit dem Hinweis auf lymphoproliferative
Erkrankungen nicht mit in ihre Klassifikation übernommen wurde, erscheint in der
ATS/ERS-Klassifikation erneut, da sie in seltenen Fällen als idiopathische Variante
vorkommt [6].
2.3 Klinische Manifestation
2.3.1 Idiopathische pulmonale Fibrose (IPF)
Für die Diagnose einer IPF ohne Vorliegen eines histologischen Befundes hat die ATS-
Arbeitsgruppe 4 Haupt- und 4 Nebenkriterien definiert. Die Hauptkriterien sind: 1.
Ausschluss von anderen bekannten Ursachen wie Medikamente, Umwelteinflüsse und
Kollagenosen, 2. Restriktion und eingeschränkter Gasaustausch in den
Lungenfunktionstests, 3. retikuläre Veränderungen und minimale milchglasartige
Veränderungen im HRCT sowie 4. eine TBB oder BAL, die auf keine andere alternative
Diagnose hinweisen. Nebenkriterien sind ein Alter von über 50 Jahren, schleichender
Beginn ohne Hinweis auf eine andere Erkrankung, Dauer der Symptome von über 3
Monaten und inspiratorisches Knistern bei der Auskultation [6]. Symptome sind dabei
insbesondere Husten und Dyspnoe, häufig auch Trommelschlegelfinger und seltener
Gelenkbeschwerden. Die Prognose ist schlecht, nur wenige Patienten sprechen gut auf
die Therapie an [22, 33, 34, 35, 36, 37, 38].

13
2.3.2 Desquamative interstitielle Pneumonie (DIP)
Die DIP ist ebenfalls eine chronisch verlaufende Erkrankung. Die Patienten sind etwa
10 Jahre jünger als die IPF-Patienten, stellen sich aber mit ähnlichen Symptomen,
nämlich Dyspnoe und Husten vor. Es werden auch Zyanose, Uhrglasnägel und
Trommelschlegelfinger beobachtet. Zumeist handelt es sich bei den Patienten um
Raucher. Im Röntgenbild sind retikuläre oder retikulo-noduläre und milchglasartige
Veränderungen zu sehen. Im Allgemeinen sprechen diese Patienten gut auf eine
Steroid-Therapie an, eine Vollremission ist möglich [5, 19, 20, 22, 27, 35].
2.3.3 Respiratorische Bronchiolitis mit interstitieller Lungenerkrankung
(RBILD)
Im Unterschied zur DIP sind bei der RBILD alle Patienten Raucher. Männer sind
doppelt so häufig betroffen wie Frauen, wobei das Durchschnittsalter noch unter dem
der DIP liegt. Die Symptome sind bei den meisten Patienten nur geringfügig vorhanden,
können jedoch auch stark ausgeprägt sein. Es liegen dann zumeist eine Dyspnoe, die
langsam zugenommen hat, und ein neu aufgetretener bzw. veränderter Husten vor,
wobei weitere Hypoxiezeichen fehlen. Die Röntgenbilder zeigen fein-retikuläre oder
noduläre Verschattungen bei normalem Lungenvolumen. Nach Aufhören des
Zigarettenkonsums kommt es gewöhnlich zu einer Rückbildung der Symptome,
ansonsten können diese gut mit Steroiden therapiert werden [5, 22, 27, 39, 40, 41].
2.3.4 Akute interstitielle Pneumonie (AIP)
Die AIP ist eine fulminant verlaufende Form mit schnell zunehmender Dyspnoe, Husten
und Zyanose, die zuvor gesunde Patienten aller Altersgruppen betrifft. Voran geht oft
eine grippeähnliche Krankheit, dann tritt starker Husten mit Fieber und Dyspnoe auf.
Von einer AIP sollte nur dann gesprochen werden, wenn zuvor alle möglichen Ursachen
einer Lungenschädigung, wie hohe Sauerstoffkonzentrationen, virale Pneumonien oder
Medikamentennebenwirkungen ausgeschlossen worden sind, um die AIP gegen ein
ARDS abzugrenzen. Im Röntgenbild entwickeln sich schnell diffuse, bilaterale Infiltrate
[5, 8, 19, 23, 29].

14
2.3.5 Nicht-spezifische interstitielle Pneumonie (NSIP)
Patienten mit einer NSIP stellen sich ebenfalls mit Dyspnoe und produktivem Husten
vor. Die Patienten sind im Vergleich mit der IPF um 8 bis 10 Jahre jünger und hier
herrscht eine ausgewogene Geschlechtsverteilung. Die Beschwerden können subakut
einsetzen oder schleichend zunehmen. Die häufigste Veränderung in den
Thoraxaufnahmen sind bilaterale fleckige Verdichtungen oder ähnliche Befunde wie bei
der IPF. Die Prognose ist nicht schlecht, die meisten Symptome verschwinden unter
Steroid-Therapie ganz oder bleiben stabil, falls es sich um den zellulären Typ handelt.
Der fibrotische Typ steht bezüglich der Prognose zwischen der zellulären NSIP und der
IPF [26, 34, 37, 38, 41, 42, 43, 44].
2.3.6 Bronchiolitis obliterans mit organisierender Pneumonie (BOOP)
Klinisch beginnt die BOOP häufig mit grippeähnlichen Symptomen, die sich unter
Antibiotikatherapie nicht bessern, gefolgt von Husten, zunehmender Dyspnoe,
Gewichtsverlust, Arthralgien und Fieber. Die Patienten sind im Mittel 50 Jahre alt, es
besteht kein Geschlechtsunterschied. Typisch sind bilaterale fleckförmige Infiltrate, die
die Lokalisation wechseln können. Außerdem treten auch flächigere Verdichtungen und
retikuläre Infiltrate auf. Die meisten Patienten genesen unter Steroiden vollständig, aber
nach zu frühem Absetzten kommt es zu Remissionen [15, 45, 46, 47, 48, 49, 50, 51, 52].
2.3.7 Lymphozytäre interstitielle Pneumonie (LIP)
Die Gruppe der LIP lässt sich in vier Untergruppen aufteilen: zum einen kann die LIP
mit Kollagenosen assoziiert sein, zum anderen mit Immunschwäche, mit bestimmten
weiteren Krankheiten oder auch idiopathisch sein. Die Patienten sind im Schnitt 56
Jahre alt, klagen über unproduktiven Husten, Dyspnoe, Brustschmerzen und
Gewichtsverlust. Röntgenologisch sind fleckige bilaterale Infiltrate oder
retikulonoduläre Verschattungen zu sehen. Der klinische Verlauf ist unterschiedlich:
etwa gleich viele Patienten genesen, bleiben stabil oder versterben. Tritt eine LIP bei
HIV-postitiven Kindern auf, so ist dies als Manifestation von AIDS anzusehen [6, 25,
53, 54].
15
Tab. 6: Übersicht klinische Kriterien der einzelnen Entitäten
Diagnose Klinische Zeichen
IPF - Lungenfunktion: restriktive Veränderungen
- HRCT: retikuläre Veränderungen, wenig Milchglas
- Alter > 50 Jahre, Symptomdauer > 3 Monate
- Husten, Dyspnoe, Trommelschlegelfinger
- Schlechte Prognose, schlechtes Steroidansprechen
DIP - eher jüngere Patienten
- ähnliche Symptome wie IPF
- Zigarettenraucher
- Ansprechen auf Steroid-Therapie
RBILD - immer Zigarettenraucher
- ♂:♀ = 2:1
- milde Symptomatik
- Nikotinkarenz bringt Besserung
AIP - plötzliches Einsetzen von Husten, Dyspnoe, Zyanose bei sonst
Gesunden
- Bild wie ARDS ohne bekannte auslösende Noxe
NSIP - subakuter Verlauf mit Dyspnoe, produktiver Husten
- gemischtes Kollektiv
- Ansprechen auf Steroid-Therapie
BOOP - grippeähnliche Symptome, dann Husten, Dyspnoe, Arthralgien,
Fieber
- 50 Jahre, ♂:♀ = 1:1
- günstige Prognose
LIP - Durchschnittsalter 56 Jahre
- unproduktiver Husten, Dyspnoe, Brustschmerzen, Gewichtsverlust
- variabler Verlauf

16
3. Methodik
3.1 Patientengut
Aus der Dokumentation der Abteilung für Allgemeine und Spezielle Pathologie der
Ruhr-Universität Bochum wurden alle Fälle interstitieller Pneumonien und
Lungenfibrosen herausgesucht, die aus der Ruhrlandklinik Essen im Zeitraum von 1993
bis 2000 zur histologischen Begutachtung eingeschickt wurden. Wir wählten nur
Patienten aus, die sich zur Diagnosefindung bzw. Diagnosesicherung einer offenen
Lungenbiopsie unterziehen mussten und schlossen Fälle mit transbronchialen Biopsien
oder Lappenresektionen aus. Ebenso wurden von vornherein solche Fälle
ausgeschlossen, bei denen auf dem Anforderungsschein eine mögliche Ätiologie der
Erkrankung dokumentiert war. Stellte sich erst bei der Durchsicht der Krankenakten im
Nachhinein eine Ursache für die Erkrankung heraus, so wurden diese Fälle als
sekundäre Läsionen zusammengefasst.
Schnitte von allen vorhandenen Blöcken wurden mindestens mit Hämatoxylin-Eosin
(HE) gefärbt, daneben wurde zumindest ein Schnitt in der Elastica-van-Giesson-
Färbung, Periodic-Acid-Schiff-Färbung und Berliner-Blau-Färbung angefertigt und
untersucht. Zum Nachweis bzw. Ausschluss einer Langerhanszell’ Histiozytosis wurden
routinemäßig bei klinischem bzw. histologischem Verdacht immunhistochemische
Markierungen mit monoklonalen Antikörpern gegen CD 1a und S-100-Protein (DAKO
Hamburg) nach der ABC-Methode durchgeführt.
3.2 Histologische Auswertung
Die Präparate wurden aus dem Archiv des Pathologischen Institutes herausgesucht.
Meistens waren die HE-, Elastica-van-Giesson-, und Periodic-Acid-Schiff-Färbung
vorhanden. Bei Bedarf wurden neue Schnitte angefertigt. Zur Begutachtung lag jeweils
ausreichend viel Lungengewebe mit ein bis vier Lungenkeilen, im Durchschnitt 1,4
Keile vor. Die Keile wurden bis auf Ausnahmen aus der Lingula, S8 oder S9
entnommen.
Anschließend wurden die Präparate unter dem Lichtmikroskop betrachtet und anhand
der Klassifikation nach Katzenstein [5, 6, 19, 22] eingeteilt. Die erste histologische

17
Bewertung erfolgte ohne Kenntnis klinischer Angaben über die jeweiligen Patienten.
Falls bei der ersten Durchsicht keine sichere Zuordnung möglich war, erfolgte die
Benennung von Differentialdiagnosen und eine erneute Bewertung in Kenntnis der
klinischen Befunde.
Auf die exakte Klassifikation gemäß der Einteilung nach Katzenstein wurde größte
Sorgfalt angewandt.
Nach der Aufarbeitung der klinischen Daten wurden die Proben erneut betrachtet und in
Zusammenschau der klinischen Befunde die Diagnose überprüft und teilweise geändert.
Um die Verlässlichkeit der histologischen Diagnose zu beschreiben, wurden die
Schnitte zusätzlich mit den Begriffen „sicher“, „am ehesten“ und „möglich“
beschrieben.
3.3 Datenerhebung
Die Krankenakten wurden im Archiv der Ruhrlandklinik vollständig und einheitlich
durchgesehen und die Daten mit Hilfe des Statistikprogrammes SPSS Version 10.0
erfasst. Weiterhin kam die Patientenkartei der Ambulanz zur Auswertung, sofern die
Patienten im Anschluss an den stationären Aufenthalt weiterhin zu ambulanten
Kontrollen erschienen. Waren die Patienten bei einem niedergelassenen Arzt in
weiterer Betreuung, so wurden Verlaufsdaten durch telefonische Anfragen erhoben. Um
die Überlebenszeiten auch bei Patienten berechnen zu können, deren Krankengeschichte
nicht weiter zu verfolgen war, wurden teilweise auch Informationen über die jeweiligen
Krankenkassen gesammelt.
Anschließend erfolgte eine fallspezifische Durchsicht aller Daten zusammen mit Prof.
Dr. Costabel, Ruhrlandklinik Essen, zur Überprüfung der Plausibilität der
histologischen bzw. klinischen Diagnose. Es wurden zunächst die klinischen Angaben
durchgesehen, insbesondere im Hinblick auf die Anamnese, Röntgenbilder, die BAL,
und den Krankheitsverlauf. Es wurde viel Wert darauf gelegt, mögliche Ursachen für
die Lungenveränderungen zu finden.

18
Die Krankheitsverläufe wurden zuletzt zusammen mit Prof. Costabel unter
Berücksichtigung aller Informationen in die Kategorien „Vollremission“, „besser“,
„stabil“, „schlechter“, und „Patient verstorben“ unterteilt.
3.4 Auswertung der Daten
Die Auswertung der Daten erfolgte mit dem Statistikprogramm SPSS 11.0 für
Windows.
Statistische Beratungen fanden vor Beginn und nach Abschluss der Datenerfassung am
Institut für Biomathematik der Ruhr-Universität Bochum statt.
Zur Darstellung der Daten wurden übliche Verfahren der deskriptiven Statistik
verwendet (Häufigkeitsverteilungen, Mittelwert, Standardabweichung,
Variationsbreite). Weiterhin kamen die Varianzanalyse ANOVA, der t-Test, der Chi-
Quadrattest und das Kaplan-Meier-Verfahren mit Log-Rank Test für die Erstellung der
Überlebenskurven zur Anwendung.

19
4. Ergebnisse
4.1 Darstellung des Gesamtkollektivs
4.1.1 Allgemeines
Basierend auf einer Datenbankrecherche wurden in den Jahren von 1993 bis 2000 103
Patienten mit der Diagnose interstitielle Pneumonie bzw. Lungenfibrose gefunden.
Davon wurden 11 Patienten aus folgenden Gründen direkt ausgeschlossen: 3 Patienten
hatten ein Adeno-Carzinom, eine Patientin hatte eine Neurofibromatose und ein
weiterer Patient eine Asbestose. Bei 6 Patienten stellte sich heraus, dass das Kriterium
der offenen Lungenbiopsie nicht erfüllt war. Davon war bei 4 Patienten eine
Lobektomie und bei 2 Patienten eine transbronchiale Biopsie durchgeführt worden. Es
blieben also 92 Fälle, die histologisch untersucht und deren klinische Daten erhoben
wurden. Sie stellen das Gesamtkollektiv dar. Aus dem Gesamtkollektiv waren bei 4
Patienten die Veränderungen nicht interstitiell, hierbei handelte es sich um 3 eitrige
Pneumonien und eine idiopathische reine Bronchiolitis obliterans.
Bei der Gesamtbetrachtung aller Aspekte ließ sich bei 8 Patienten eine Ursache für die
Fibrose finden: ein Fall mit Zustand nach Knochenmarkstransplantation und ein Fall mit
nachgewiesenen Asbestkörperchen in der BAL mit jeweils NSIP-ähnlichen Histologie,
einer mit großbullösem Emphysem (BOOP-ähnliche Histologie), einer mit assoziierter
Colitis ulcerosa (BOOP-ähnliche Histologie), einer Leber-Zirrhose (BOOP-ähnliche
Histologie), eine rheumatische Arthritis (AIP-ähnliche Histologie), einer mit
assoziiertem Jo-1-Antikörper-Syndrom (UIP-ähnliche Histologie), und ein Fall, der in
Zusammenhang mit der Einnahme von ACE-Hemmern gesehen werden muss (UIP-
ähnliche Histologie). Diese Fälle werden als sekundäre Pneumonien bezeichnet.
Außerdem lag bei 10 Patienten eine EAA und bei 7 Patienten eine HX vor.
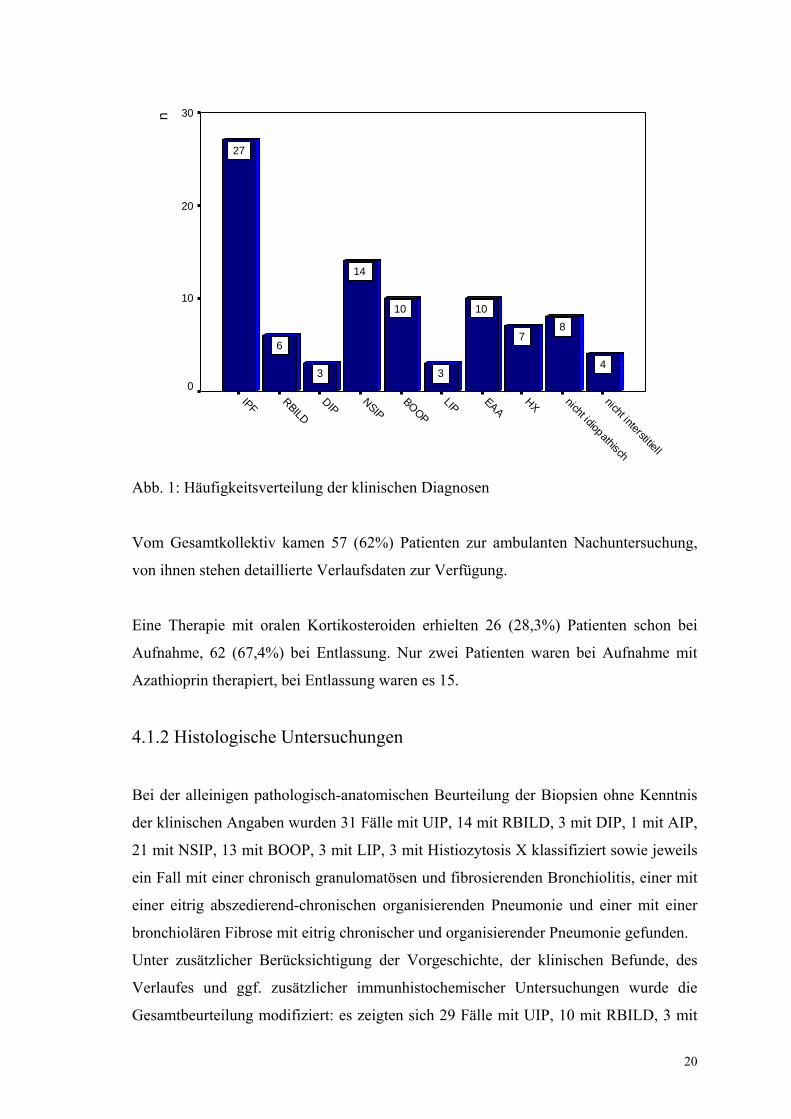
Nachdem alle Informationen zusammengetragen waren, wurden klinisch folgende
Diagnosen gestellt: 27 Fälle mit UIP, 6 mit RBILD, 3 mit DIP, 14 mit NSIP, 10 mit
BOOP und 3 mit LIP, insgesamt also 63 Fälle nach der ATS-Klassifikation.
20
nicht interstitiell
nicht idiopathisch
HXEAALIPBOOP
NSIPDIP
RBILDIPF
n 30
20
10
0
4
87
10
3
10
14
3
6
27
Abb. 1: Häufigkeitsverteilung der klinischen Diagnosen
Vom Gesamtkollektiv kamen 57 (62%) Patienten zur ambulanten Nachuntersuchung,
von ihnen stehen detaillierte Verlaufsdaten zur Verfügung.
Eine Therapie mit oralen Kortikosteroiden erhielten 26 (28,3%) Patienten schon bei
Aufnahme, 62 (67,4%) bei Entlassung. Nur zwei Patienten waren bei Aufnahme mit
Azathioprin therapiert, bei Entlassung waren es 15.
4.1.2 Histologische Untersuchungen
Bei der alleinigen pathologisch-anatomischen Beurteilung der Biopsien ohne Kenntnis
der klinischen Angaben wurden 31 Fälle mit UIP, 14 mit RBILD, 3 mit DIP, 1 mit AIP,
21 mit NSIP, 13 mit BOOP, 3 mit LIP, 3 mit Histiozytosis X klassifiziert sowie jeweils
ein Fall mit einer chronisch granulomatösen und fibrosierenden Bronchiolitis, einer mit
einer eitrig abszedierend-chronischen organisierenden Pneumonie und einer mit einer
bronchiolären Fibrose mit eitrig chronischer und organisierender Pneumonie gefunden.
Unter zusätzlicher Berücksichtigung der Vorgeschichte, der klinischen Befunde, des
Verlaufes und ggf. zusätzlicher immunhistochemischer Untersuchungen wurde die
Gesamtbeurteilung modifiziert: es zeigten sich 29 Fälle mit UIP, 10 mit RBILD, 3 mit
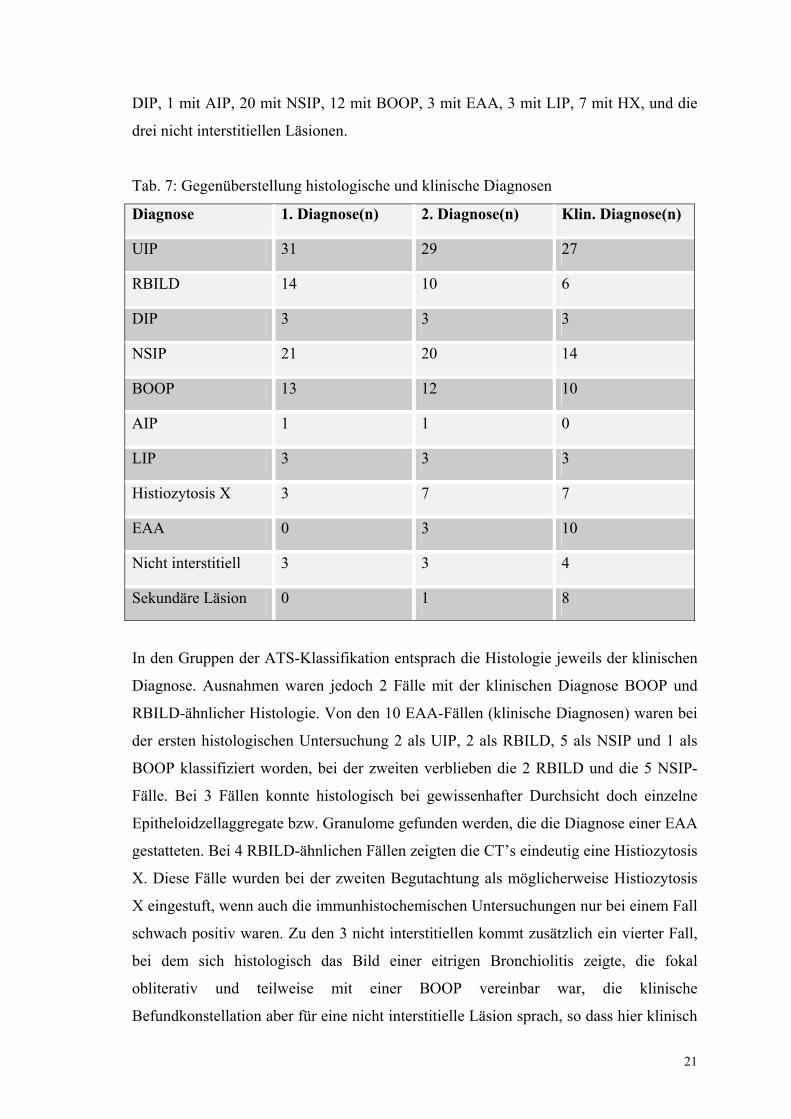
21
DIP, 1 mit AIP, 20 mit NSIP, 12 mit BOOP, 3 mit EAA, 3 mit LIP, 7 mit HX, und die
drei nicht interstitiellen Läsionen.
Tab. 7: Gegenüberstellung histologische und klinische Diagnosen
Diagnose 1. Diagnose(n) 2. Diagnose(n) Klin. Diagnose(n)
UIP 31 29 27
RBILD 14 10 6
DIP 3 3 3
NSIP 21 20 14
BOOP 13 12 10
AIP 1 1 0
LIP 3 3 3
Histiozytosis X 3 7 7
EAA 0 3 10
Nicht interstitiell 3 3 4
Sekundäre Läsion 0 1 8
In den Gruppen der ATS-Klassifikation entsprach die Histologie jeweils der klinischen
Diagnose. Ausnahmen waren jedoch 2 Fälle mit der klinischen Diagnose BOOP und
RBILD-ähnlicher Histologie. Von den 10 EAA-Fällen (klinische Diagnosen) waren bei
der ersten histologischen Untersuchung 2 als UIP, 2 als RBILD, 5 als NSIP und 1 als
BOOP klassifiziert worden, bei der zweiten verblieben die 2 RBILD und die 5 NSIP-
Fälle. Bei 3 Fällen konnte histologisch bei gewissenhafter Durchsicht doch einzelne
Epitheloidzellaggregate bzw. Granulome gefunden werden, die die Diagnose einer EAA
gestatteten. Bei 4 RBILD-ähnlichen Fällen zeigten die CT’s eindeutig eine Histiozytosis
X. Diese Fälle wurden bei der zweiten Begutachtung als möglicherweise Histiozytosis
X eingestuft, wenn auch die immunhistochemischen Untersuchungen nur bei einem Fall
schwach positiv waren. Zu den 3 nicht interstitiellen kommt zusätzlich ein vierter Fall,
bei dem sich histologisch das Bild einer eitrigen Bronchiolitis zeigte, die fokal
obliterativ und teilweise mit einer BOOP vereinbar war, die klinische
Befundkonstellation aber für eine nicht interstitielle Läsion sprach, so dass hier klinisch
22
Befundkonstellation aber für eine nicht interstitielle Läsion sprach, so dass hier klinisch
von einer reinen Bronchiolitis obliterans ohne organisierende Pneumonie ausgegangen
wurde. Unter der Gruppe der sekundären Läsionen subsummieren sich 2 Fälle mit einer
UIP-ähnlichen, 2 mit einer NSIP-ähnlichen, 3 mit einer BOOP-ähnlichen und 1 mit
einer AIP-ähnlichen Histologie.
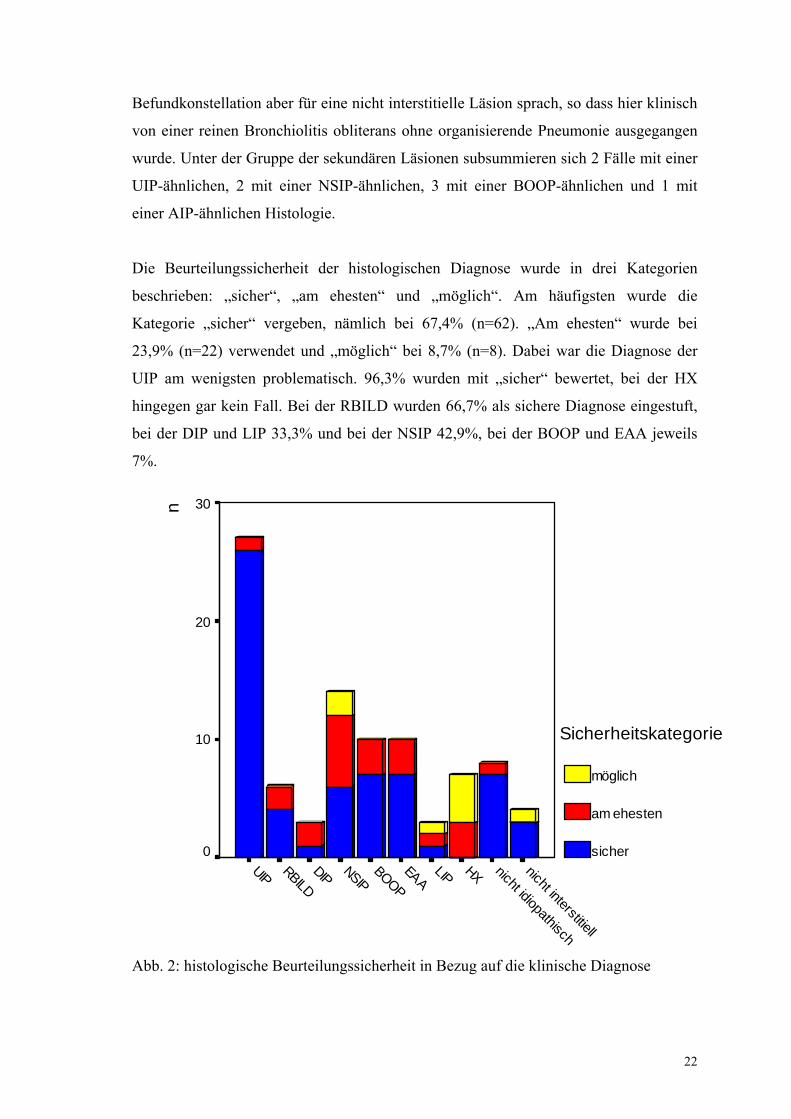
Die Beurteilungssicherheit der histologischen Diagnose wurde in drei Kategorien
beschrieben: „sicher“, „am ehesten“ und „möglich“. Am häufigsten wurde die
Kategorie „sicher“ vergeben, nämlich bei 67,4% (n=62). „Am ehesten“ wurde bei
23,9% (n=22) verwendet und „möglich“ bei 8,7% (n=8). Dabei war die Diagnose der
UIP am wenigsten problematisch. 96,3% wurden mit „sicher“ bewertet, bei der HX
hingegen gar kein Fall. Bei der RBILD wurden 66,7% als sichere Diagnose eingestuft,
bei der DIP und LIP 33,3% und bei der NSIP 42,9%, bei der BOOP und EAA jeweils
7%.
nicht interstitiell
nicht idiopathisch
HXLIPEAABOOP
NSIPDIPRBILD
UIP
n 30
20
10
0
Sicherheitskategorie
möglich
am ehesten
sicher
Abb. 2: histologische Beurteilungssicherheit in Bezug auf die klinische Diagnose
23
4.1.2.1 Gewöhnliche interstitielle Pneumonie
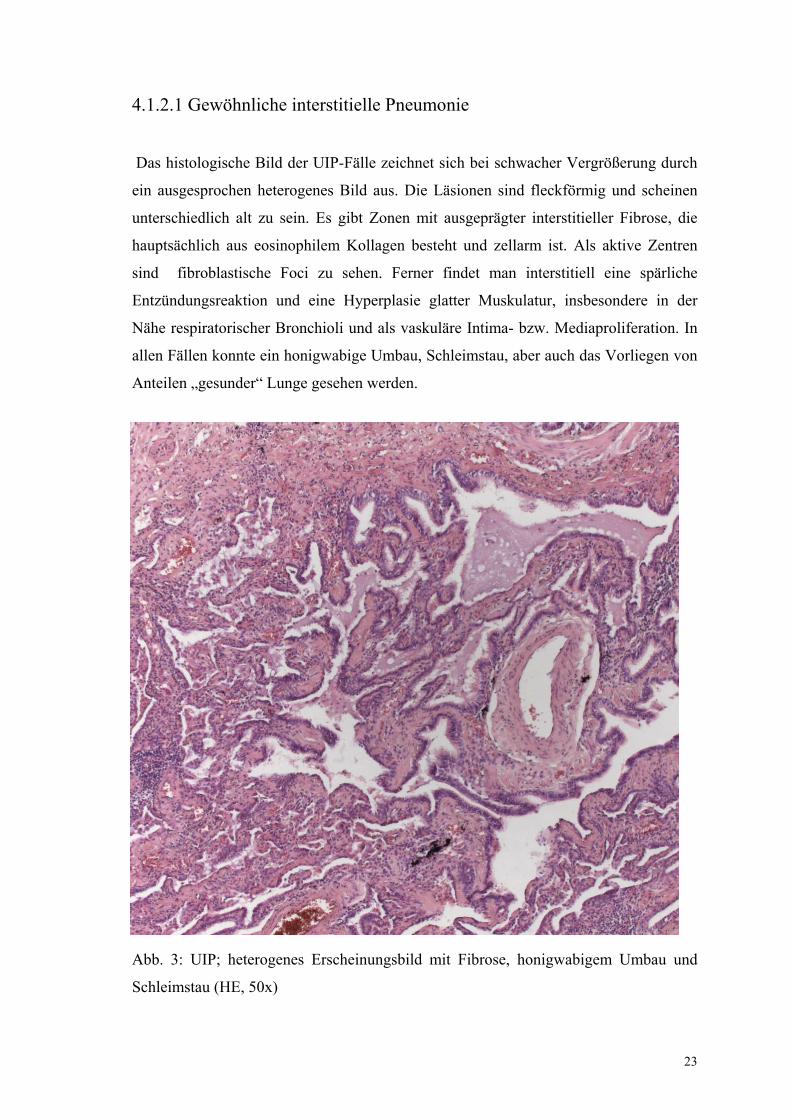
Das histologische Bild der UIP-Fälle zeichnet sich bei schwacher Vergrößerung durch
ein ausgesprochen heterogenes Bild aus. Die Läsionen sind fleckförmig und scheinen
unterschiedlich alt zu sein. Es gibt Zonen mit ausgeprägter interstitieller Fibrose, die
hauptsächlich aus eosinophilem Kollagen besteht und zellarm ist. Als aktive Zentren
sind fibroblastische Foci zu sehen. Ferner findet man interstitiell eine spärliche
Entzündungsreaktion und eine Hyperplasie glatter Muskulatur, insbesondere in der
Nähe respiratorischer Bronchioli und als vaskuläre Intima- bzw. Mediaproliferation. In
allen Fällen konnte ein honigwabige Umbau, Schleimstau, aber auch das Vorliegen von
Anteilen „gesunder“ Lunge gesehen werden.
Abb. 3: UIP; heterogenes Erscheinungsbild mit Fibrose, honigwabigem Umbau und
Schleimstau (HE, 50x)

24
Bei stärkerer Vergrößerung sind die Veränderungen deutlicher sichtbar. Die
fibroblastischen Foci fallen jedoch bereits in kleineren Vergrößerungen auf.
Abb. 4: UIP; Nachweis mehrerer fibroblastischer Foci sowie eines geringen
fleckförmigen Rundzellinfiltrates (HE, 100x)
Die fibroblastischen Foci bestehen aus glatt begrenzten polsterförmigen
Fibroblastenproliferationen. Dazwischen findet sich reichlich Kollagen. Die Foci sind
lumenwärts mit hyperplastischem Alveolarepithel überzogen.
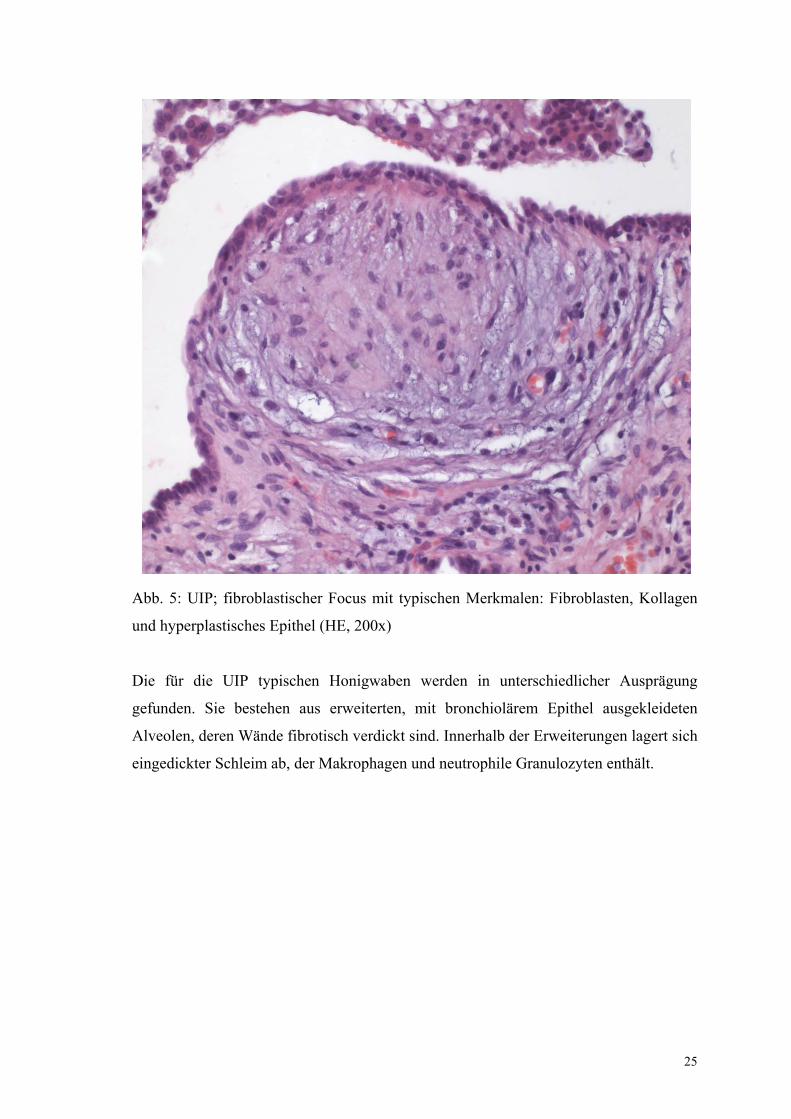
25
Abb. 5: UIP; fibroblastischer Focus mit typischen Merkmalen: Fibroblasten, Kollagen
und hyperplastisches Epithel (HE, 200x)
Die für die UIP typischen Honigwaben werden in unterschiedlicher Ausprägung
gefunden. Sie bestehen aus erweiterten, mit bronchiolärem Epithel ausgekleideten
Alveolen, deren Wände fibrotisch verdickt sind. Innerhalb der Erweiterungen lagert sich
eingedickter Schleim ab, der Makrophagen und neutrophile Granulozyten enthält.
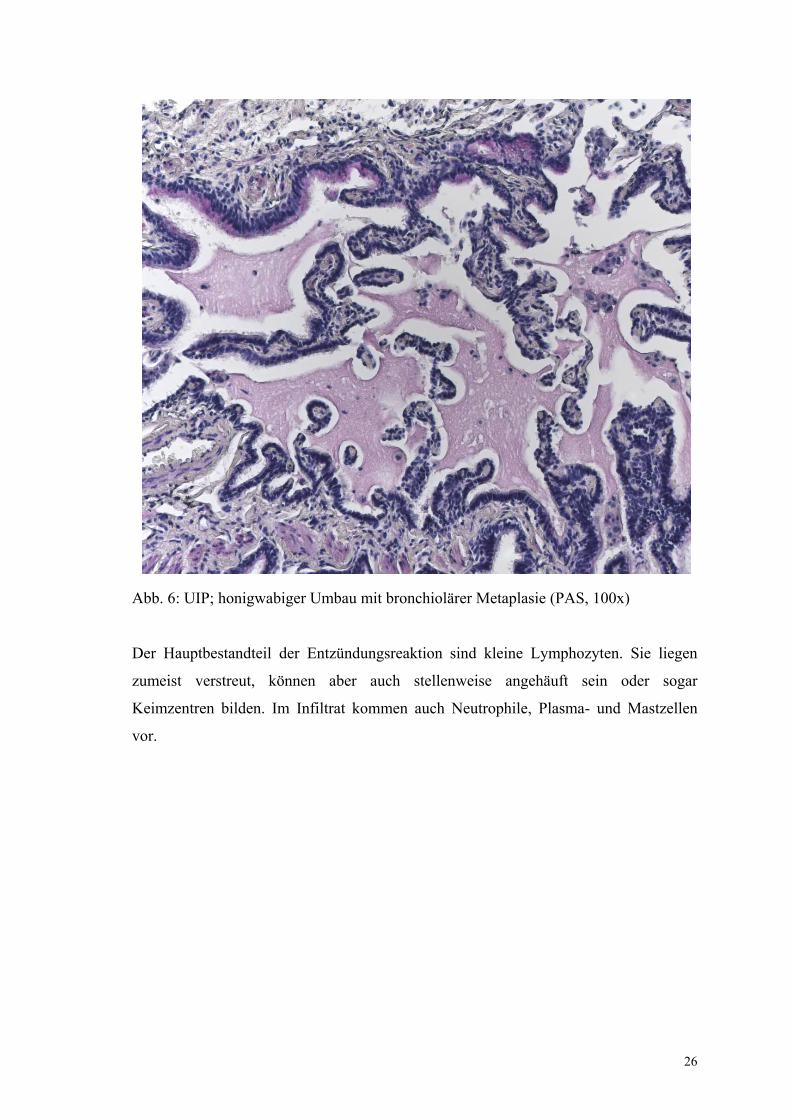
26
Abb. 6: UIP; honigwabiger Umbau mit bronchiolärer Metaplasie (PAS, 100x)
Der Hauptbestandteil der Entzündungsreaktion sind kleine Lymphozyten. Sie liegen
zumeist verstreut, können aber auch stellenweise angehäuft sein oder sogar
Keimzentren bilden. Im Infiltrat kommen auch Neutrophile, Plasma- und Mastzellen
vor.

27
Abb. 7: UIP; Infiltration des Interstitiums mit Lymphozyten (HE, 200x)
Ausschlusskriterium für das Vorliegen einer UIP sind epitheloidzellige Granulome.
28
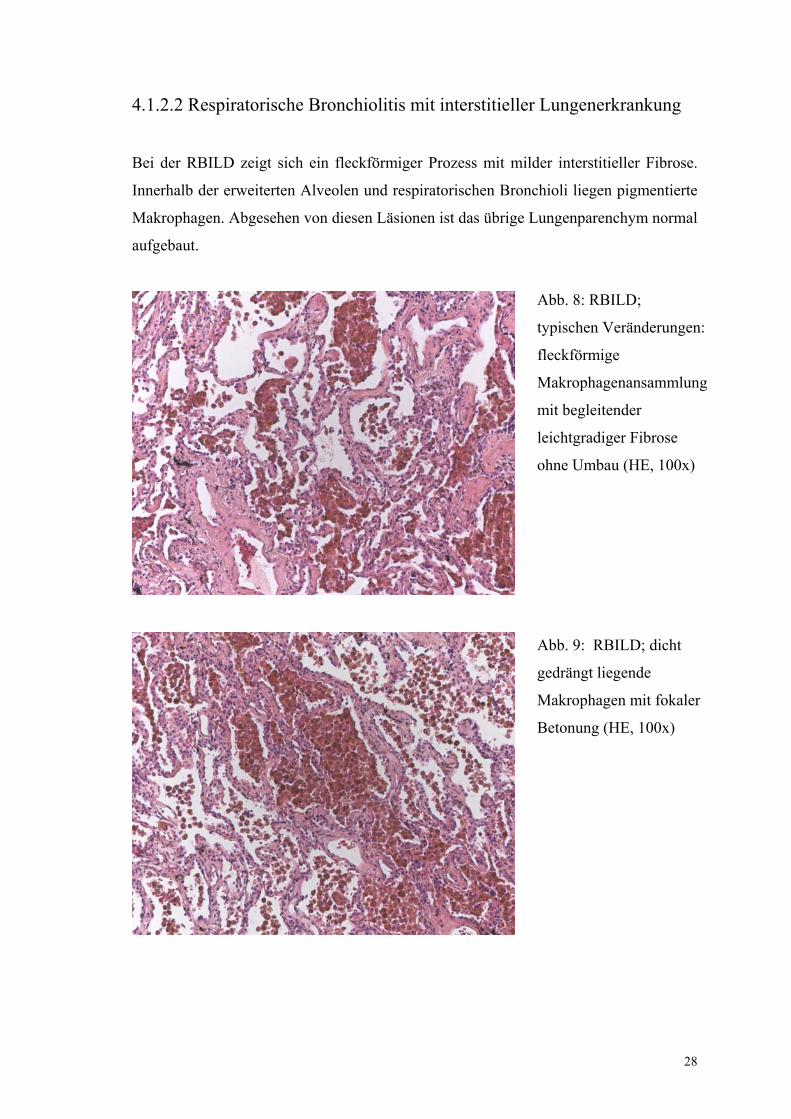
4.1.2.2 Respiratorische Bronchiolitis mit interstitieller Lungenerkrankung
Bei der RBILD zeigt sich ein fleckförmiger Prozess mit milder interstitieller Fibrose.
Innerhalb der erweiterten Alveolen und respiratorischen Bronchioli liegen pigmentierte
Makrophagen. Abgesehen von diesen Läsionen ist das übrige Lungenparenchym normal
aufgebaut.
Abb. 8: RBILD;
typischen Veränderungen:
fleckförmige
Makrophagenansammlung
mit begleitender
leichtgradiger Fibrose
ohne Umbau (HE, 100x)
Abb. 9: RBILD; dicht
gedrängt liegende
Makrophagen mit fokaler
Betonung (HE, 100x)

29
Abb. 10: RBILD; die
Makrophagen sind
bräunlich pigmentiert
(HE, 400x)
Abb. 11: RBILD;
Die Makrophagen sind in
der Berliner Blau Färbung
deutlich positiv (Fe, 400x)
30
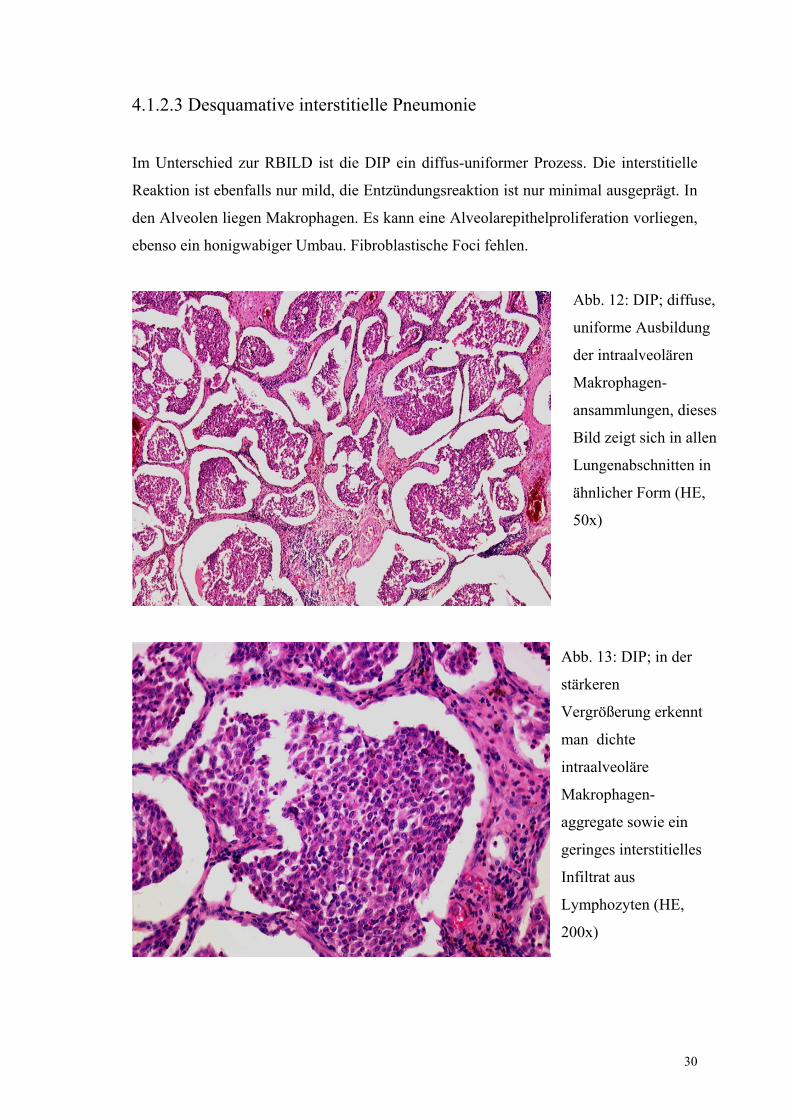
4.1.2.3 Desquamative interstitielle Pneumonie
Im Unterschied zur RBILD ist die DIP ein diffus-uniformer Prozess. Die interstitielle
Reaktion ist ebenfalls nur mild, die Entzündungsreaktion ist nur minimal ausgeprägt. In
den Alveolen liegen Makrophagen. Es kann eine Alveolarepithelproliferation vorliegen,
ebenso ein honigwabiger Umbau. Fibroblastische Foci fehlen.
Abb. 12: DIP; diffuse,
uniforme Ausbildung
der intraalveolären
Makrophagen-
ansammlungen, dieses
Bild zeigt sich in allen
Lungenabschnitten in
ähnlicher Form (HE,
50x)
Abb. 13: DIP; in der
stärkeren
Vergrößerung erkennt
man dichte
intraalveoläre
Makrophagen-
aggregate sowie ein
geringes interstitielles
Infiltrat aus
Lymphozyten (HE,
200x)
31
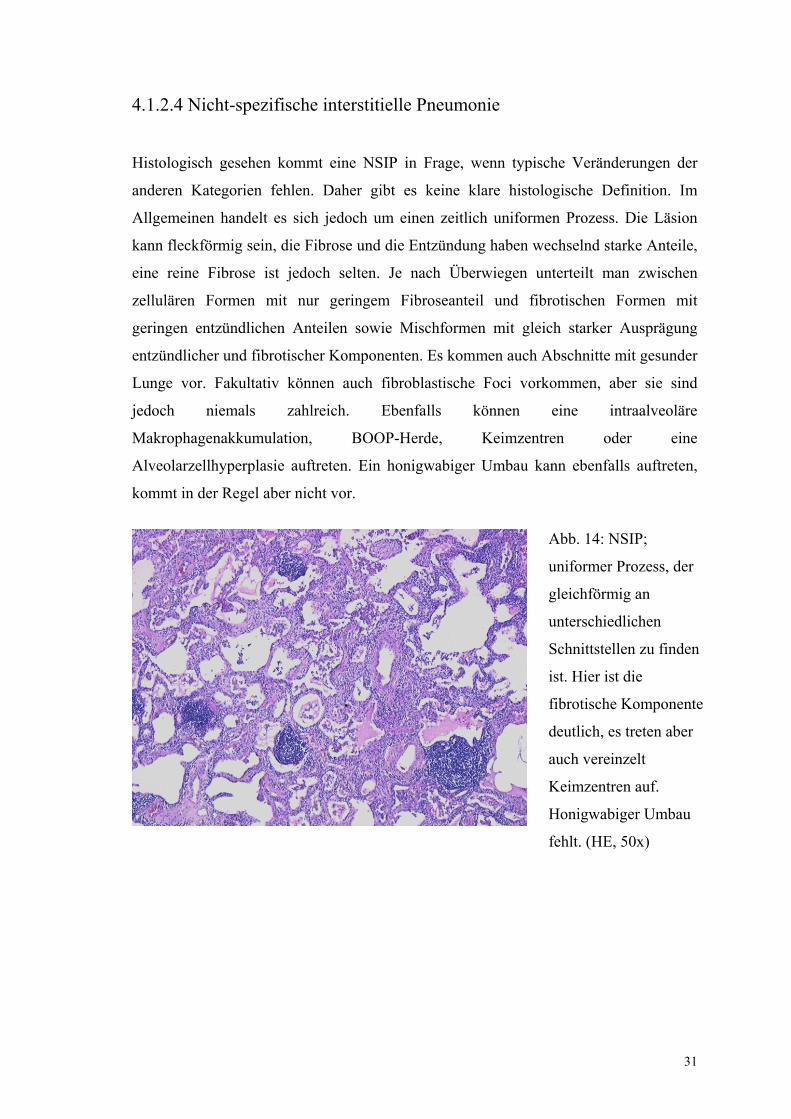
4.1.2.4 Nicht-spezifische interstitielle Pneumonie
Histologisch gesehen kommt eine NSIP in Frage, wenn typische Veränderungen der
anderen Kategorien fehlen. Daher gibt es keine klare histologische Definition. Im
Allgemeinen handelt es sich jedoch um einen zeitlich uniformen Prozess. Die Läsion
kann fleckförmig sein, die Fibrose und die Entzündung haben wechselnd starke Anteile,
eine reine Fibrose ist jedoch selten. Je nach Überwiegen unterteilt man zwischen
zellulären Formen mit nur geringem Fibroseanteil und fibrotischen Formen mit
geringen entzündlichen Anteilen sowie Mischformen mit gleich starker Ausprägung
entzündlicher und fibrotischer Komponenten. Es kommen auch Abschnitte mit gesunder
Lunge vor. Fakultativ können auch fibroblastische Foci vorkommen, aber sie sind
jedoch niemals zahlreich. Ebenfalls können eine intraalveoläre
Makrophagenakkumulation, BOOP-Herde, Keimzentren oder eine
Alveolarzellhyperplasie auftreten. Ein honigwabiger Umbau kann ebenfalls auftreten,
kommt in der Regel aber nicht vor.
Abb. 14: NSIP;
uniformer Prozess, der
gleichförmig an
unterschiedlichen
Schnittstellen zu finden
ist. Hier ist die
fibrotische Komponente
deutlich, es treten aber
auch vereinzelt
Keimzentren auf.
Honigwabiger Umbau
fehlt. (HE, 50x)
32
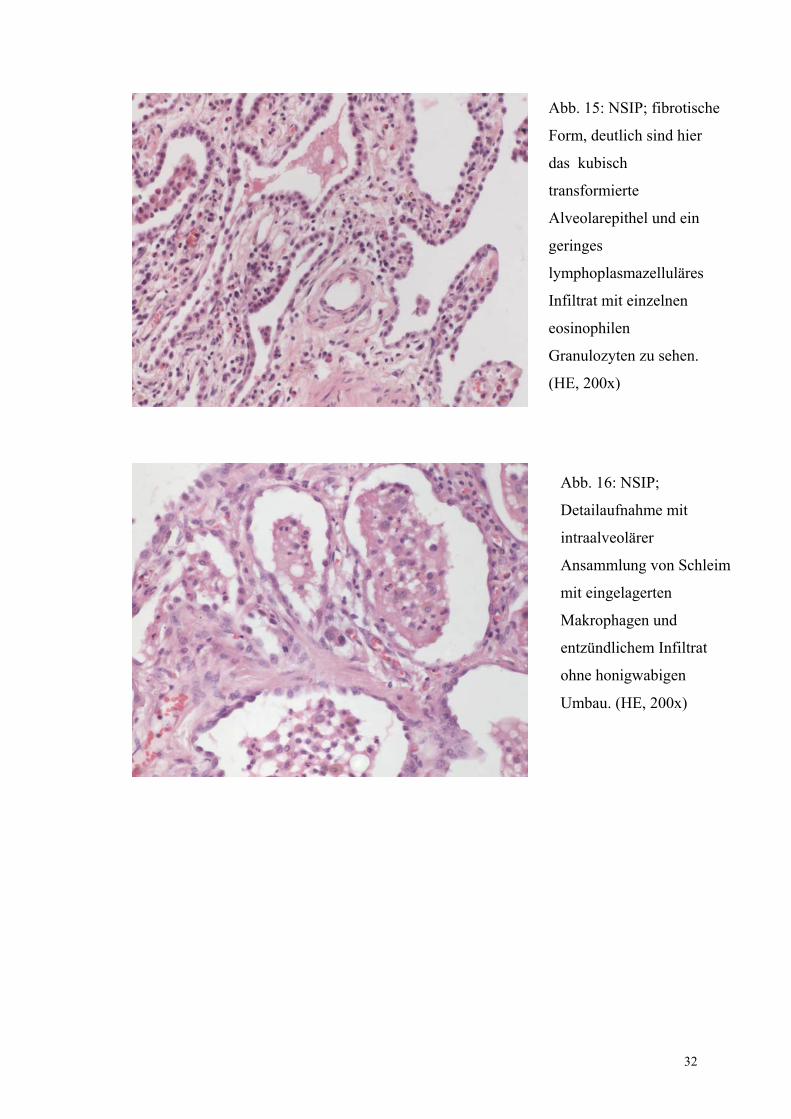
Abb. 15: NSIP; fibrotische
Form, deutlich sind hier
das kubisch
transformierte
Alveolarepithel und ein
geringes
lymphoplasmazelluläres
Infiltrat mit einzelnen
eosinophilen
Granulozyten zu sehen.
(HE, 200x)
Abb. 16: NSIP;
Detailaufnahme mit
intraalveolärer
Ansammlung von Schleim
mit eingelagerten
Makrophagen und
entzündlichem Infiltrat
ohne honigwabigen
Umbau. (HE, 200x)
33
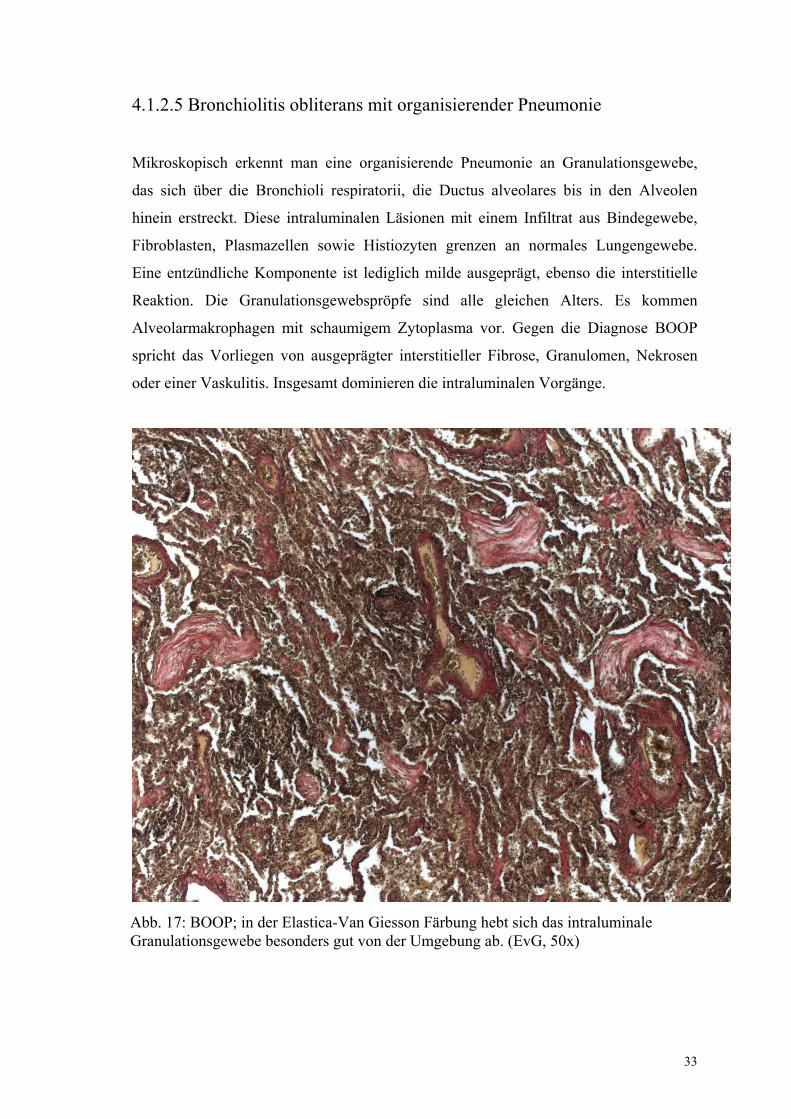
4.1.2.5 Bronchiolitis obliterans mit organisierender Pneumonie
Mikroskopisch erkennt man eine organisierende Pneumonie an Granulationsgewebe,
das sich über die Bronchioli respiratorii, die Ductus alveolares bis in den Alveolen
hinein erstreckt. Diese intraluminalen Läsionen mit einem Infiltrat aus Bindegewebe,
Fibroblasten, Plasmazellen sowie Histiozyten grenzen an normales Lungengewebe.
Eine entzündliche Komponente ist lediglich milde ausgeprägt, ebenso die interstitielle
Reaktion. Die Granulationsgewebspröpfe sind alle gleichen Alters. Es kommen
Alveolarmakrophagen mit schaumigem Zytoplasma vor. Gegen die Diagnose BOOP
spricht das Vorliegen von ausgeprägter interstitieller Fibrose, Granulomen, Nekrosen
oder einer Vaskulitis. Insgesamt dominieren die intraluminalen Vorgänge.
Abb. 17: BOOP; in der Elastica-Van Giesson Färbung hebt sich das intraluminale Granulationsgewebe besonders gut von der Umgebung ab. (EvG, 50x)
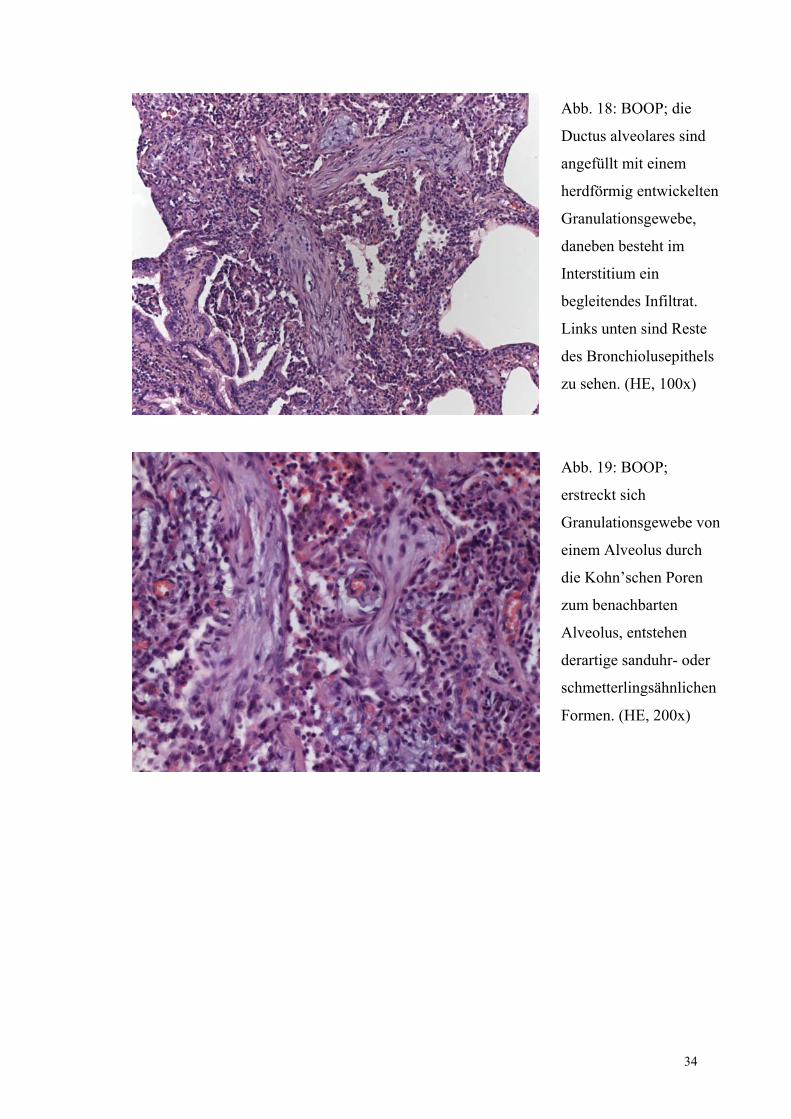
34
Abb. 18: BOOP; die
Ductus alveolares sind
angefüllt mit einem
herdförmig entwickelten
Granulationsgewebe,
daneben besteht im
Interstitium ein
begleitendes Infiltrat.
Links unten sind Reste
des Bronchiolusepithels
zu sehen. (HE, 100x)
Abb. 19: BOOP;
erstreckt sich
Granulationsgewebe von
einem Alveolus durch
die Kohn’schen Poren
zum benachbarten
Alveolus, entstehen
derartige sanduhr- oder
schmetterlingsähnlichen
Formen. (HE, 200x)
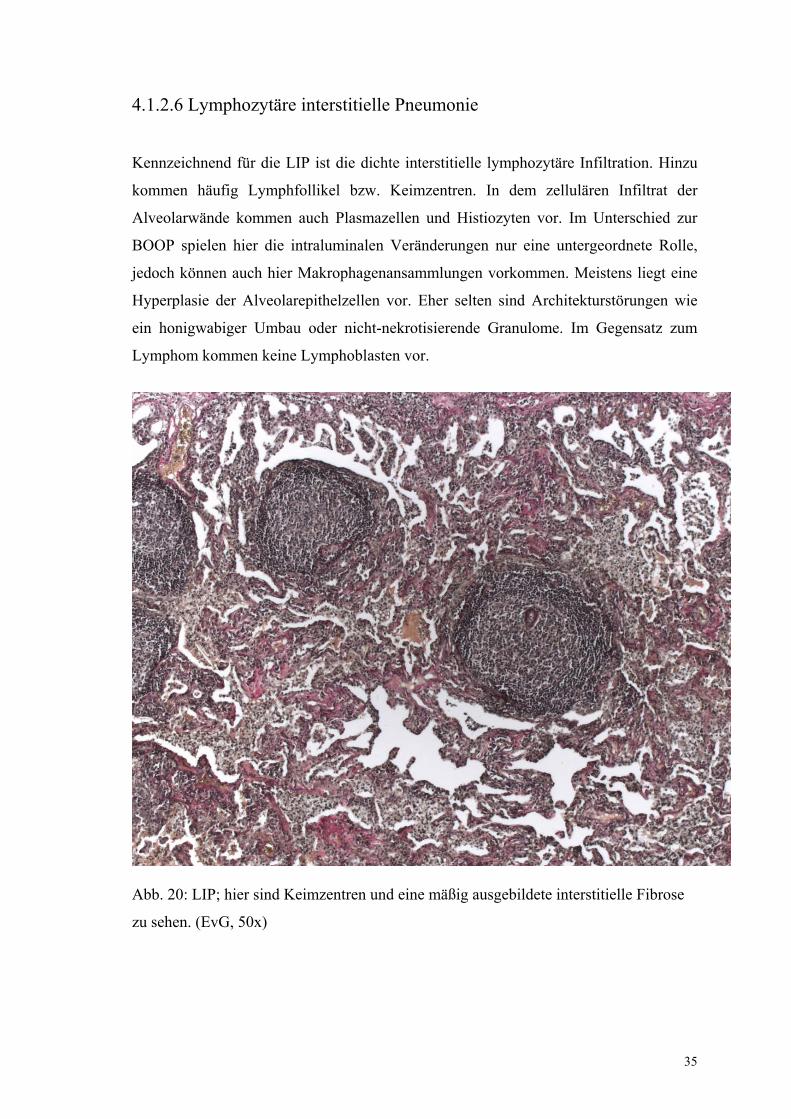
35
4.1.2.6 Lymphozytäre interstitielle Pneumonie
Kennzeichnend für die LIP ist die dichte interstitielle lymphozytäre Infiltration. Hinzu
kommen häufig Lymphfollikel bzw. Keimzentren. In dem zellulären Infiltrat der
Alveolarwände kommen auch Plasmazellen und Histiozyten vor. Im Unterschied zur
BOOP spielen hier die intraluminalen Veränderungen nur eine untergeordnete Rolle,
jedoch können auch hier Makrophagenansammlungen vorkommen. Meistens liegt eine
Hyperplasie der Alveolarepithelzellen vor. Eher selten sind Architekturstörungen wie
ein honigwabiger Umbau oder nicht-nekrotisierende Granulome. Im Gegensatz zum
Lymphom kommen keine Lymphoblasten vor.
Abb. 20: LIP; hier sind Keimzentren und eine mäßig ausgebildete interstitielle Fibrose
zu sehen. (EvG, 50x)
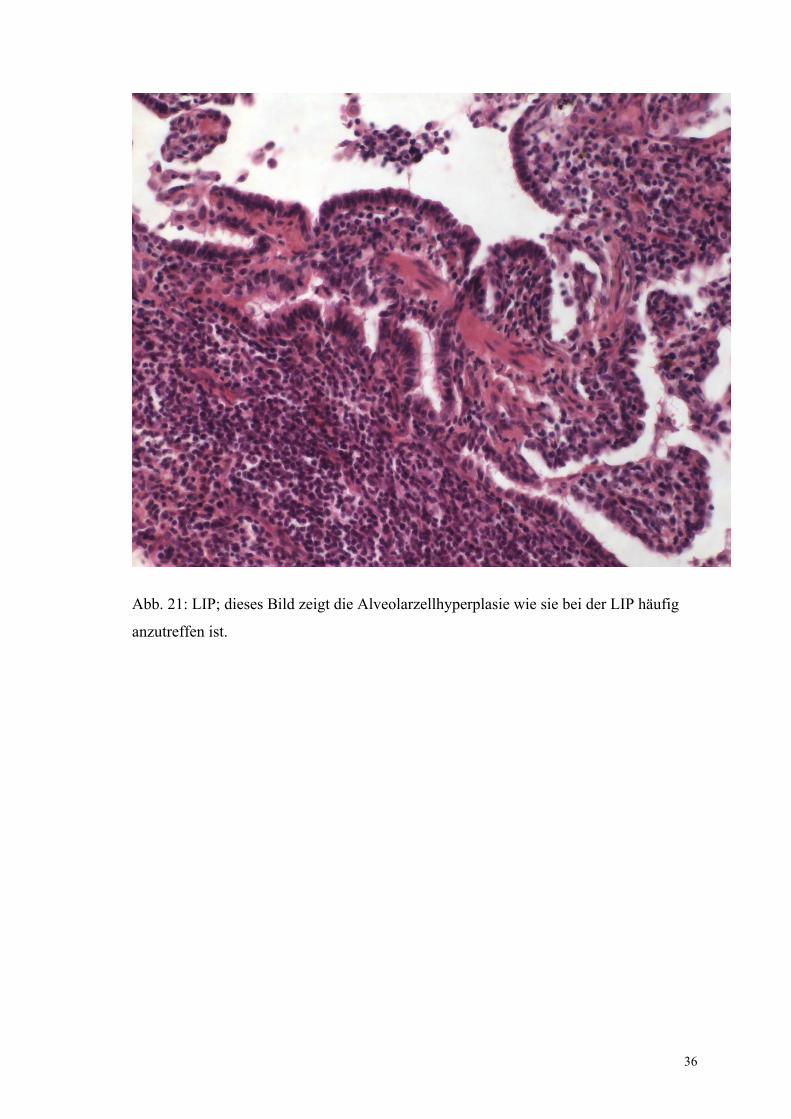
36
Abb. 21: LIP; dieses Bild zeigt die Alveolarzellhyperplasie wie sie bei der LIP häufig
anzutreffen ist.
37
4.1.3 Anamnese
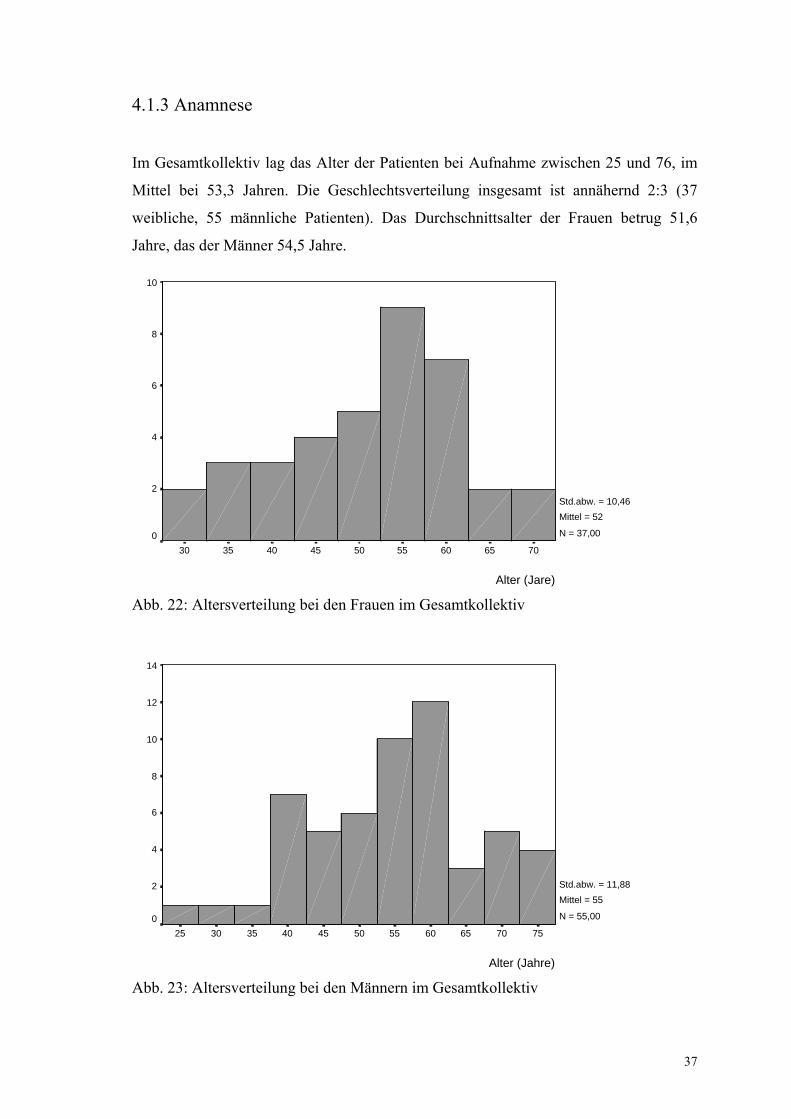
Im Gesamtkollektiv lag das Alter der Patienten bei Aufnahme zwischen 25 und 76, im
Mittel bei 53,3 Jahren. Die Geschlechtsverteilung insgesamt ist annähernd 2:3 (37
weibliche, 55 männliche Patienten). Das Durchschnittsalter der Frauen betrug 51,6
Jahre, das der Männer 54,5 Jahre.
Abb. 22: Altersverteilung bei den Frauen im Gesamtkollektiv
Abb. 23: Altersverteilung bei den Männern im Gesamtkollektiv
Alter (Jare)
706560555045403530
10
8
6
4
2
0
Std.abw. = 10,46 Mittel = 52
N = 37,00
Alter (Jahre)
7570656055504540353025
14
12
10
8
6
4
2
0
Std.abw. = 11,88 Mittel = 55
N = 55,00
38
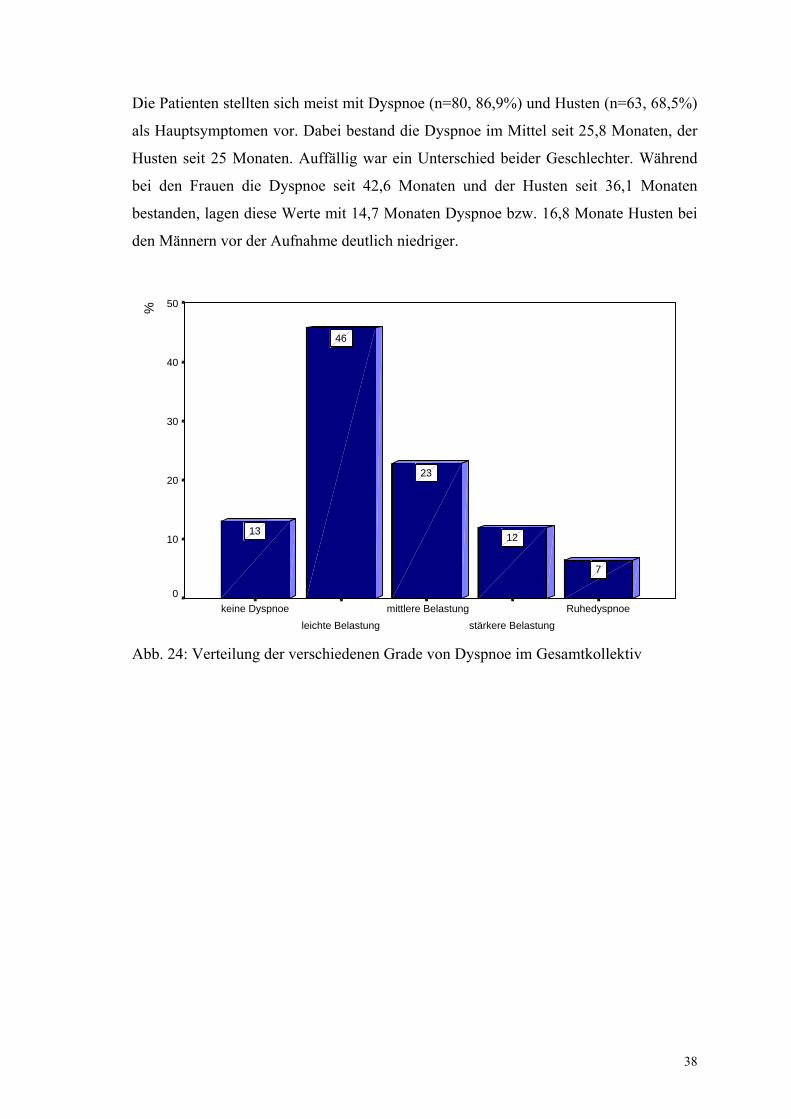
Die Patienten stellten sich meist mit Dyspnoe (n=80, 86,9%) und Husten (n=63, 68,5%)
als Hauptsymptomen vor. Dabei bestand die Dyspnoe im Mittel seit 25,8 Monaten, der
Husten seit 25 Monaten. Auffällig war ein Unterschied beider Geschlechter. Während
bei den Frauen die Dyspnoe seit 42,6 Monaten und der Husten seit 36,1 Monaten
bestanden, lagen diese Werte mit 14,7 Monaten Dyspnoe bzw. 16,8 Monate Husten bei
den Männern vor der Aufnahme deutlich niedriger.
Abb. 24: Verteilung der verschiedenen Grade von Dyspnoe im Gesamtkollektiv
Ruhedyspnoestärkere Belastung
mittlere Belastungleichte Belastung
keine Dyspnoe
% 50
40
30
20
10
0
7
12
23
46
13

39
Abb. 25: Verteilung der Symptome Husten und Auswurf im Gesamtkollektiv
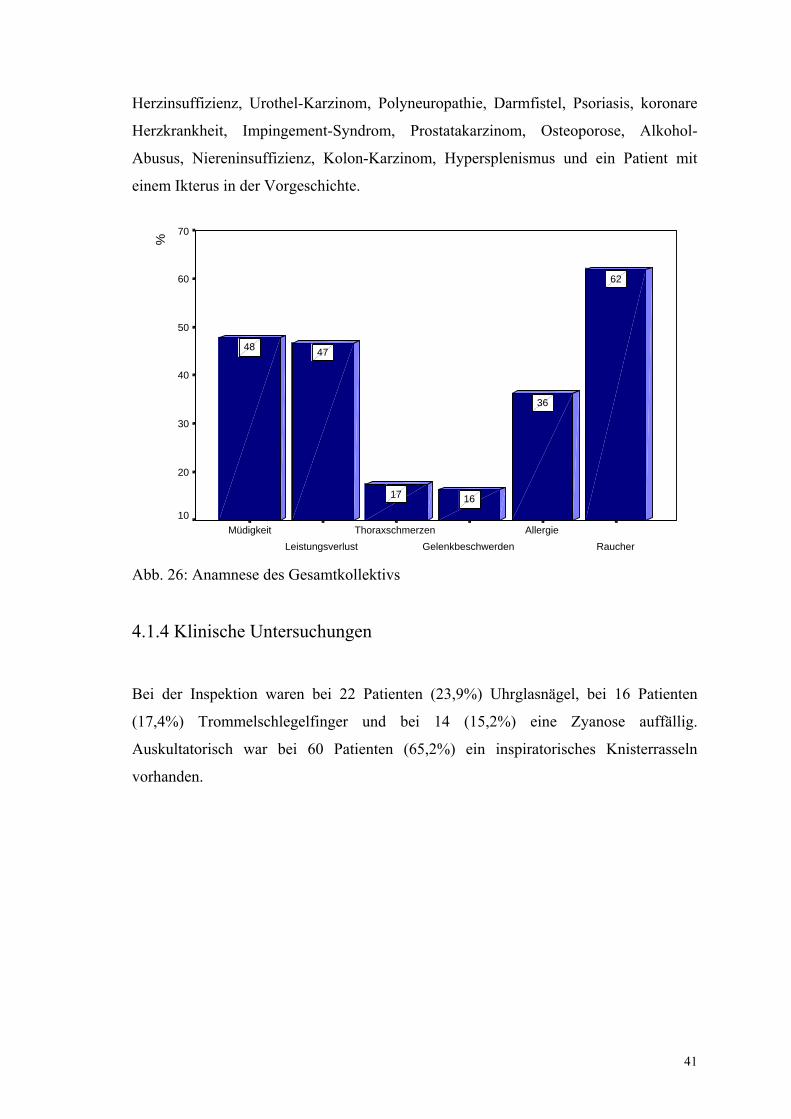
Über Müdigkeit klagten 44 Patienten (47,8%), 43 (46,7%) über Leistungsverlust.
Weitere körperliche Beschwerden waren Thoraxschmerzen (n=16, 17,4%) oder
Druckgefühl im Thoraxbereich (n=8, 8,7%), Gelenkbeschwerden (n=15, 16,3%),
Temperatur über 37,5°C (n=17, 18,5%), ungewollter Gewichtsverlust (n=21, 22,8%)
sowie ein grippaler Infekt zu Beginn der Erkrankung (n=26, 28,3%). Im
Gesamtkollektiv waren 57 (62%) Patienten Raucher, im Schnitt mit 15,15 pack-years.
Bei 33 Patienten (35,9%) war eine Allergie bekannt. Der durchschnittliche BMI beträgt
26,26 kg/m².
Bei der Umfeldanamnese gaben 18 Patienten an, Vögel innerhalb der Wohnung zu
halten oder früher gehabt zu haben (19,6%), davon 6 Patienten mit EAA, nur 4
Patienten in der Gruppe der IIP gaben aktuelle Vogelhaltung an (siehe S. 92). Insgesamt
8 Patienten hatten Kontakt zu Vögeln außerhalb der eigenen Wohnung (8,7%), davon 4
Patienten mit EAA. 9 Patienten hielten andere Haustiere wie Hunde, Katzen oder Fische
(9,8%).
Bei der Frage nach den Vorerkrankungen wurden die folgenden tabellarisch
aufgeführten Krankheiten angegeben:
produktiver HustenReizhusten + Auswurf
Reizhustennur Auswurf
kein Hustenfehlend
% 30
20
10
0
16
25
27
7
24
40
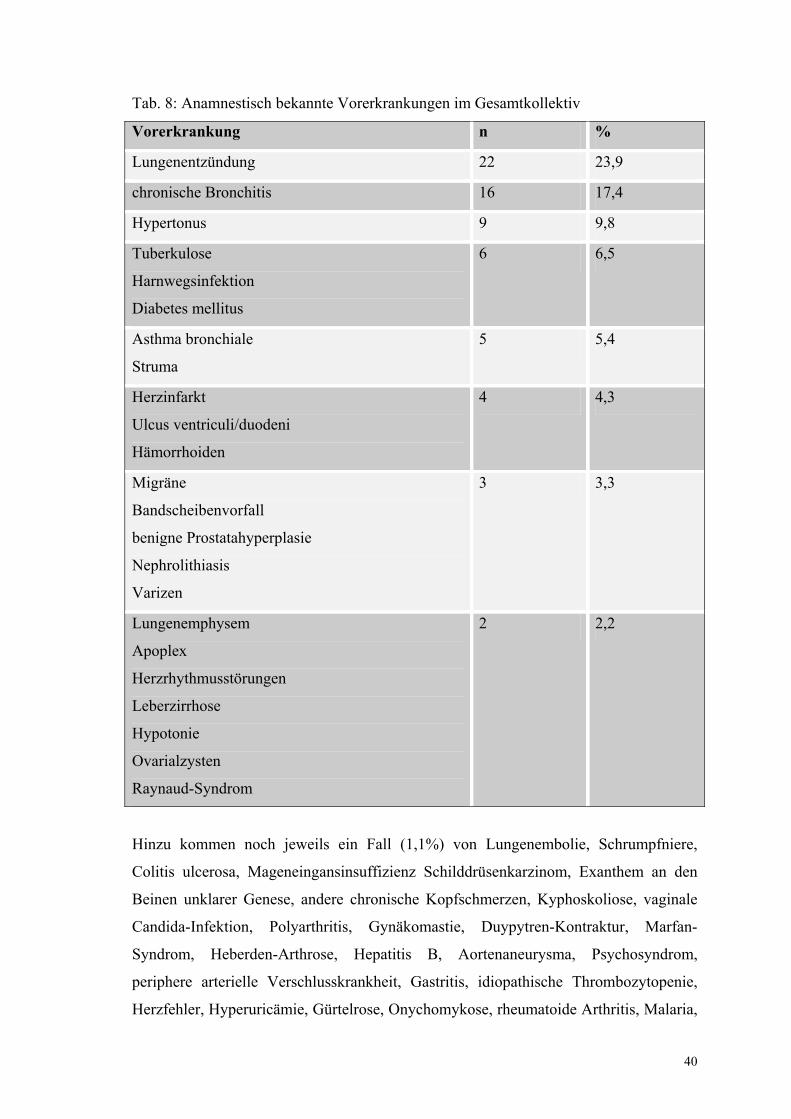
Tab. 8: Anamnestisch bekannte Vorerkrankungen im Gesamtkollektiv
Vorerkrankung n %
Lungenentzündung 22 23,9
chronische Bronchitis 16 17,4
Hypertonus 9 9,8
Tuberkulose
Harnwegsinfektion
Diabetes mellitus
6 6,5
Asthma bronchiale
Struma
5 5,4
Herzinfarkt
Ulcus ventriculi/duodeni
Hämorrhoiden
4 4,3
Migräne
Bandscheibenvorfall
benigne Prostatahyperplasie
Nephrolithiasis
Varizen
3 3,3
Lungenemphysem
Apoplex
Herzrhythmusstörungen
Leberzirrhose
Hypotonie
Ovarialzysten
Raynaud-Syndrom
2 2,2
Hinzu kommen noch jeweils ein Fall (1,1%) von Lungenembolie, Schrumpfniere,
Colitis ulcerosa, Mageneingansinsuffizienz Schilddrüsenkarzinom, Exanthem an den
Beinen unklarer Genese, andere chronische Kopfschmerzen, Kyphoskoliose, vaginale
Candida-Infektion, Polyarthritis, Gynäkomastie, Duypytren-Kontraktur, Marfan-
Syndrom, Heberden-Arthrose, Hepatitis B, Aortenaneurysma, Psychosyndrom,
periphere arterielle Verschlusskrankheit, Gastritis, idiopathische Thrombozytopenie,
Herzfehler, Hyperuricämie, Gürtelrose, Onychomykose, rheumatoide Arthritis, Malaria,
41
Herzinsuffizienz, Urothel-Karzinom, Polyneuropathie, Darmfistel, Psoriasis, koronare
Herzkrankheit, Impingement-Syndrom, Prostatakarzinom, Osteoporose, Alkohol-
Abusus, Niereninsuffizienz, Kolon-Karzinom, Hypersplenismus und ein Patient mit
einem Ikterus in der Vorgeschichte.
Abb. 26: Anamnese des Gesamtkollektivs
4.1.4 Klinische Untersuchungen
Bei der Inspektion waren bei 22 Patienten (23,9%) Uhrglasnägel, bei 16 Patienten
(17,4%) Trommelschlegelfinger und bei 14 (15,2%) eine Zyanose auffällig.
Auskultatorisch war bei 60 Patienten (65,2%) ein inspiratorisches Knisterrasseln
vorhanden.
RaucherAllergie
GelenkbeschwerdenThoraxschmerzen
LeistungsverlustMüdigkeit
% 70
60
50
40
30
20
10
62
36
1617
4748
42
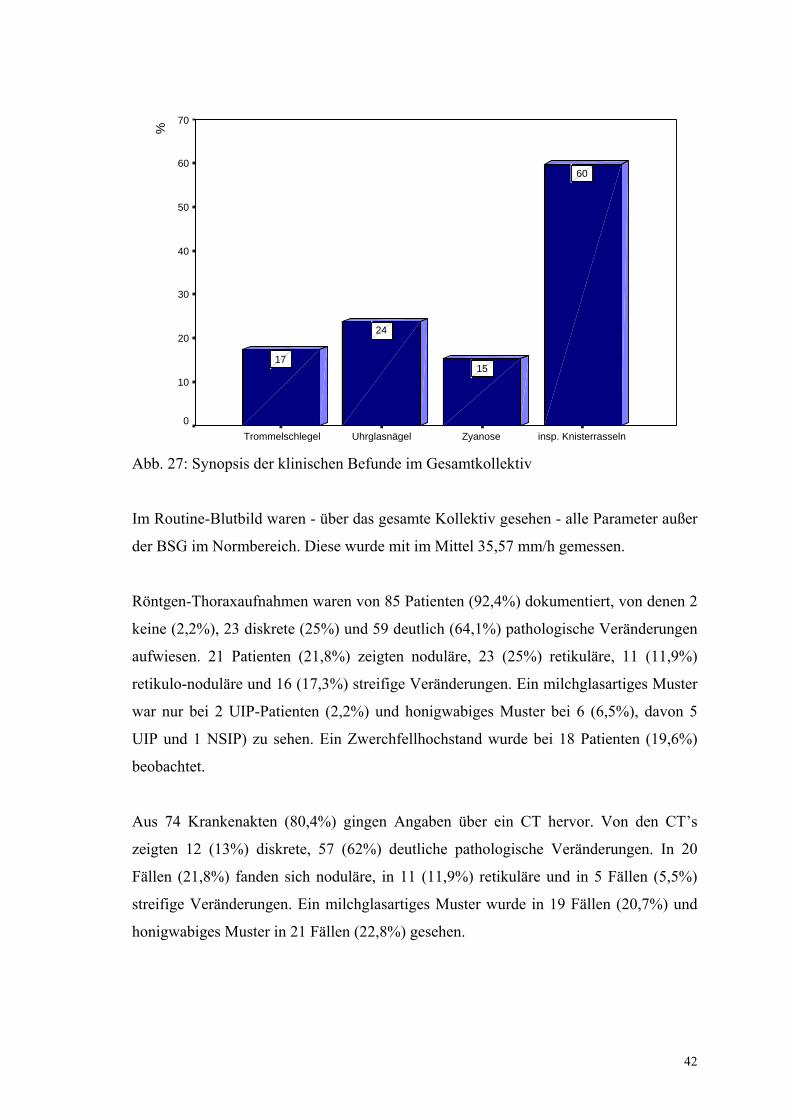
Abb. 27: Synopsis der klinischen Befunde im Gesamtkollektiv
Im Routine-Blutbild waren - über das gesamte Kollektiv gesehen - alle Parameter außer
der BSG im Normbereich. Diese wurde mit im Mittel 35,57 mm/h gemessen.
Röntgen-Thoraxaufnahmen waren von 85 Patienten (92,4%) dokumentiert, von denen 2
keine (2,2%), 23 diskrete (25%) und 59 deutlich (64,1%) pathologische Veränderungen
aufwiesen. 21 Patienten (21,8%) zeigten noduläre, 23 (25%) retikuläre, 11 (11,9%)
retikulo-noduläre und 16 (17,3%) streifige Veränderungen. Ein milchglasartiges Muster
war nur bei 2 UIP-Patienten (2,2%) und honigwabiges Muster bei 6 (6,5%), davon 5
UIP und 1 NSIP) zu sehen. Ein Zwerchfellhochstand wurde bei 18 Patienten (19,6%)
beobachtet.
Aus 74 Krankenakten (80,4%) gingen Angaben über ein CT hervor. Von den CT’s
zeigten 12 (13%) diskrete, 57 (62%) deutliche pathologische Veränderungen. In 20
Fällen (21,8%) fanden sich noduläre, in 11 (11,9%) retikuläre und in 5 Fällen (5,5%)
streifige Veränderungen. Ein milchglasartiges Muster wurde in 19 Fällen (20,7%) und
honigwabiges Muster in 21 Fällen (22,8%) gesehen.
insp. KnisterrasselnZyanoseUhrglasnägelTrommelschlegel
% 70
60
50
40
30
20
10
0
60
15
24
17
43
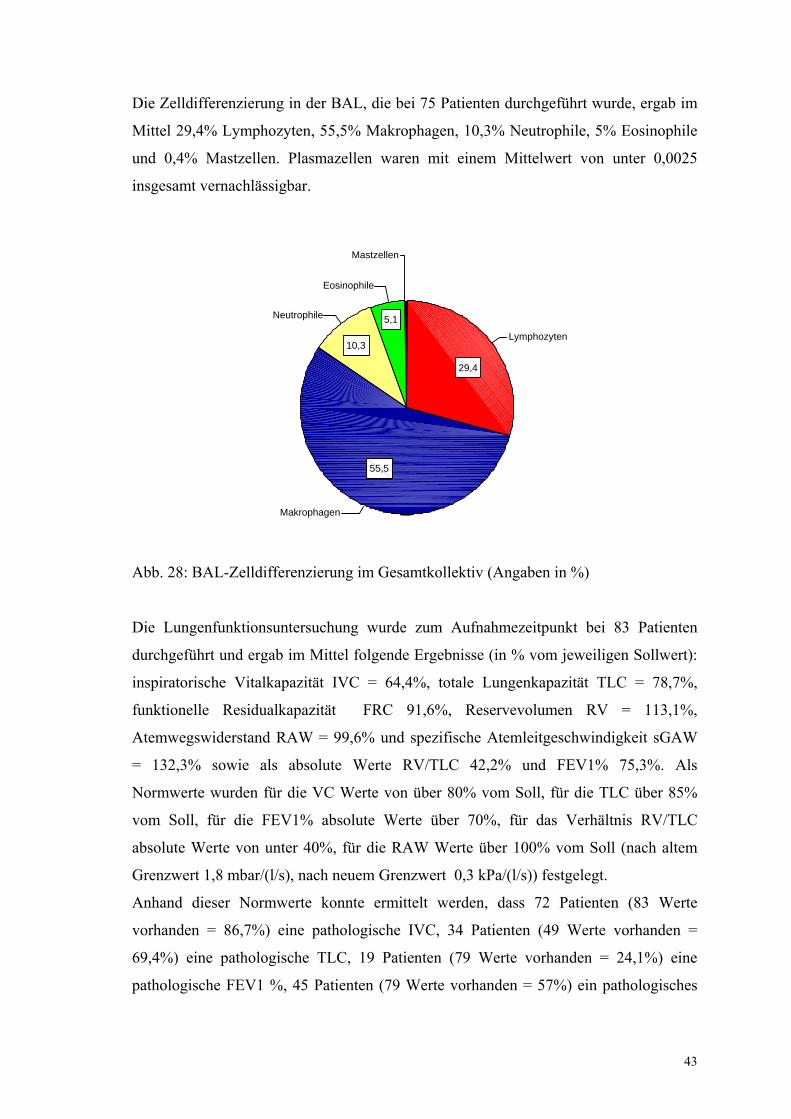
Die Zelldifferenzierung in der BAL, die bei 75 Patienten durchgeführt wurde, ergab im
Mittel 29,4% Lymphozyten, 55,5% Makrophagen, 10,3% Neutrophile, 5% Eosinophile
und 0,4% Mastzellen. Plasmazellen waren mit einem Mittelwert von unter 0,0025
insgesamt vernachlässigbar.
Abb. 28: BAL-Zelldifferenzierung im Gesamtkollektiv (Angaben in %)
Die Lungenfunktionsuntersuchung wurde zum Aufnahmezeitpunkt bei 83 Patienten
durchgeführt und ergab im Mittel folgende Ergebnisse (in % vom jeweiligen Sollwert):
inspiratorische Vitalkapazität IVC = 64,4%, totale Lungenkapazität TLC = 78,7%,
funktionelle Residualkapazität FRC 91,6%, Reservevolumen RV = 113,1%,
Atemwegswiderstand RAW = 99,6% und spezifische Atemleitgeschwindigkeit sGAW
= 132,3% sowie als absolute Werte RV/TLC 42,2% und FEV1% 75,3%. Als
Normwerte wurden für die VC Werte von über 80% vom Soll, für die TLC über 85%
vom Soll, für die FEV1% absolute Werte über 70%, für das Verhältnis RV/TLC
absolute Werte von unter 40%, für die RAW Werte über 100% vom Soll (nach altem
Grenzwert 1,8 mbar/(l/s), nach neuem Grenzwert 0,3 kPa/(l/s)) festgelegt.
Anhand dieser Normwerte konnte ermittelt werden, dass 72 Patienten (83 Werte
vorhanden = 86,7%) eine pathologische IVC, 34 Patienten (49 Werte vorhanden =
69,4%) eine pathologische TLC, 19 Patienten (79 Werte vorhanden = 24,1%) eine
pathologische FEV1 %, 45 Patienten (79 Werte vorhanden = 57%) ein pathologisches
5,1
10,3
55,5
29,4
Mastzellen
Eosinophile
Neutrophile
Makrophagen
Lymphozyten
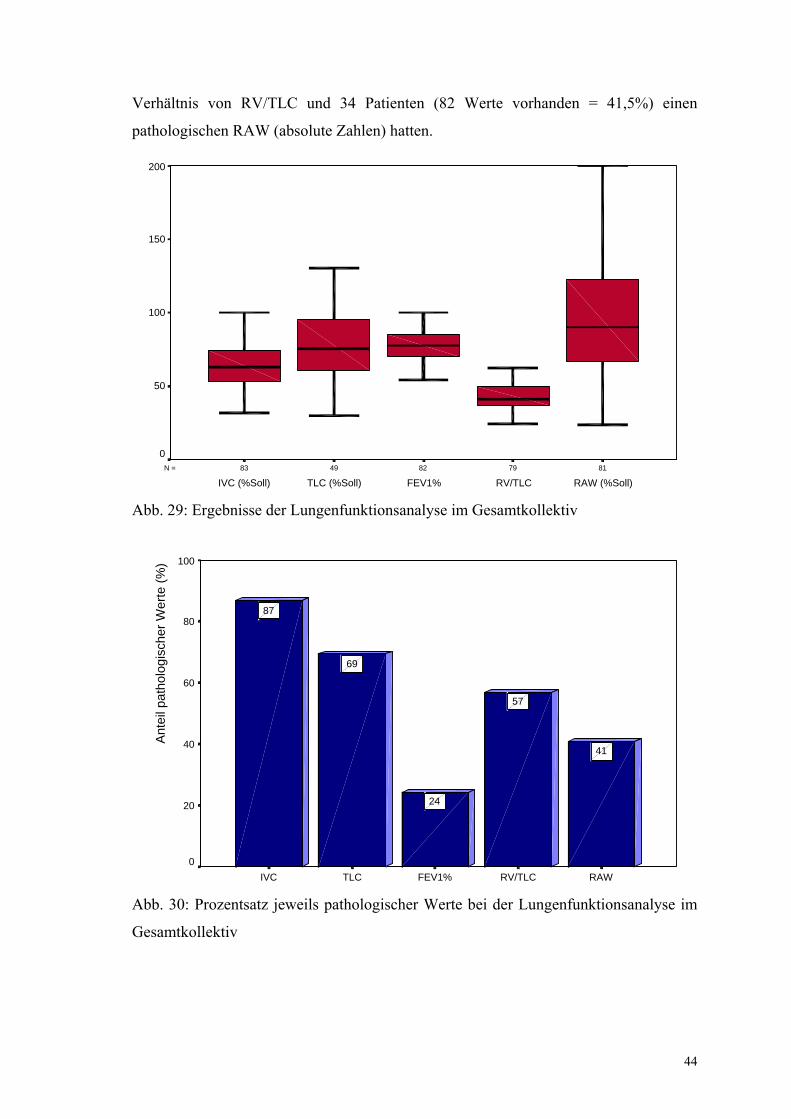
44
Verhältnis von RV/TLC und 34 Patienten (82 Werte vorhanden = 41,5%) einen
pathologischen RAW (absolute Zahlen) hatten.
Abb. 29: Ergebnisse der Lungenfunktionsanalyse im Gesamtkollektiv
Abb. 30: Prozentsatz jeweils pathologischer Werte bei der Lungenfunktionsanalyse im
Gesamtkollektiv
RAWRV/TLCFEV1%TLCIVC
Ant
eil p
atho
logi
sche
r Wer
te (%
) 100
80
60
40
20
0
41
57
24
69
87
8179824983N =
RAW (%Soll)RV/TLCFEV1%TLC (%Soll)IVC (%Soll)
200
150
100
50
0
45
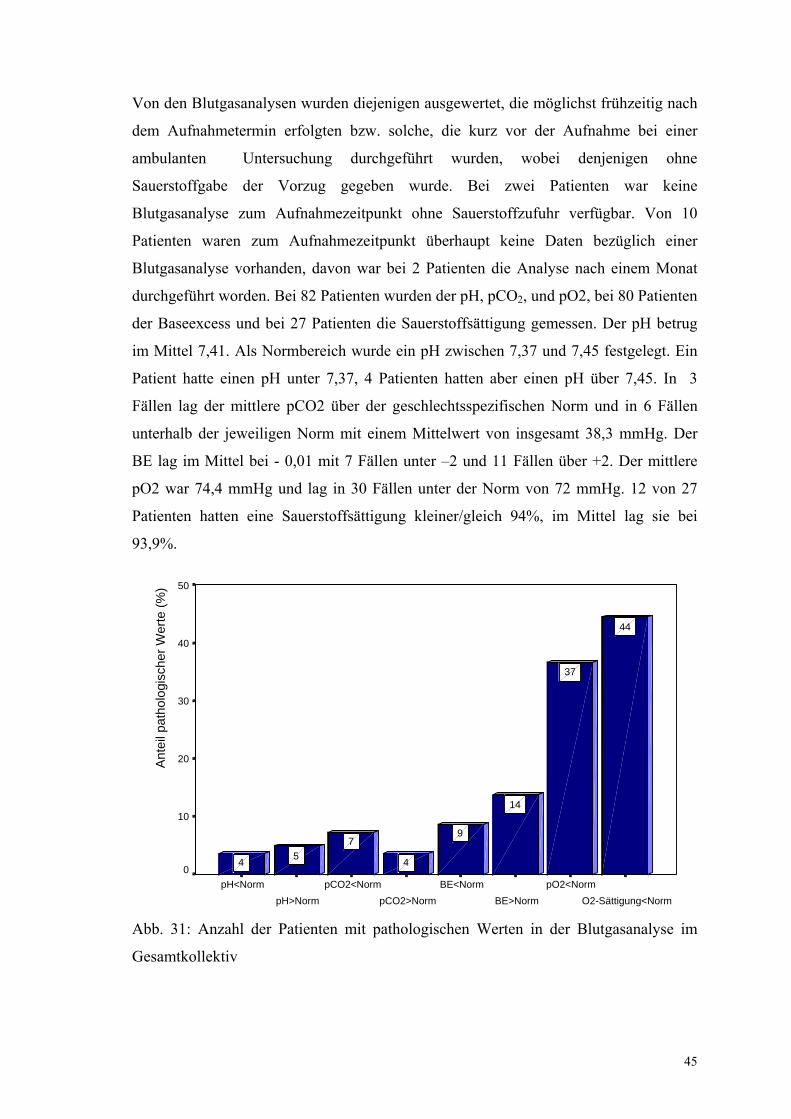
Von den Blutgasanalysen wurden diejenigen ausgewertet, die möglichst frühzeitig nach
dem Aufnahmetermin erfolgten bzw. solche, die kurz vor der Aufnahme bei einer
ambulanten Untersuchung durchgeführt wurden, wobei denjenigen ohne
Sauerstoffgabe der Vorzug gegeben wurde. Bei zwei Patienten war keine
Blutgasanalyse zum Aufnahmezeitpunkt ohne Sauerstoffzufuhr verfügbar. Von 10
Patienten waren zum Aufnahmezeitpunkt überhaupt keine Daten bezüglich einer
Blutgasanalyse vorhanden, davon war bei 2 Patienten die Analyse nach einem Monat
durchgeführt worden. Bei 82 Patienten wurden der pH, pCO2, und pO2, bei 80 Patienten
der Baseexcess und bei 27 Patienten die Sauerstoffsättigung gemessen. Der pH betrug
im Mittel 7,41. Als Normbereich wurde ein pH zwischen 7,37 und 7,45 festgelegt. Ein
Patient hatte einen pH unter 7,37, 4 Patienten hatten aber einen pH über 7,45. In 3
Fällen lag der mittlere pCO2 über der geschlechtsspezifischen Norm und in 6 Fällen
unterhalb der jeweiligen Norm mit einem Mittelwert von insgesamt 38,3 mmHg. Der
BE lag im Mittel bei - 0,01 mit 7 Fällen unter –2 und 11 Fällen über +2. Der mittlere
pO2 war 74,4 mmHg und lag in 30 Fällen unter der Norm von 72 mmHg. 12 von 27
Patienten hatten eine Sauerstoffsättigung kleiner/gleich 94%, im Mittel lag sie bei
93,9%.
Abb. 31: Anzahl der Patienten mit pathologischen Werten in der Blutgasanalyse im
Gesamtkollektiv
O2-Sättigung<NormpO2<Norm
BE>NormBE<Norm
pCO2>NormpCO2<Norm
pH>NormpH<Norm
Ant
eil p
atho
logi
sche
r Wer
te (%
) 50
40
30
20
10
0
44
37
14
9
4
754
46
4.1.5 Verlauf
Wenn die Patienten weiter zu ambulanten Untersuchungen in der Ruhrlandklinik waren,
lag zumindest der Bericht an den Hausarzt mit der Zwischenanamnese und den
klinischen Befunden vor, häufig auch Protokolle von Lungenfunktions- und
Blutgasanalysen. 18 Patienten (69,6%) kamen wiederholt zur stationären Aufnahme,
von ihnen liegen daher ebenfalls Verlaufsdaten vor.
Zum Endpunkt der Studie war von 82 Patienten bekannt, ob sie leben (n=60, 65,2%),
oder zwischenzeitlich verstorben sind (n=22, 23,9%). Diese Angaben konnten von 10
Patienten nicht erhoben werden. Die Follow-Up Dauer betrug insgesamt
durchschnittlich 3,19 Jahre.
Von 70 Patienten konnten detailliertere klinische Daten erhoben werden, so dass eine
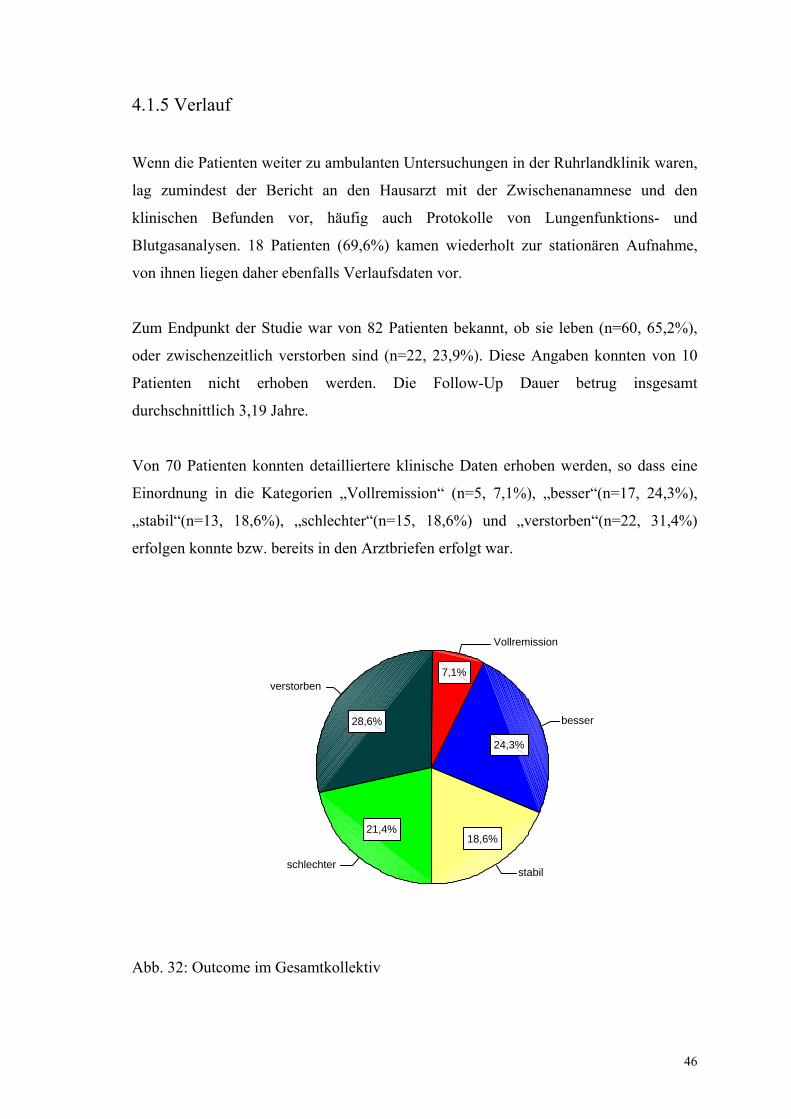
Einordnung in die Kategorien „Vollremission“ (n=5, 7,1%), „besser“(n=17, 24,3%),
„stabil“(n=13, 18,6%), „schlechter“(n=15, 18,6%) und „verstorben“(n=22, 31,4%)
erfolgen konnte bzw. bereits in den Arztbriefen erfolgt war.
Abb. 32: Outcome im Gesamtkollektiv
28,6%
21,4%18,6%
24,3%
7,1%verstorben
schlechterstabil
besser
Vollremission
47
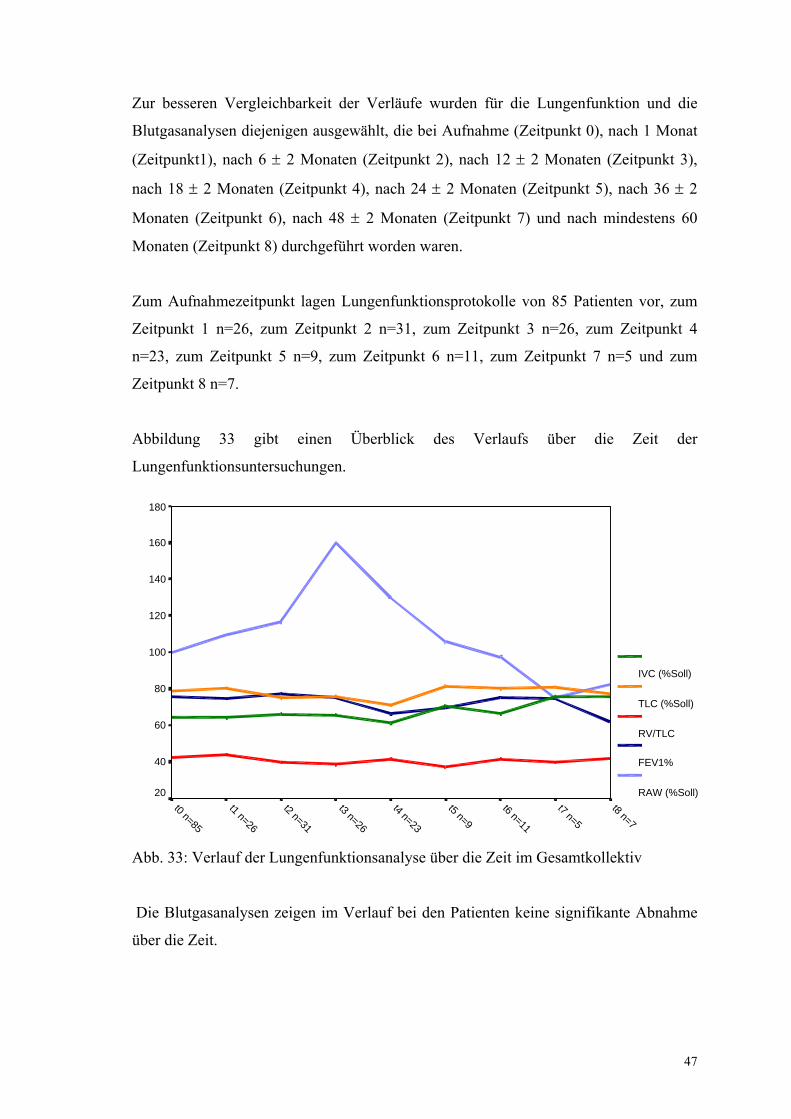
Zur besseren Vergleichbarkeit der Verläufe wurden für die Lungenfunktion und die
Blutgasanalysen diejenigen ausgewählt, die bei Aufnahme (Zeitpunkt 0), nach 1 Monat
(Zeitpunkt1), nach 6 ± 2 Monaten (Zeitpunkt 2), nach 12 ± 2 Monaten (Zeitpunkt 3),
nach 18 ± 2 Monaten (Zeitpunkt 4), nach 24 ± 2 Monaten (Zeitpunkt 5), nach 36 ± 2
Monaten (Zeitpunkt 6), nach 48 ± 2 Monaten (Zeitpunkt 7) und nach mindestens 60
Monaten (Zeitpunkt 8) durchgeführt worden waren.
Zum Aufnahmezeitpunkt lagen Lungenfunktionsprotokolle von 85 Patienten vor, zum
Zeitpunkt 1 n=26, zum Zeitpunkt 2 n=31, zum Zeitpunkt 3 n=26, zum Zeitpunkt 4
n=23, zum Zeitpunkt 5 n=9, zum Zeitpunkt 6 n=11, zum Zeitpunkt 7 n=5 und zum
Zeitpunkt 8 n=7.
Abbildung 33 gibt einen Überblick des Verlaufs über die Zeit der
Lungenfunktionsuntersuchungen.
Abb. 33: Verlauf der Lungenfunktionsanalyse über die Zeit im Gesamtkollektiv
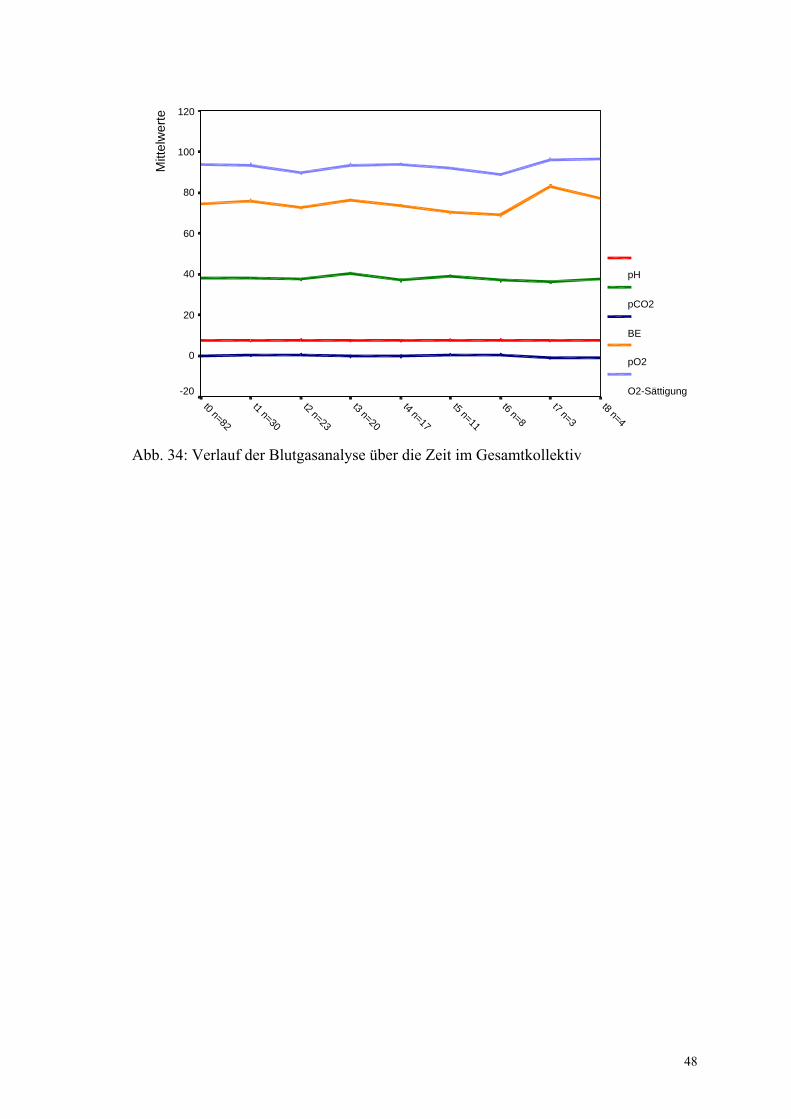
Die Blutgasanalysen zeigen im Verlauf bei den Patienten keine signifikante Abnahme
über die Zeit.
t8 n=7t7 n=5
t6 n=11
t5 n=9t4 n=23
t3 n=26
t2 n=31
t1 n=26
t0 n=85
180
160
140
120
100
80
60
40
20
IVC (%Soll)
TLC (%Soll)
RV/TLC
FEV1%
RAW (%Soll)
48
Abb. 34: Verlauf der Blutgasanalyse über die Zeit im Gesamtkollektiv
t8 n=4t7 n=3
t6 n=8t5 n=11
t4 n=17
t3 n=20
t2 n=23
t1 n=30
t0 n=82
Mitt
elw
erte 120
100
80
60
40
20
0
-20
pH
pCO2
BE
pO2
O2-Sättigung

49
4.2 Idiopathische pulmonale Fibrose
4.2.1 Anamnese
Der typische Patient mit einer IPF ist männlich (Geschlechtsverteilung m:w = 4,4:1,
Männer n=22, Frauen n=5), im Mittel 59,4 Jahre alt und klagt über Dyspnoe (n = 26,
96,3%) und Husten (n = 21, 77,7%). Im Durchschnitt bestehen sie seit 18,8 bzw. 21,1
Monaten.
Über Auswurf klagten 12 Patienten (44,4%), wobei dieser zumeist weiß oder gelblich
gefärbt war. Müdigkeit und Leistungsverlust waren bei 13 (48,1%) Patienten ein
Problem, bei 6 (22,2%) war ein grippaler Infekt vorausgegangen, 8 (29,6%) beschrieben
Thoraxschmerzen, 3 (11,1%) Gelenkbeschwerden. Diese waren bei einem Patienten
isoliert vorhanden, bei den beiden anderen in Kombination mit Thoraxschmerzen
aufgetreten. Nur 2 Patienten (7,4%) hatten eine Körpertemperatur von 37,6°C, alle
anderen Temperaturen lagen unter 37,5°C.
11 (40,1%) gaben an, an einer Allergie (Medikamente, Farbstoffe, Tierhaare,
Hausstaub, Pollen) zu leiden. Raucher waren 18 (66,7%) Patienten mit einem
durchschnittlichen Konsum von 17,2 pack years. Der mittlere BMI lag bei 28,2 kg/m²,
wobei 3 Patienten (11,1%) einen ungewollten Gewichtsverlust von 2, 5 und 6 kg
angaben. 4 Patienten verzeichneten eine Gewichtszunahme (14,6%). In der
Krankengeschichte berichteten bezüglich Vorerkrankungen der Atemwege 3 Patienten
(11,1%) von einer Pneumonie, ebenfalls 3 von einem chronischen Asthma, 2 von einer
chronischen Bronchitis (7,4%) und einer von einer Tuberkulose im Kindesalter.
Ansonsten wurde zweimal ein Ulcus ventriculi, ein Hypertonus, eine benigne
Prostatahyperplasie, Herzrhythmusstörungen und jeweils einmal eine postoperative
Lungenembolie angegeben, ein Herzinfarkt, eine Struma, eine Hepatitis B, ein
Aortenaneurysma, eine periphere arterielle Verschlusskrankheit, eine koronare
Herzkrankheit, ein Prostatakarzinom, ein Kolonkarzinom sowie eine Psoriasis.
33,3% (n = 9) der Patienten waren bei Aufnahme mit Steroiden antherapiert.
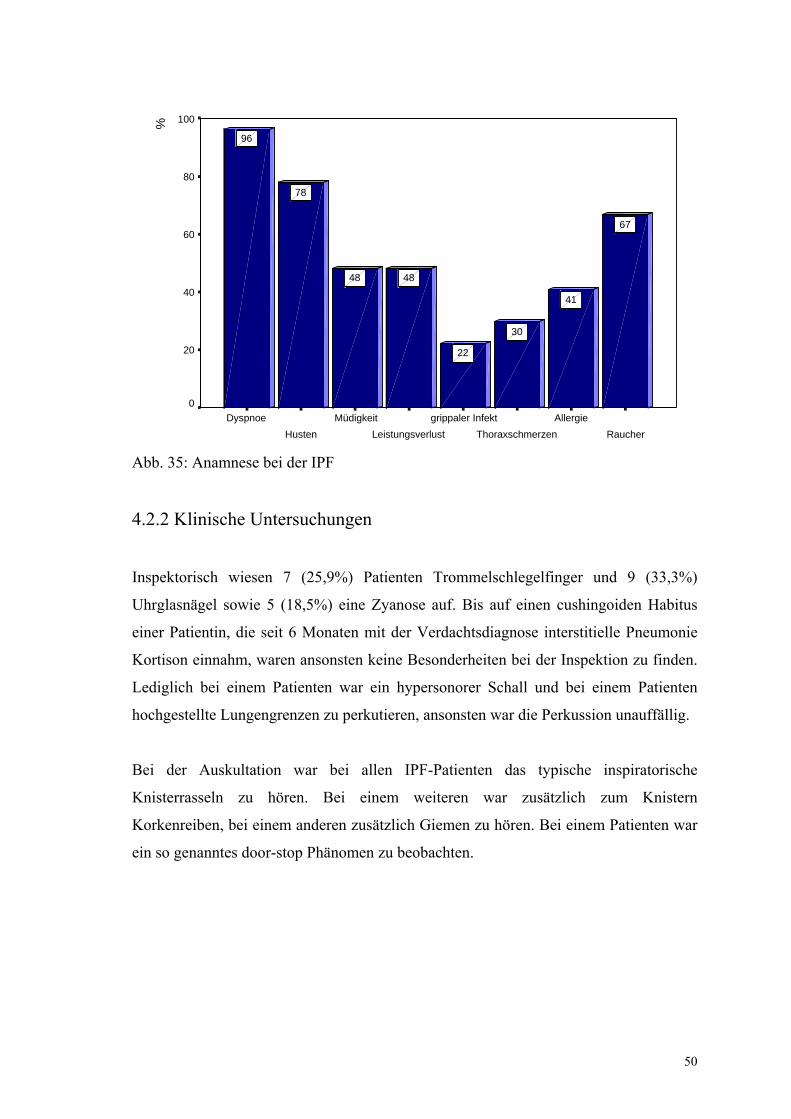
50
Abb. 35: Anamnese bei der IPF
4.2.2 Klinische Untersuchungen
Inspektorisch wiesen 7 (25,9%) Patienten Trommelschlegelfinger und 9 (33,3%)
Uhrglasnägel sowie 5 (18,5%) eine Zyanose auf. Bis auf einen cushingoiden Habitus
einer Patientin, die seit 6 Monaten mit der Verdachtsdiagnose interstitielle Pneumonie
Kortison einnahm, waren ansonsten keine Besonderheiten bei der Inspektion zu finden.
Lediglich bei einem Patienten war ein hypersonorer Schall und bei einem Patienten
hochgestellte Lungengrenzen zu perkutieren, ansonsten war die Perkussion unauffällig.
Bei der Auskultation war bei allen IPF-Patienten das typische inspiratorische
Knisterrasseln zu hören. Bei einem weiteren war zusätzlich zum Knistern
Korkenreiben, bei einem anderen zusätzlich Giemen zu hören. Bei einem Patienten war
ein so genanntes door-stop Phänomen zu beobachten.
RaucherAllergie
Thoraxschmerzengrippaler Infekt
LeistungsverlustMüdigkeit
HustenDyspnoe
% 100
80
60
40
20
0
67
41
30
22
4848
78
96
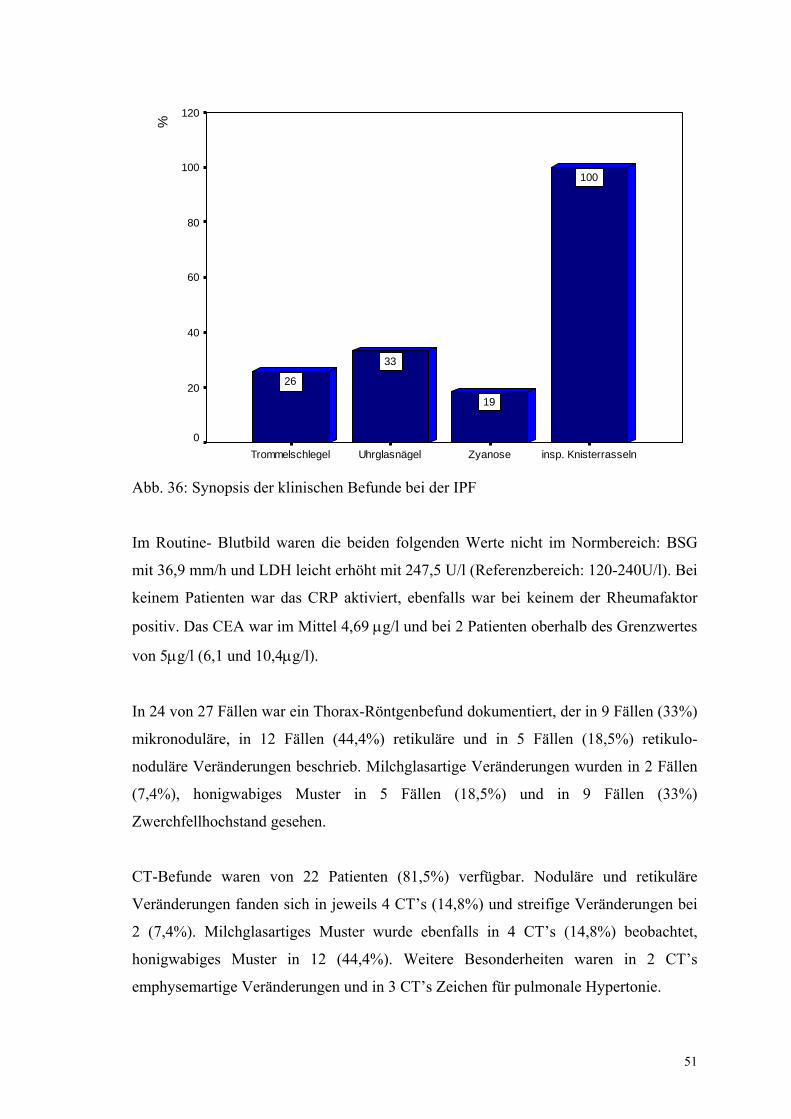
51
insp. KnisterrasselnZyanoseUhrglasnägelTrommelschlegel
% 120
100
80
60
40
20
0
100
19
33
26
Abb. 36: Synopsis der klinischen Befunde bei der IPF
Im Routine- Blutbild waren die beiden folgenden Werte nicht im Normbereich: BSG
mit 36,9 mm/h und LDH leicht erhöht mit 247,5 U/l (Referenzbereich: 120-240U/l). Bei
keinem Patienten war das CRP aktiviert, ebenfalls war bei keinem der Rheumafaktor
positiv. Das CEA war im Mittel 4,69 μg/l und bei 2 Patienten oberhalb des Grenzwertes
von 5μg/l (6,1 und 10,4μg/l).
In 24 von 27 Fällen war ein Thorax-Röntgenbefund dokumentiert, der in 9 Fällen (33%)
mikronoduläre, in 12 Fällen (44,4%) retikuläre und in 5 Fällen (18,5%) retikulo-
noduläre Veränderungen beschrieb. Milchglasartige Veränderungen wurden in 2 Fällen
(7,4%), honigwabiges Muster in 5 Fällen (18,5%) und in 9 Fällen (33%)
Zwerchfellhochstand gesehen.
CT-Befunde waren von 22 Patienten (81,5%) verfügbar. Noduläre und retikuläre
Veränderungen fanden sich in jeweils 4 CT’s (14,8%) und streifige Veränderungen bei
2 (7,4%). Milchglasartiges Muster wurde ebenfalls in 4 CT’s (14,8%) beobachtet,
honigwabiges Muster in 12 (44,4%). Weitere Besonderheiten waren in 2 CT’s
emphysemartige Veränderungen und in 3 CT’s Zeichen für pulmonale Hypertonie.
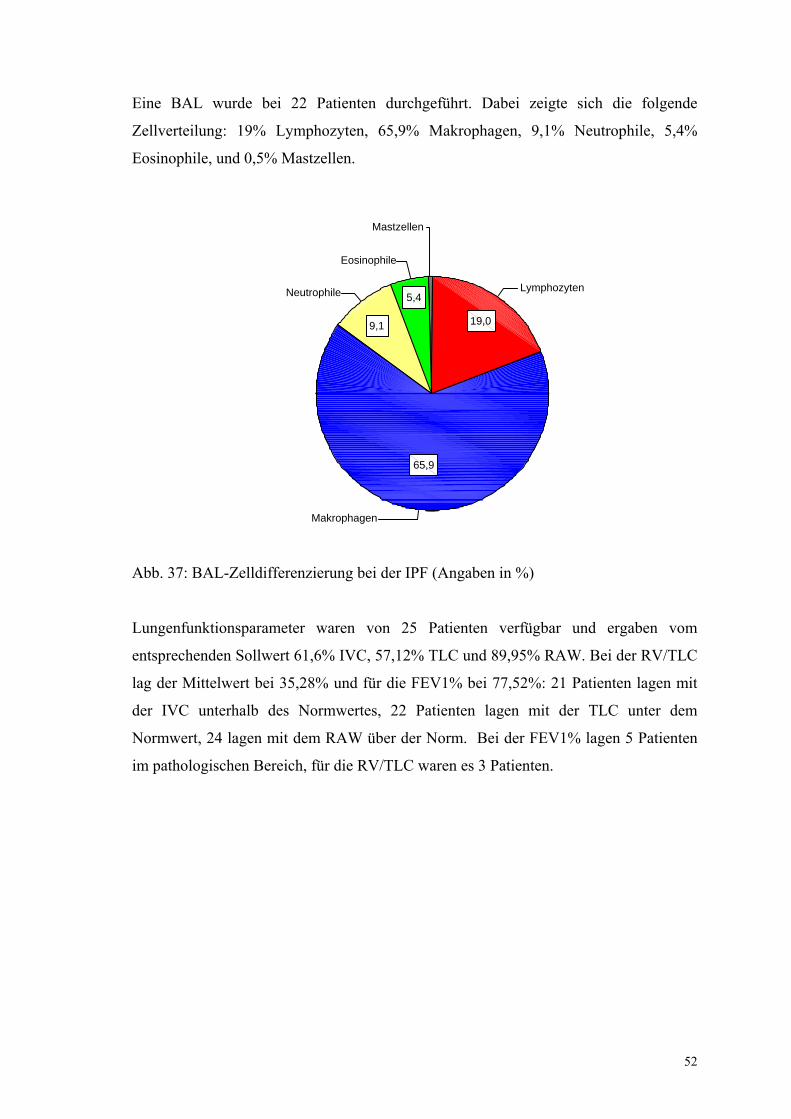
52
Eine BAL wurde bei 22 Patienten durchgeführt. Dabei zeigte sich die folgende
Zellverteilung: 19% Lymphozyten, 65,9% Makrophagen, 9,1% Neutrophile, 5,4%
Eosinophile, und 0,5% Mastzellen.
Abb. 37: BAL-Zelldifferenzierung bei der IPF (Angaben in %)
Lungenfunktionsparameter waren von 25 Patienten verfügbar und ergaben vom
entsprechenden Sollwert 61,6% IVC, 57,12% TLC und 89,95% RAW. Bei der RV/TLC
lag der Mittelwert bei 35,28% und für die FEV1% bei 77,52%: 21 Patienten lagen mit
der IVC unterhalb des Normwertes, 22 Patienten lagen mit der TLC unter dem
Normwert, 24 lagen mit dem RAW über der Norm. Bei der FEV1% lagen 5 Patienten
im pathologischen Bereich, für die RV/TLC waren es 3 Patienten.
5,4
9,1
65,9
19,0
Mastzellen
Eosinophile
Neutrophile
Makrophagen
Lymphozyten
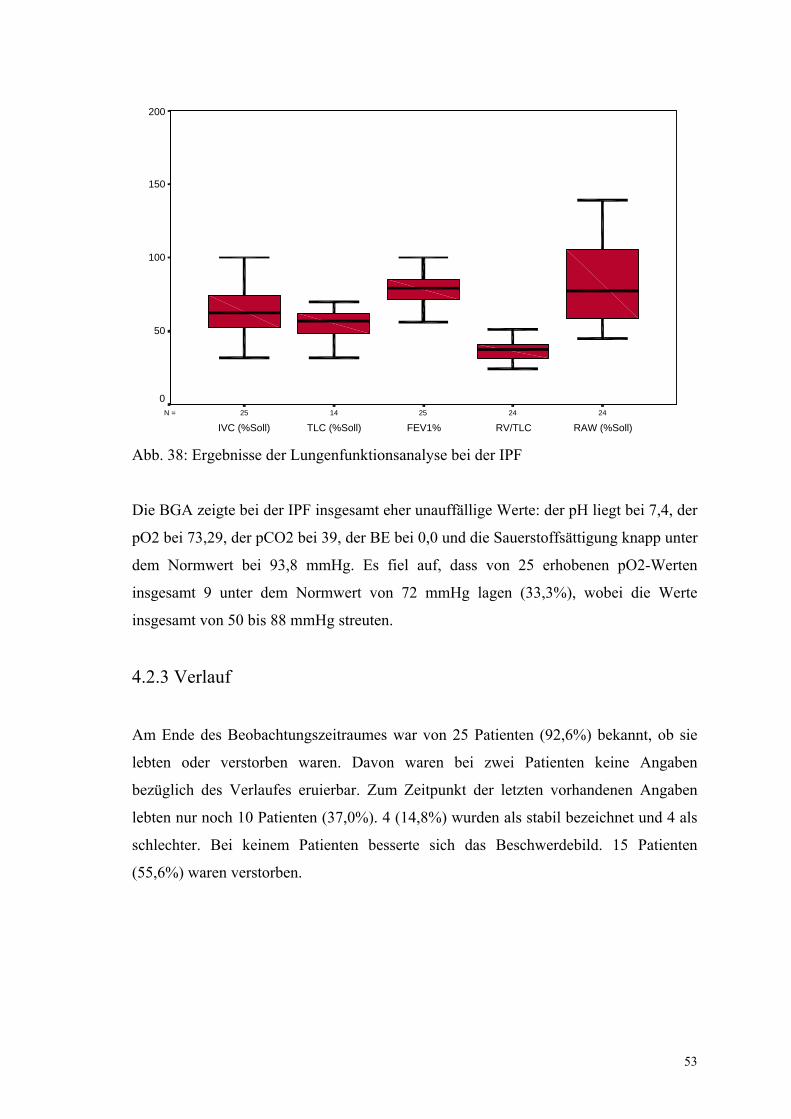
53
Abb. 38: Ergebnisse der Lungenfunktionsanalyse bei der IPF
Die BGA zeigte bei der IPF insgesamt eher unauffällige Werte: der pH liegt bei 7,4, der
pO2 bei 73,29, der pCO2 bei 39, der BE bei 0,0 und die Sauerstoffsättigung knapp unter
dem Normwert bei 93,8 mmHg. Es fiel auf, dass von 25 erhobenen pO2-Werten
insgesamt 9 unter dem Normwert von 72 mmHg lagen (33,3%), wobei die Werte
insgesamt von 50 bis 88 mmHg streuten.
4.2.3 Verlauf
Am Ende des Beobachtungszeitraumes war von 25 Patienten (92,6%) bekannt, ob sie
lebten oder verstorben waren. Davon waren bei zwei Patienten keine Angaben
bezüglich des Verlaufes eruierbar. Zum Zeitpunkt der letzten vorhandenen Angaben
lebten nur noch 10 Patienten (37,0%). 4 (14,8%) wurden als stabil bezeichnet und 4 als
schlechter. Bei keinem Patienten besserte sich das Beschwerdebild. 15 Patienten
(55,6%) waren verstorben.
2424251425N =
RAW (%Soll)RV/TLCFEV1%TLC (%Soll)IVC (%Soll)
200
150
100
50
0
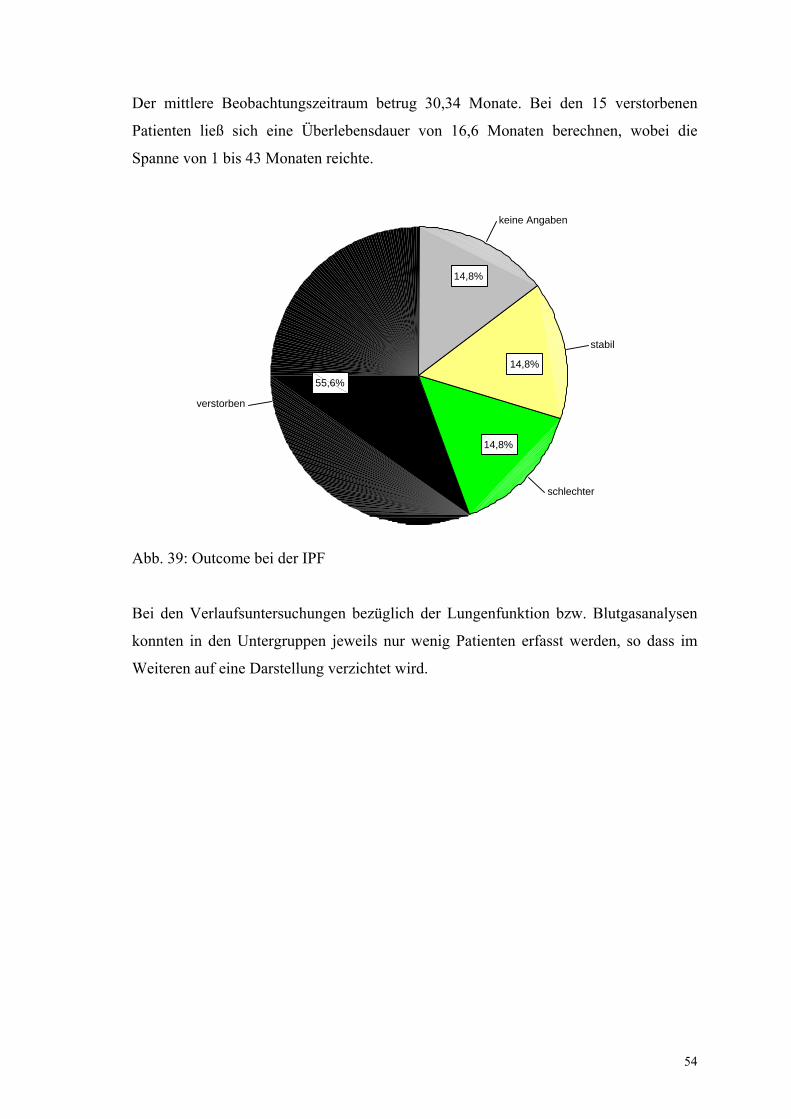
54
Der mittlere Beobachtungszeitraum betrug 30,34 Monate. Bei den 15 verstorbenen
Patienten ließ sich eine Überlebensdauer von 16,6 Monaten berechnen, wobei die
Spanne von 1 bis 43 Monaten reichte.
Abb. 39: Outcome bei der IPF
Bei den Verlaufsuntersuchungen bezüglich der Lungenfunktion bzw. Blutgasanalysen
konnten in den Untergruppen jeweils nur wenig Patienten erfasst werden, so dass im
Weiteren auf eine Darstellung verzichtet wird.
55,6%
14,8%
14,8%
14,8%
verstorben
schlechter
stabil
keine Angaben

55
4.3 Respiratorische Bronchiolitis mit interstitieller Lungenerkrankung
4.3.1 Anamnese
Die Geschlechtsverteilung der 6 Patienten mit RBILD beträgt m:w = 5:1. Das
Durchschnittsalter liegt bei 44,6 Jahren. Das Beschwerdebild zeichnet sich durch
Dyspnoe (n=4, 66,7%), Husten (n=3, 50%), Auswurf (n=4, 66,7%), Müdigkeit (n=4,
66,7%) und Leistungsverlust (n=2, 33,3%) aus. Weiterhin klagten zwei Patienten
(33,3%) über einen grippalen Infekt zu Beginn der Erkrankung, ebenfalls zwei über
erhöhte Temperatur bis maximal 38,2°C (33,3%), ein Patient (16,7%) hatte
Thoraxschmerzen, zwei (33,3%) Gelenkbeschwerden und einer (16,7%) litt an einer
Allergie gegen Penicillin. Die Dyspnoe bestand im Mittel seit 14,5 Monaten und der
Husten seit 26,5 Monaten. Wie bei der RBILD typisch, waren alle Patienten Raucher.
Der Durchschnittliche Zigarettenverzehr lag bei 26,6 pack years. Der BMI betrug im
Schnitt 25,7 kg/m². Anamnestisch war weiterhin bei 2 Patienten eine Pneumonie
(33,3%) bekannt, ein Patient gab ein Ulcus ventriculi, Kopfschmerzen und
Hämorrhoiden an (16,7%), bei einem anderen war ein Karzinom der Schilddrüse
bekannt, ein anderer Patient hatte eine Skoliose, ein anderer berichtete über eine
Hüftgelenksarthrose und eine Bandscheiben-OP, ein weiterer Patient hatte eine
Leberzirrhose aufgrund einer chronischer Hepatitis sowie eine Nephrolithiasis, und ein
anderer hatte bereits eine Myokarditis durchgemacht, außerdem bestanden HWS/BWS-
Beschwerden, Psoriasis und Kopfschmerzen.
Ein Patient hatte zum Zeitpunkt der Aufnahme eine orale Steroidmedikation.
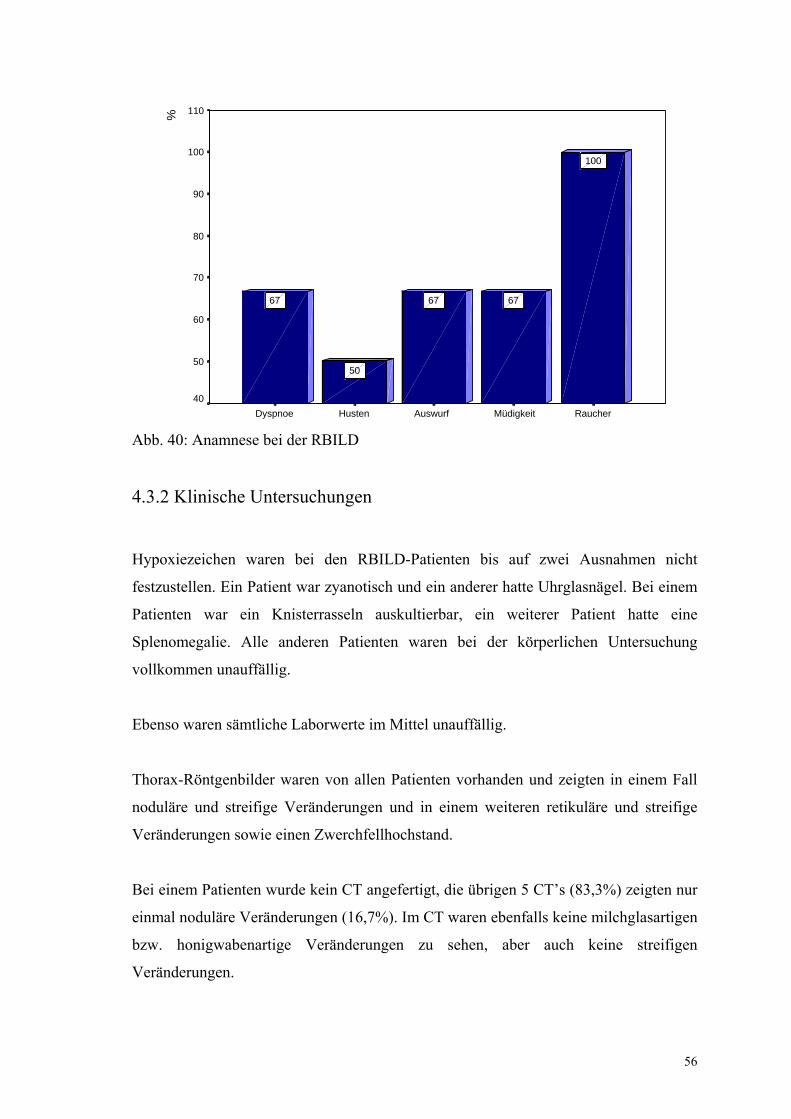
56
Abb. 40: Anamnese bei der RBILD
4.3.2 Klinische Untersuchungen
Hypoxiezeichen waren bei den RBILD-Patienten bis auf zwei Ausnahmen nicht
festzustellen. Ein Patient war zyanotisch und ein anderer hatte Uhrglasnägel. Bei einem
Patienten war ein Knisterrasseln auskultierbar, ein weiterer Patient hatte eine
Splenomegalie. Alle anderen Patienten waren bei der körperlichen Untersuchung
vollkommen unauffällig.
Ebenso waren sämtliche Laborwerte im Mittel unauffällig.
Thorax-Röntgenbilder waren von allen Patienten vorhanden und zeigten in einem Fall
noduläre und streifige Veränderungen und in einem weiteren retikuläre und streifige
Veränderungen sowie einen Zwerchfellhochstand.
Bei einem Patienten wurde kein CT angefertigt, die übrigen 5 CT’s (83,3%) zeigten nur
einmal noduläre Veränderungen (16,7%). Im CT waren ebenfalls keine milchglasartigen
bzw. honigwabenartige Veränderungen zu sehen, aber auch keine streifigen
Veränderungen.
RaucherMüdigkeitAuswurfHustenDyspnoe
% 110
100
90
80
70
60
50
40
100
6767
50
67
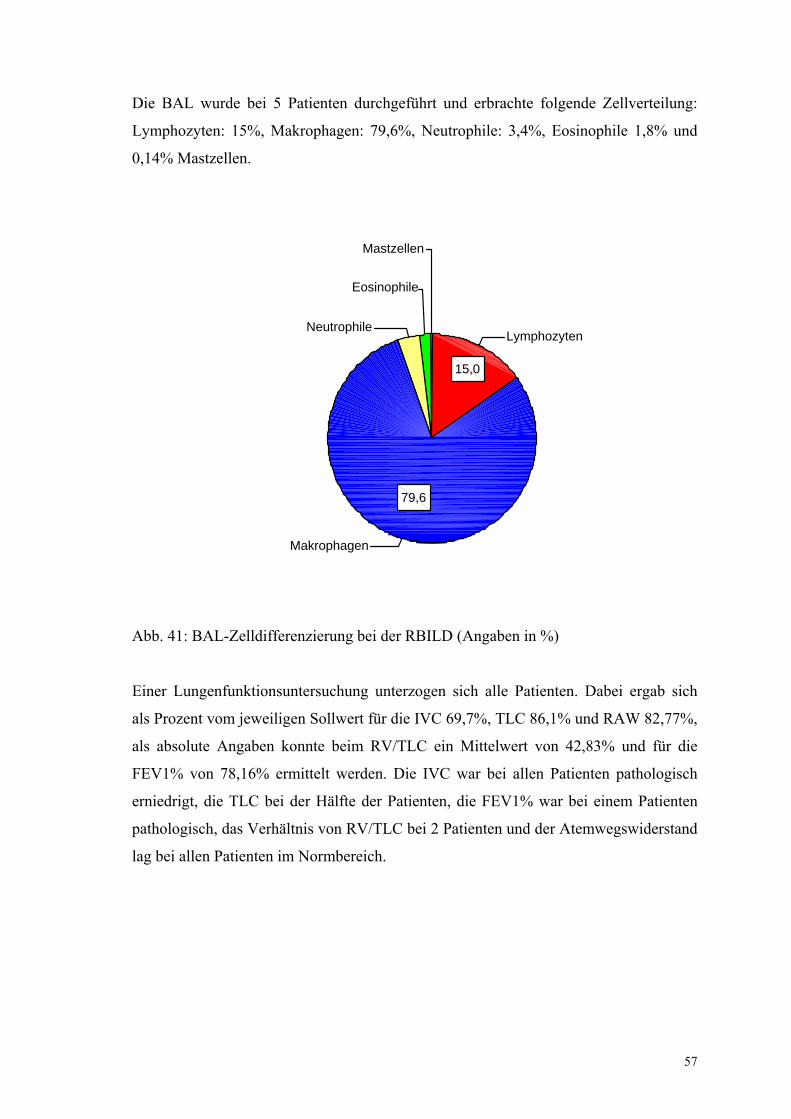
57
Die BAL wurde bei 5 Patienten durchgeführt und erbrachte folgende Zellverteilung:
Lymphozyten: 15%, Makrophagen: 79,6%, Neutrophile: 3,4%, Eosinophile 1,8% und
0,14% Mastzellen.
Abb. 41: BAL-Zelldifferenzierung bei der RBILD (Angaben in %)
Einer Lungenfunktionsuntersuchung unterzogen sich alle Patienten. Dabei ergab sich
als Prozent vom jeweiligen Sollwert für die IVC 69,7%, TLC 86,1% und RAW 82,77%,
als absolute Angaben konnte beim RV/TLC ein Mittelwert von 42,83% und für die
FEV1% von 78,16% ermittelt werden. Die IVC war bei allen Patienten pathologisch
erniedrigt, die TLC bei der Hälfte der Patienten, die FEV1% war bei einem Patienten
pathologisch, das Verhältnis von RV/TLC bei 2 Patienten und der Atemwegswiderstand
lag bei allen Patienten im Normbereich.
79,6
15,0
Mastzellen
Eosinophile
Neutrophile
Makrophagen
Lymphozyten

58
Abb. 42: Ergebnisse der Lungenfunktionsanalyse bei der RBILD
Für die BGA zeigten sich im Mittel Normwerte, einzelne Patienten unterschritten aber
die Normwerte. Ein Patient hatte einen BE von –3, der pO2-Wert war einmal bei 62 und
einmal bei 66 mmHg und ein Patient hatte eine Sättigung von 92,2%.
4.3.3 Verlauf
Am Ende der Studie war über die RBILD-Patienten bekannt, dass alle 6 Patienten leben.
Die mittlere Follow-up Dauer war dabei 4,9 Jahre. Genauere Angaben waren nur bei 3
Patienten vorhanden, von denen 1 (16,7%) unter Steroidtherapie als besser und 2
(33,3%) als stabil eingeordnet wurden. Einer dieser Patienten erhielt direkt eine
Steroidtherapie und war damit stabil. Der andere Patient war zunächst ohne Therapie
stabil, eine kurze Episode mit Verschlechterung konnte mit Steroiden abgefangen
werden. Es ist nur über diesen Patienten bekannt, dass er seinen Tabakkonsum
fortführte, bei den anderen war über den Raucherstatus im Follow-up nichts
dokumentiert.
Von den RBILD-Patienten kamen nur die drei genannten zu ambulanten Kontrollen.
33333N =
RAW (%Soll)FEV1%RV/TLCTLC (%Soll)IVC (%Soll)
120
100
80
60
40
20
0

59
4.4 Desquamative interstitielle Pneumonie
4.4.1 Anamnese
In der Gruppe der DIP waren von den 3 Patienten zwei männlich. Das
Durchschnittsalter liegt bei 50,5 Jahren. Ein Patient klagte über Reizhusten in
Zusammenhang mit einem bronchialen Infekt, der bereits seit einem Monat bestand.
Dieser Patient gab außerdem Gelenkbeschwerden und Fieber an, das CRP war auf 10,5
mg/l aktiviert, zudem bestand eine leichte Leukozytose. Bei einem weiteren Patienten
bestand eine Ruhedyspnoe, die sich schleichend entwickelt hatte, der andere Patient
hatte eine Dyspnoe bei alltäglicher Belastung, die schon seit 23 Jahren in
Zusammenhang mit einem Asthma bronchiale bestand. Über Auswurf klagte keiner der
DIP-Patienten, hingegen litten alle unter Müdigkeit und Leistungsverlust. Weitere
Erkrankungen waren bei einem Patienten ein Urothel-Karzinom, einer hatte eine
chronische Bronchitits und der dritte Patient litt unter einer Hypertension und
Kopfschmerzen.
Raucher waren zwei Patienten mit 8 pack years. Der BMI lag bei 31,8 kg/m². Ein
Patient war mit Steroiden antherapiert.
4.4.2 Klinische Untersuchungen
Bei der DIP war sowohl die Inspektion als auch die Perkussion völlig unauffällig.
Lediglich der Patient mit dem bronchialen Infekt bot in der Auskultation ein
inspiratorisches Knisterrasseln.
Das Blutbild war bis auf eine BSG-Beschleunigung von 46 mm/h sowie diskret erhöhte
Leukozyten (10.130/μl) unauffällig.
Bei der DIP waren in allen drei Fällen Röntgenbilder verfügbar, die nur in einem Fall
retikuläre (16,7%) und in zwei Fällen streifige Veränderungen (33,3%) aufwiesen, ein
weiterer Patient hatte einen Zwerchfellhochstand (33,3%).
CT-Befunde waren bei zwei Patienten verfügbar. Ein Fall zeigte noduläre (33,3%),
keiner retikuläre Veränderungen. Milchglasartige Veränderungen waren nicht zu sehen,

60
dafür aber bei einem Patienten ein honigwabiges Muster (33,3%) und in einem weiteren
Fall streifige Veränderungen (33,3%).
Von zwei Patienten lagen vollständige Lavagen vor. Der dritte Patient wurde wegen
einer auffälligen BAL eingewiesen, von der aber nur die Neutrophilen und Eosinophilen
aus einem mitgebrachten Vorbefund dokumentiert waren.
In der BAL fand sich durchschnittlich folgende Konstellation: 67% Makrophagen, 11%
Lymphozyten, 13,3% Neutrophile, und 8,9% Eosinophile.
Tab. 9: BAL-Zelldifferenzierung bei der DIP (Daten in %)
BAL Pat.-Nr. 34 Pat.-Nr. 35 Pat.-Nr. 36 Mittelwert
Makrophagen 59 75 fehlt 67
Lymphozyten 14 8 fehlt 11
Neutrophile 26 12 2,0 13,3
Eosinophile 1,6 5,0 20 8,9
Lungenfunktionsanalytisch wurde eine IVC von 59,1%, ein TLC von 69% und ein
RAW von 86,1% vom jeweiligen Soll sowie als absolute Werte ein RV/TLC von 39,0%
und ein FEV1% von 68,5% gefunden, wobei Daten von lediglich zwei Patienten
dokumentiert waren.
Bei der Blutgasanalyse war lediglich eine Erniedrigung des pO2 auf 68,3 mmHg zu
beobachten. Bei einer Fallzahl von 3 hatte ein Patient einen pO2 von 56 und ein anderer
von 65 mmHg.
4.4.3 Verlauf
Von den drei Patienten mit der Diagnose DIP konnten über alle Verlaufsdaten erhoben
werden. Ein Patient verstarb 54 Monate, nachdem die Diagnose DIP gestellt wurde. 8
Monate bevor er verstarb, wurde bei diesem Patienten ein Bronchialkarzinom
diagnostiziert. Die übrigen 2 wurden als „stabil“ klassifiziert. Bei diesen beiden
Patienten war der Beobachtungszeitraum mit 12 und 22 Monaten kürzer.
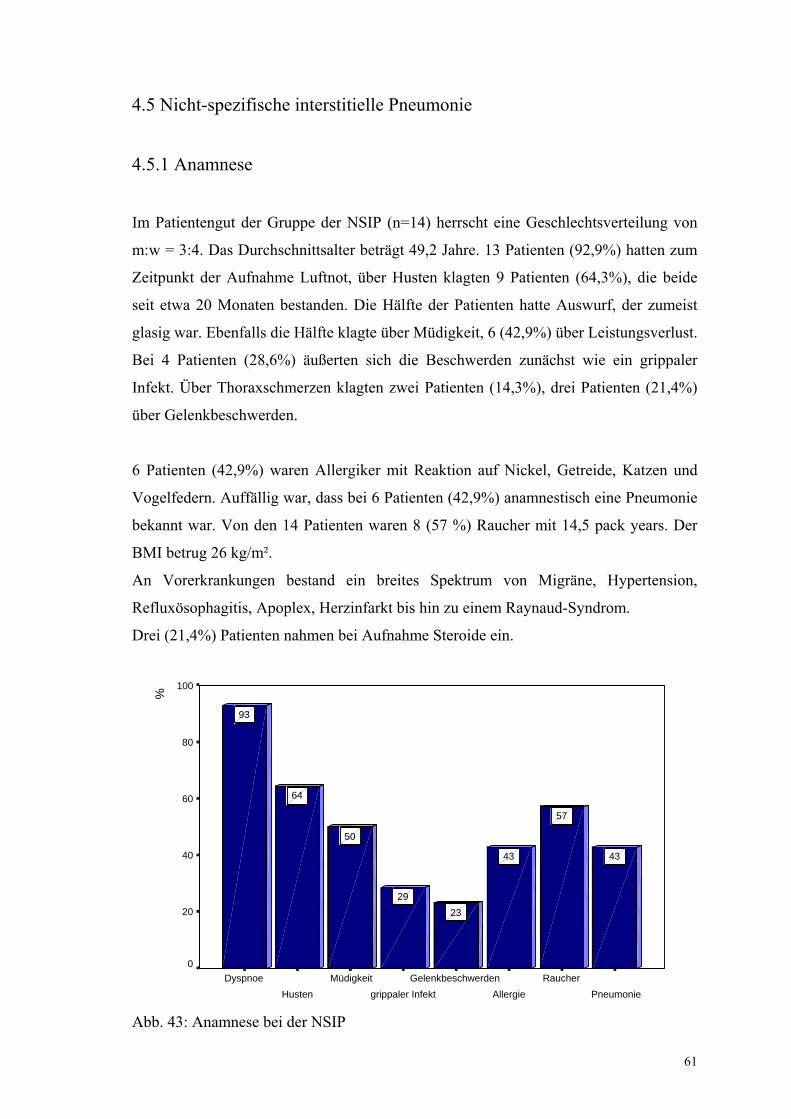
61
4.5 Nicht-spezifische interstitielle Pneumonie
4.5.1 Anamnese
Im Patientengut der Gruppe der NSIP (n=14) herrscht eine Geschlechtsverteilung von
m:w = 3:4. Das Durchschnittsalter beträgt 49,2 Jahre. 13 Patienten (92,9%) hatten zum
Zeitpunkt der Aufnahme Luftnot, über Husten klagten 9 Patienten (64,3%), die beide
seit etwa 20 Monaten bestanden. Die Hälfte der Patienten hatte Auswurf, der zumeist
glasig war. Ebenfalls die Hälfte klagte über Müdigkeit, 6 (42,9%) über Leistungsverlust.
Bei 4 Patienten (28,6%) äußerten sich die Beschwerden zunächst wie ein grippaler
Infekt. Über Thoraxschmerzen klagten zwei Patienten (14,3%), drei Patienten (21,4%)
über Gelenkbeschwerden.
6 Patienten (42,9%) waren Allergiker mit Reaktion auf Nickel, Getreide, Katzen und
Vogelfedern. Auffällig war, dass bei 6 Patienten (42,9%) anamnestisch eine Pneumonie
bekannt war. Von den 14 Patienten waren 8 (57 %) Raucher mit 14,5 pack years. Der
BMI betrug 26 kg/m².
An Vorerkrankungen bestand ein breites Spektrum von Migräne, Hypertension,
Refluxösophagitis, Apoplex, Herzinfarkt bis hin zu einem Raynaud-Syndrom.
Drei (21,4%) Patienten nahmen bei Aufnahme Steroide ein.
Abb. 43: Anamnese bei der NSIP
PneumonieRaucher
AllergieGelenkbeschwerden
grippaler InfektMüdigkeit
HustenDyspnoe
% 100
80
60
40
20
0
43
57
43
2329
50
64
93

62
4.5.2 Klinische Untersuchungen
Inspektorisch war bei 2 Patienten (21,4%) eine Zyanose und bei jeweils 3 (28,6%)
Patienten Trommelschlegelfinger zu sehen. Außerdem wirkte ein Patient stark
vorgealtert.
Bei einem Patienten (7,1%) ließ sich ein Schenkelschall perkutieren.
Die Auskultation erbrachte bei 9 Patienten (64,3%) Knisterrasseln.
insp. KnisterrasselnZyanoseUhrglasnägelTrommelschlegel
% 70
60
50
40
30
20
10
64
21
2929
Abb. 44: Synopsis der klinischen Befunde bei der NSIP
Im Routine-Labor war eine BSG-Beschleunigung von 40,9 mm/h auffällig, außerdem
war bei zwei Patienten (14,3%) der Rheumafaktor positiv. Alle übrigen Werte waren im
Normbereich.
Röntgenbefunde waren von 13 Patienten (92,9%) vorhanden. 3 mal wurden noduläre
Veränderungen gefunden (21,4%), 4 mal retikuläre (28,5%), 2 mal retikulo-noduläre (
14,2%) und 2 mal streifige Veränderungen (14,3%). Ein Patient hatte darüber hinaus
deutlich honigwabigen Umbau (71,%). Zwei Patienten hatten einen
Zwerchfellhochstand (14,3%).
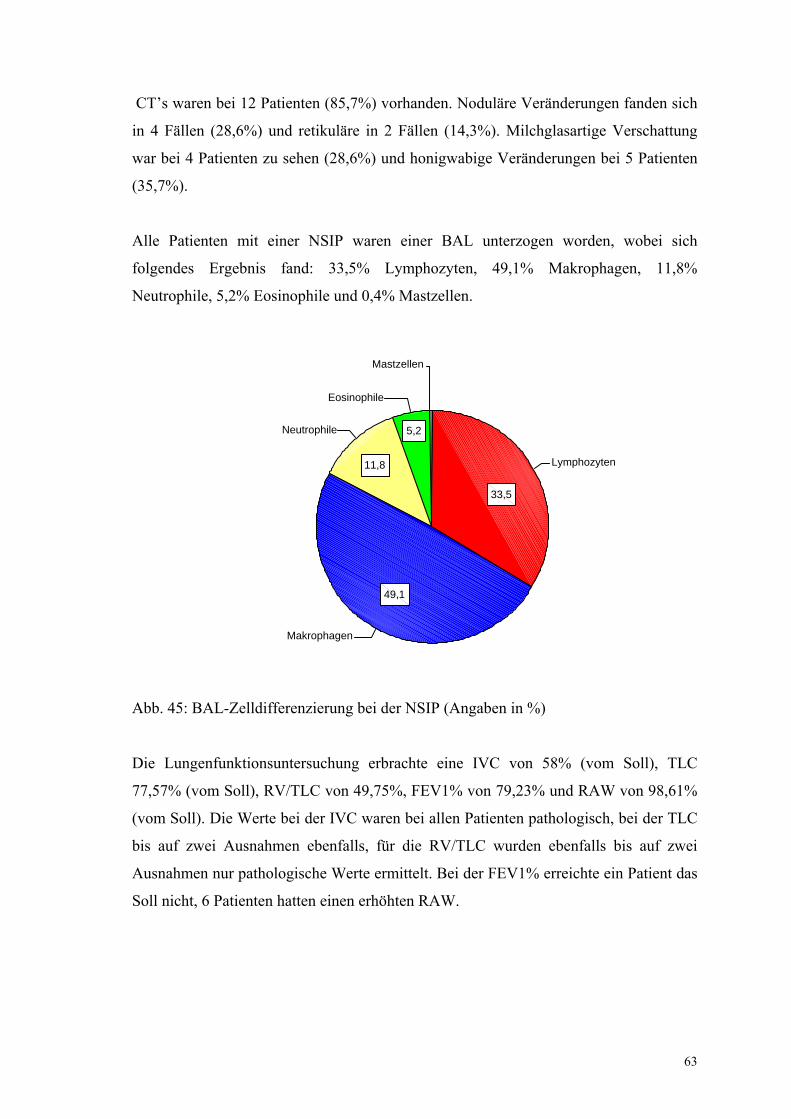
63
CT’s waren bei 12 Patienten (85,7%) vorhanden. Noduläre Veränderungen fanden sich
in 4 Fällen (28,6%) und retikuläre in 2 Fällen (14,3%). Milchglasartige Verschattung
war bei 4 Patienten zu sehen (28,6%) und honigwabige Veränderungen bei 5 Patienten
(35,7%).
Alle Patienten mit einer NSIP waren einer BAL unterzogen worden, wobei sich
folgendes Ergebnis fand: 33,5% Lymphozyten, 49,1% Makrophagen, 11,8%
Neutrophile, 5,2% Eosinophile und 0,4% Mastzellen.
Abb. 45: BAL-Zelldifferenzierung bei der NSIP (Angaben in %)
Die Lungenfunktionsuntersuchung erbrachte eine IVC von 58% (vom Soll), TLC
77,57% (vom Soll), RV/TLC von 49,75%, FEV1% von 79,23% und RAW von 98,61%
(vom Soll). Die Werte bei der IVC waren bei allen Patienten pathologisch, bei der TLC
bis auf zwei Ausnahmen ebenfalls, für die RV/TLC wurden ebenfalls bis auf zwei
Ausnahmen nur pathologische Werte ermittelt. Bei der FEV1% erreichte ein Patient das
Soll nicht, 6 Patienten hatten einen erhöhten RAW.
5,2
11,8
49,1
33,5
Mastzellen
Eosinophile
Neutrophile
Makrophagen
Lymphozyten
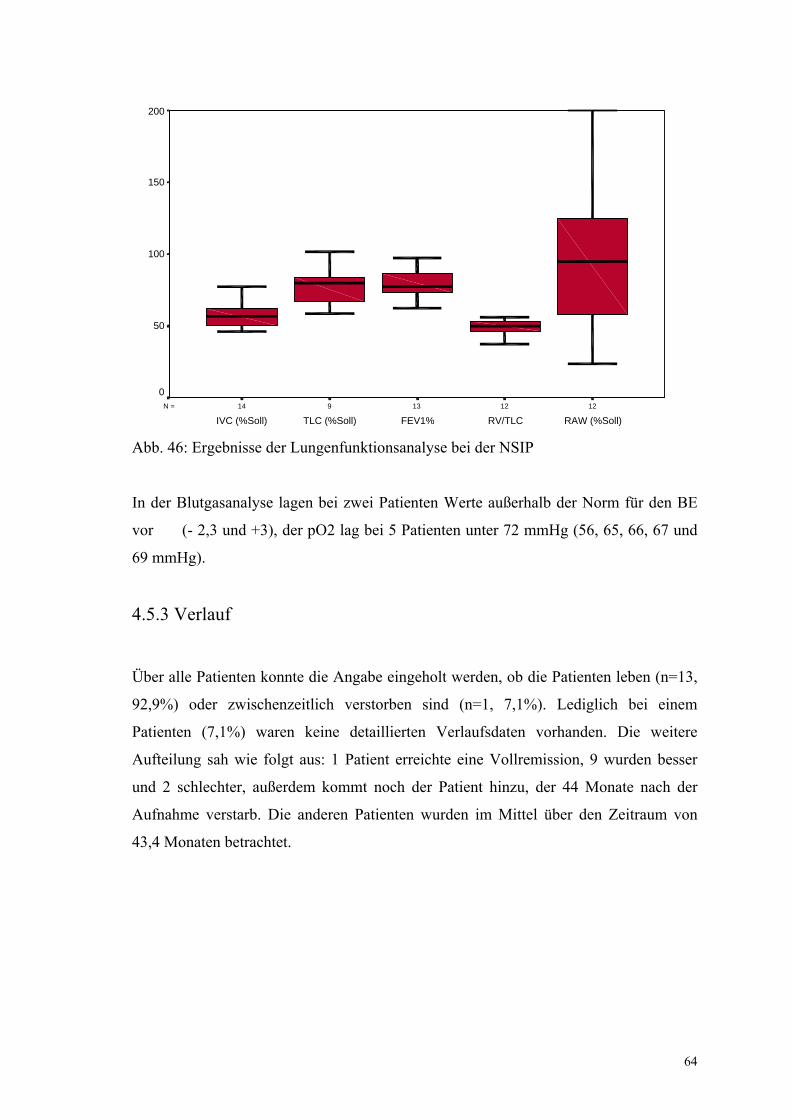
64
Abb. 46: Ergebnisse der Lungenfunktionsanalyse bei der NSIP
In der Blutgasanalyse lagen bei zwei Patienten Werte außerhalb der Norm für den BE
vor (- 2,3 und +3), der pO2 lag bei 5 Patienten unter 72 mmHg (56, 65, 66, 67 und
69 mmHg).
4.5.3 Verlauf
Über alle Patienten konnte die Angabe eingeholt werden, ob die Patienten leben (n=13,
92,9%) oder zwischenzeitlich verstorben sind (n=1, 7,1%). Lediglich bei einem
Patienten (7,1%) waren keine detaillierten Verlaufsdaten vorhanden. Die weitere
Aufteilung sah wie folgt aus: 1 Patient erreichte eine Vollremission, 9 wurden besser
und 2 schlechter, außerdem kommt noch der Patient hinzu, der 44 Monate nach der
Aufnahme verstarb. Die anderen Patienten wurden im Mittel über den Zeitraum von
43,4 Monaten betrachtet.
121213914N =
RAW (%Soll)RV/TLCFEV1%TLC (%Soll)IVC (%Soll)
200
150
100
50
0
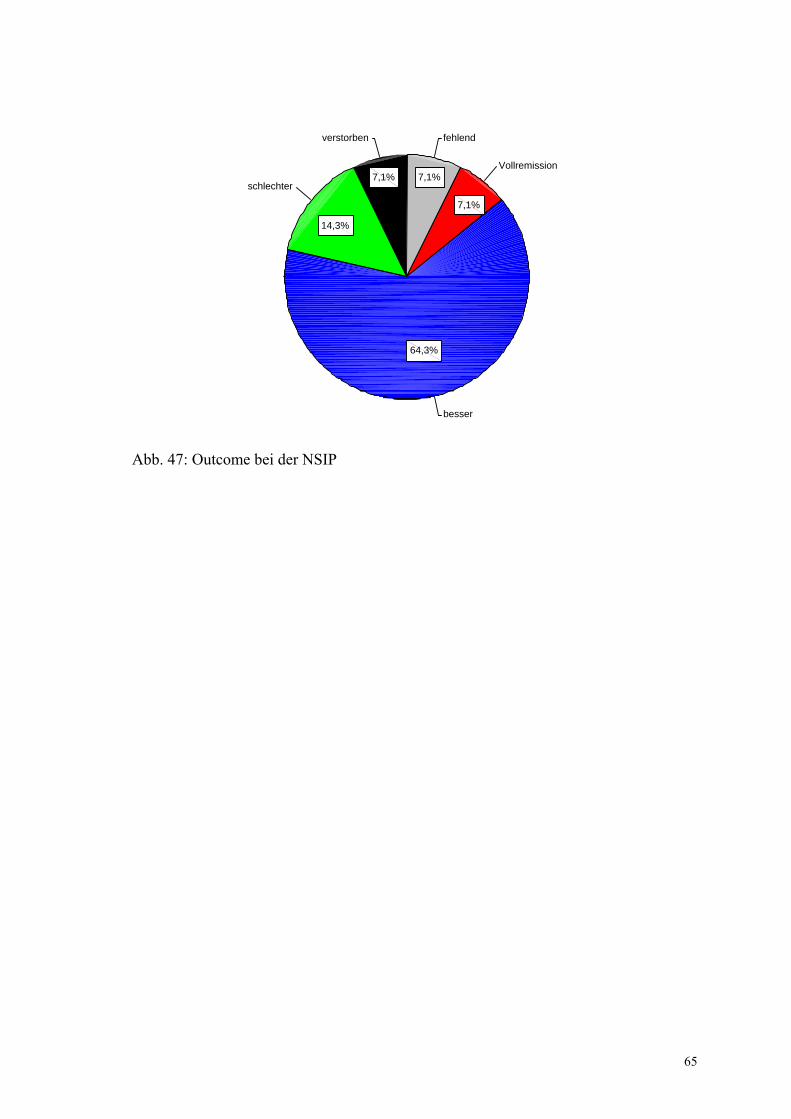
65
Abb. 47: Outcome bei der NSIP
7,1%
14,3%
64,3%
7,1%
7,1%
verstorben
schlechter
besser
Vollremission
fehlend
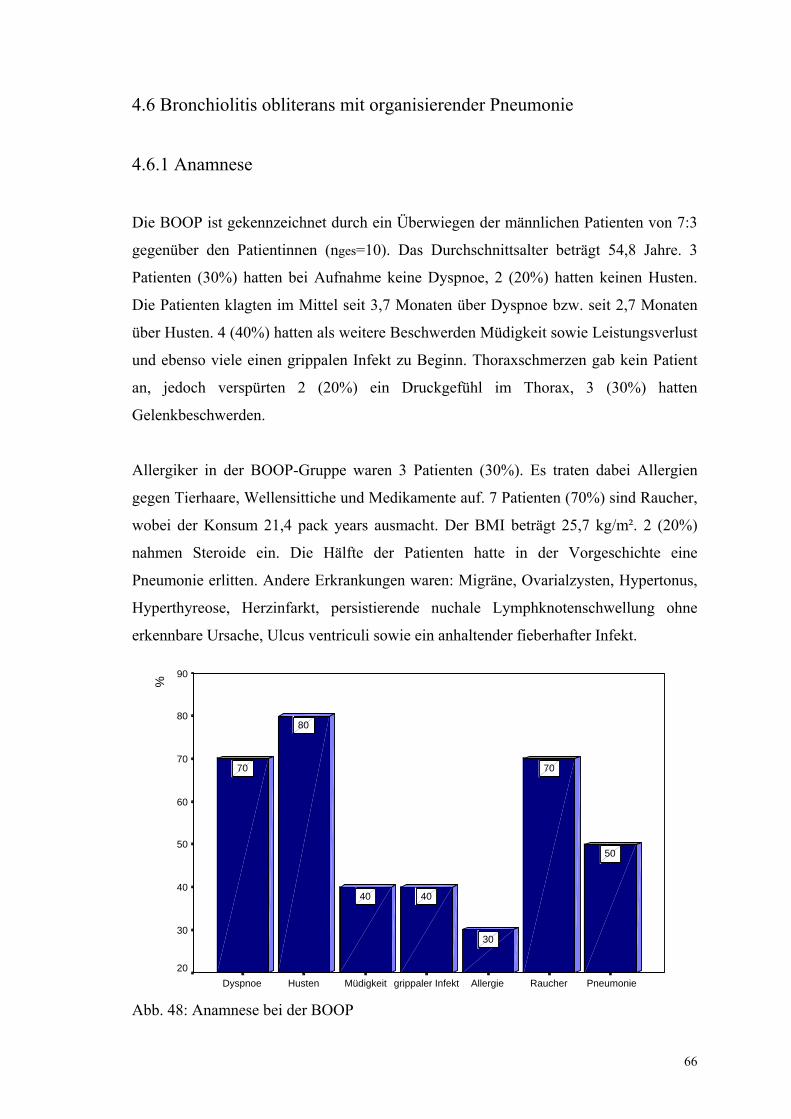
66
4.6 Bronchiolitis obliterans mit organisierender Pneumonie
4.6.1 Anamnese
Die BOOP ist gekennzeichnet durch ein Überwiegen der männlichen Patienten von 7:3
gegenüber den Patientinnen (nges=10). Das Durchschnittsalter beträgt 54,8 Jahre. 3
Patienten (30%) hatten bei Aufnahme keine Dyspnoe, 2 (20%) hatten keinen Husten.
Die Patienten klagten im Mittel seit 3,7 Monaten über Dyspnoe bzw. seit 2,7 Monaten
über Husten. 4 (40%) hatten als weitere Beschwerden Müdigkeit sowie Leistungsverlust
und ebenso viele einen grippalen Infekt zu Beginn. Thoraxschmerzen gab kein Patient
an, jedoch verspürten 2 (20%) ein Druckgefühl im Thorax, 3 (30%) hatten
Gelenkbeschwerden.
Allergiker in der BOOP-Gruppe waren 3 Patienten (30%). Es traten dabei Allergien
gegen Tierhaare, Wellensittiche und Medikamente auf. 7 Patienten (70%) sind Raucher,
wobei der Konsum 21,4 pack years ausmacht. Der BMI beträgt 25,7 kg/m². 2 (20%)
nahmen Steroide ein. Die Hälfte der Patienten hatte in der Vorgeschichte eine
Pneumonie erlitten. Andere Erkrankungen waren: Migräne, Ovarialzysten, Hypertonus,
Hyperthyreose, Herzinfarkt, persistierende nuchale Lymphknotenschwellung ohne
erkennbare Ursache, Ulcus ventriculi sowie ein anhaltender fieberhafter Infekt.
Abb. 48: Anamnese bei der BOOP
PneumonieRaucherAllergiegrippaler InfektMüdigkeitHustenDyspnoe
% 90
80
70
60
50
40
30
20
50
70
30
4040
80
70
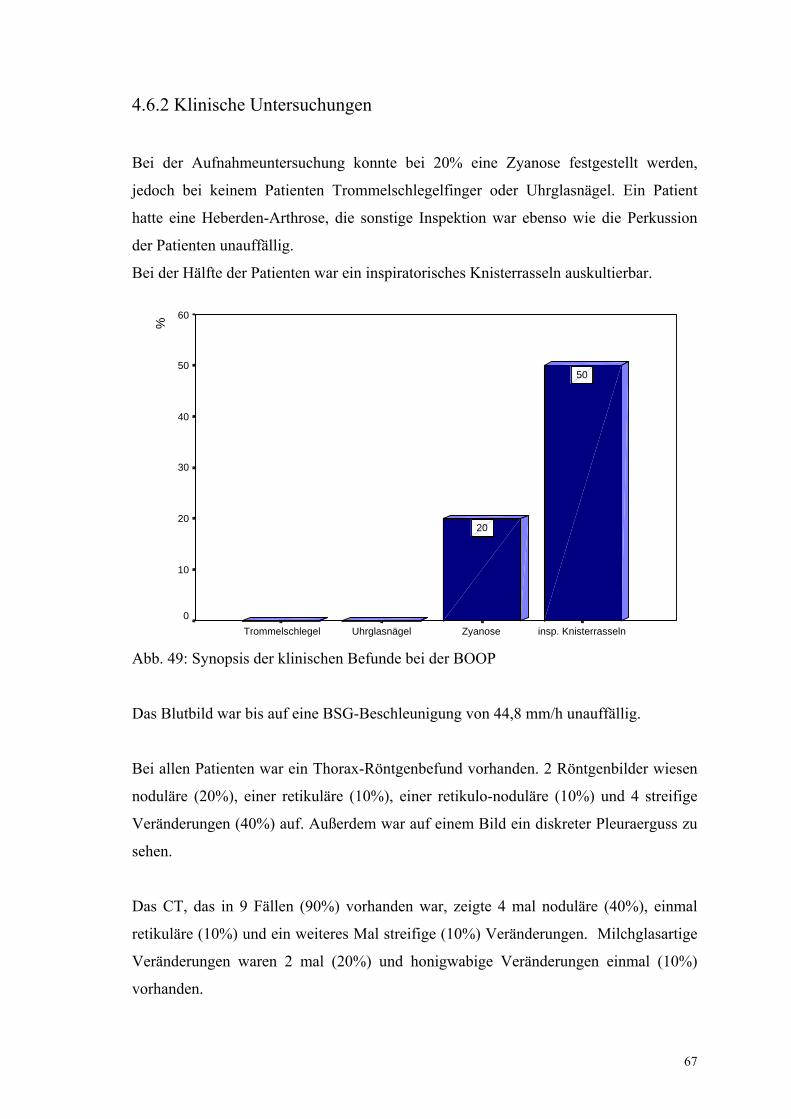
67
4.6.2 Klinische Untersuchungen
Bei der Aufnahmeuntersuchung konnte bei 20% eine Zyanose festgestellt werden,
jedoch bei keinem Patienten Trommelschlegelfinger oder Uhrglasnägel. Ein Patient
hatte eine Heberden-Arthrose, die sonstige Inspektion war ebenso wie die Perkussion
der Patienten unauffällig.
Bei der Hälfte der Patienten war ein inspiratorisches Knisterrasseln auskultierbar.
Abb. 49: Synopsis der klinischen Befunde bei der BOOP
Das Blutbild war bis auf eine BSG-Beschleunigung von 44,8 mm/h unauffällig.
Bei allen Patienten war ein Thorax-Röntgenbefund vorhanden. 2 Röntgenbilder wiesen
noduläre (20%), einer retikuläre (10%), einer retikulo-noduläre (10%) und 4 streifige
Veränderungen (40%) auf. Außerdem war auf einem Bild ein diskreter Pleuraerguss zu
sehen.
Das CT, das in 9 Fällen (90%) vorhanden war, zeigte 4 mal noduläre (40%), einmal
retikuläre (10%) und ein weiteres Mal streifige (10%) Veränderungen. Milchglasartige
Veränderungen waren 2 mal (20%) und honigwabige Veränderungen einmal (10%)
vorhanden.
insp. KnisterrasselnZyanoseUhrglasnägelTrommelschlegel
% 60
50
40
30
20
10
0
50
20

68
Die BAL wurde bei 9 Patienten durchgeführt. Das Ergebnis der Untersuchung war
folgende Verteilung: 41% Lymphozyten, 44,5% Makrophagen, 8,4% Neutrophile,
5,2% Eosinophile und 0,3% Mastzellen.
Abb. 50: BAL-Zelldifferenzierung bei der BOOP (Angaben in %)
In der Lungenfunktionsuntersuchung wurden folgende Werte ermittelt: 71,1% IVC,
90,4% TLC, 89,1% RAW vom Sollwert, 42,3% RV/TLC und 80,3% FEV1% als
absolute Werte.
Abb. 51: Ergebnisse der Lungenfunktionsanalyse bei der BOOP
101010610N =
RAW (%Soll)RV/TLCFEV1%TLC (%Soll)IVC (%Soll)
200
150
100
50
0
5,2
8,4
44,5
41,0
Mastzellen
Eosinophile
Neutrophile
Makrophagen
Lymphozyten
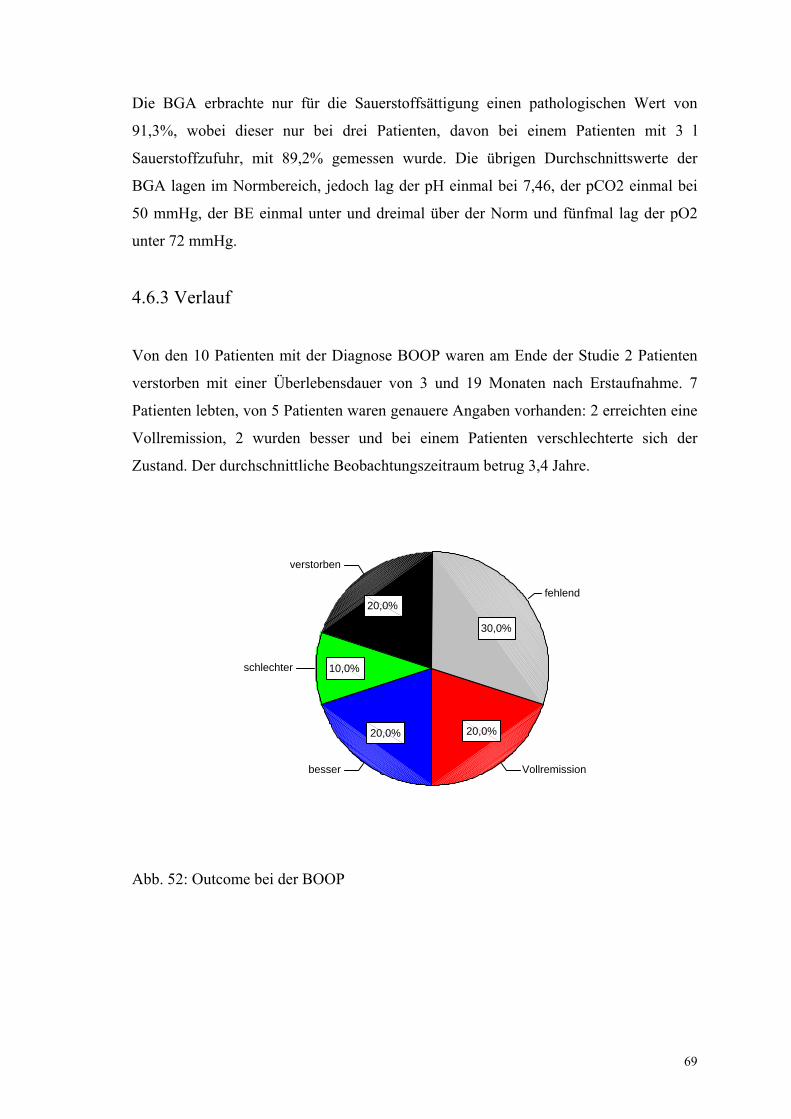
69
Die BGA erbrachte nur für die Sauerstoffsättigung einen pathologischen Wert von
91,3%, wobei dieser nur bei drei Patienten, davon bei einem Patienten mit 3 l
Sauerstoffzufuhr, mit 89,2% gemessen wurde. Die übrigen Durchschnittswerte der
BGA lagen im Normbereich, jedoch lag der pH einmal bei 7,46, der pCO2 einmal bei
50 mmHg, der BE einmal unter und dreimal über der Norm und fünfmal lag der pO2
unter 72 mmHg.
4.6.3 Verlauf
Von den 10 Patienten mit der Diagnose BOOP waren am Ende der Studie 2 Patienten
verstorben mit einer Überlebensdauer von 3 und 19 Monaten nach Erstaufnahme. 7
Patienten lebten, von 5 Patienten waren genauere Angaben vorhanden: 2 erreichten eine
Vollremission, 2 wurden besser und bei einem Patienten verschlechterte sich der
Zustand. Der durchschnittliche Beobachtungszeitraum betrug 3,4 Jahre.
Abb. 52: Outcome bei der BOOP
20,0%
10,0%
20,0% 20,0%
30,0%
verstorben
schlechter
besser Vollremission
fehlend

70
4.7 Lymphozytäre interstitielle Pneumonie
4.7.1 Anamnese
Die drei Patienten in der LIP-Gruppe sind allesamt männlich. Das Durchschnittsalter
liegt bei 55,9 Jahren. Zwei Patienten litten bei mittlerer Belastung unter Luftnot und
Husten, der in einem Fall mit weißlichem Auswurf einherging. Die Beschwerden
bestanden seit 9 Monaten. Zwei Patienten klagten über Müdigkeit und einer über
Leistungsverlust. Die sonstige Anamnese war blande. Kein Patient hatte weitere
Beschwerden, keiner war Allergiker, keiner war Raucher und keiner nahm zur
Aufnahme bereits Steroide ein. Anamnestisch war eine Gürtelrose sowie eine
Cholezystolithiasis bekannt.
4.7.2 Klinische Untersuchungen
Auch die körperliche Untersuchung verlief im Allgemeinen blande. Bei zwei Patienten
war auskultatorisch ein Knistern zu hören.
In der Blutuntersuchung war eine BSG-Beschleunigung von 75,7 mm/h und
Thrombozyten von 484.000/μl festzustellen.
Eine BGA war nur bei zwei Patienten vorhanden. das Ergebnis erbrachte einmal einen
pCO2 von 32 und einmal von 34 mmHg, die übrige BGA war unauffällig.
Röntgenbilder des Thorax waren bei allen LIP-Fällen vorhanden, zeigten aber nur bei
zwei Patienten noduläre Veränderungen (66,6%).
CT’s waren bei 2 Patienten vorhanden (66,7%), aber nur einmal zeigten sich noduläre
Veränderungen.
Ebenfalls war nur bei zwei Patienten eine BAL durchgeführt worden. Die Werte waren
bei den Patienten folgende:
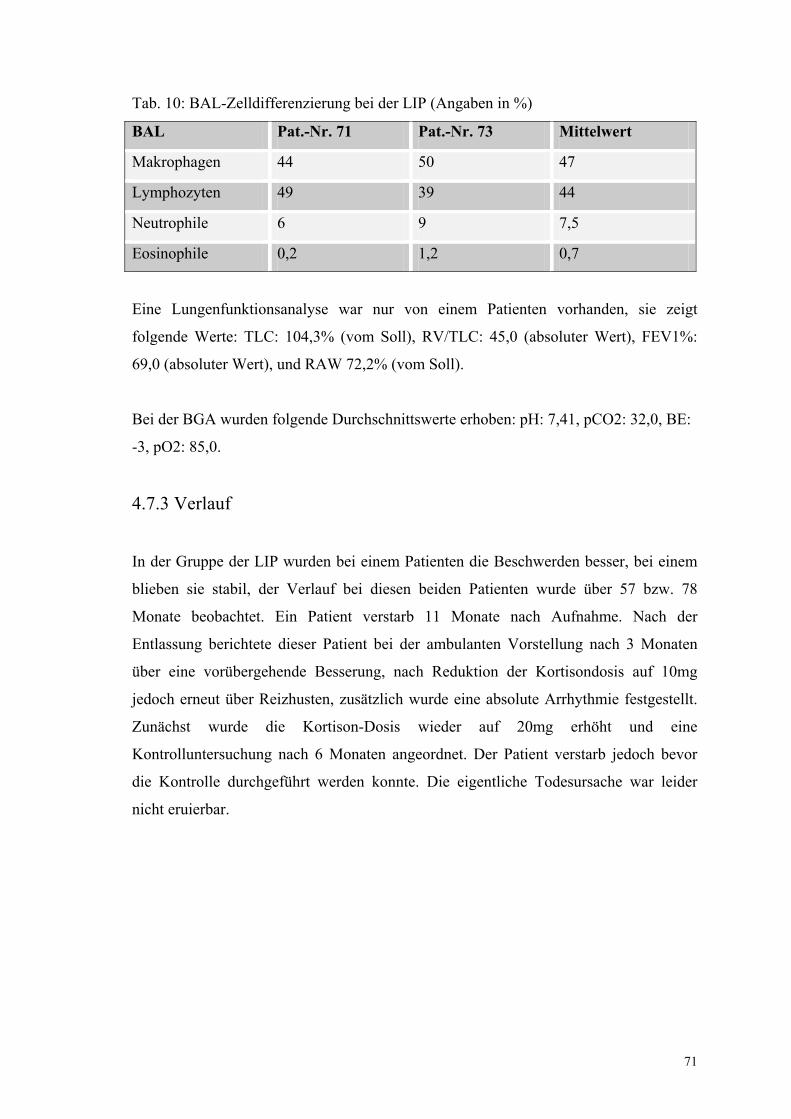
71
Tab. 10: BAL-Zelldifferenzierung bei der LIP (Angaben in %)
BAL Pat.-Nr. 71 Pat.-Nr. 73 Mittelwert
Makrophagen 44 50 47
Lymphozyten 49 39 44
Neutrophile 6 9 7,5
Eosinophile 0,2 1,2 0,7
Eine Lungenfunktionsanalyse war nur von einem Patienten vorhanden, sie zeigt
folgende Werte: TLC: 104,3% (vom Soll), RV/TLC: 45,0 (absoluter Wert), FEV1%:
69,0 (absoluter Wert), und RAW 72,2% (vom Soll).
Bei der BGA wurden folgende Durchschnittswerte erhoben: pH: 7,41, pCO2: 32,0, BE:
-3, pO2: 85,0.
4.7.3 Verlauf
In der Gruppe der LIP wurden bei einem Patienten die Beschwerden besser, bei einem
blieben sie stabil, der Verlauf bei diesen beiden Patienten wurde über 57 bzw. 78
Monate beobachtet. Ein Patient verstarb 11 Monate nach Aufnahme. Nach der
Entlassung berichtete dieser Patient bei der ambulanten Vorstellung nach 3 Monaten
über eine vorübergehende Besserung, nach Reduktion der Kortisondosis auf 10mg
jedoch erneut über Reizhusten, zusätzlich wurde eine absolute Arrhythmie festgestellt.
Zunächst wurde die Kortison-Dosis wieder auf 20mg erhöht und eine
Kontrolluntersuchung nach 6 Monaten angeordnet. Der Patient verstarb jedoch bevor
die Kontrolle durchgeführt werden konnte. Die eigentliche Todesursache war leider
nicht eruierbar.
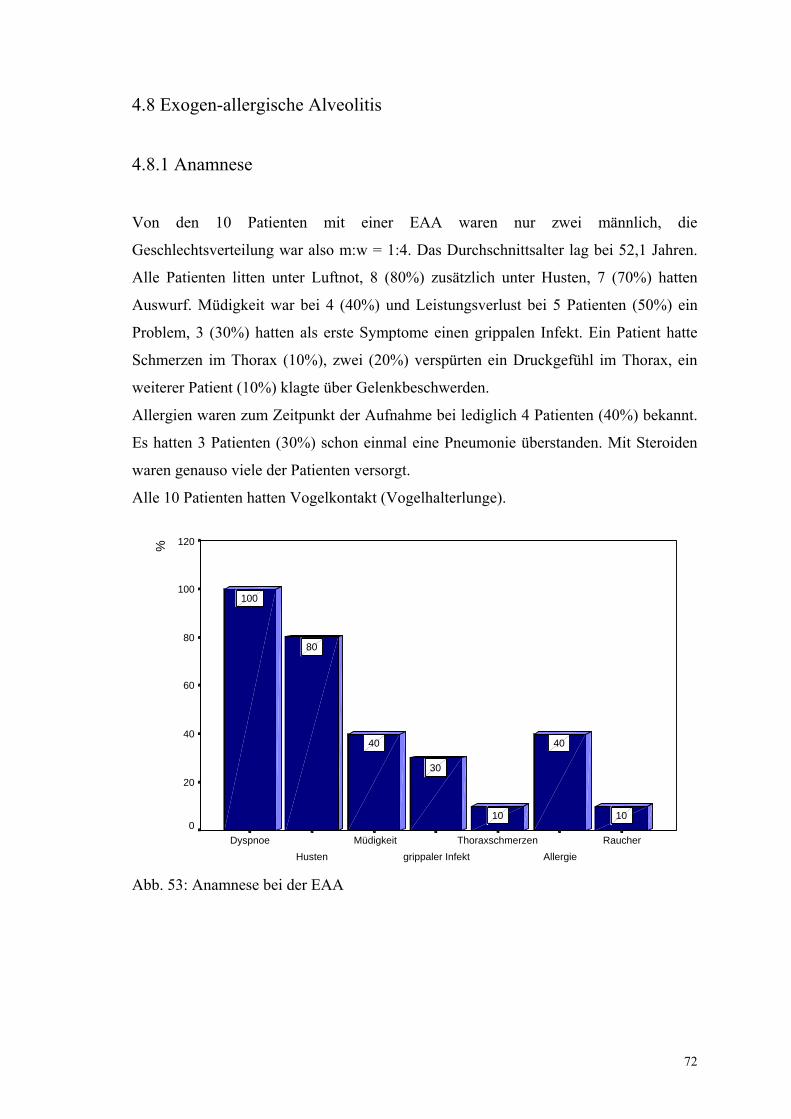
72
4.8 Exogen-allergische Alveolitis
4.8.1 Anamnese
Von den 10 Patienten mit einer EAA waren nur zwei männlich, die
Geschlechtsverteilung war also m:w = 1:4. Das Durchschnittsalter lag bei 52,1 Jahren.
Alle Patienten litten unter Luftnot, 8 (80%) zusätzlich unter Husten, 7 (70%) hatten
Auswurf. Müdigkeit war bei 4 (40%) und Leistungsverlust bei 5 Patienten (50%) ein
Problem, 3 (30%) hatten als erste Symptome einen grippalen Infekt. Ein Patient hatte
Schmerzen im Thorax (10%), zwei (20%) verspürten ein Druckgefühl im Thorax, ein
weiterer Patient (10%) klagte über Gelenkbeschwerden.
Allergien waren zum Zeitpunkt der Aufnahme bei lediglich 4 Patienten (40%) bekannt.
Es hatten 3 Patienten (30%) schon einmal eine Pneumonie überstanden. Mit Steroiden
waren genauso viele der Patienten versorgt.
Alle 10 Patienten hatten Vogelkontakt (Vogelhalterlunge).
Abb. 53: Anamnese bei der EAA
RaucherAllergie
Thoraxschmerzengrippaler Infekt
MüdigkeitHusten
Dyspnoe
% 120
100
80
60
40
20
010
40
10
30
40
80
100
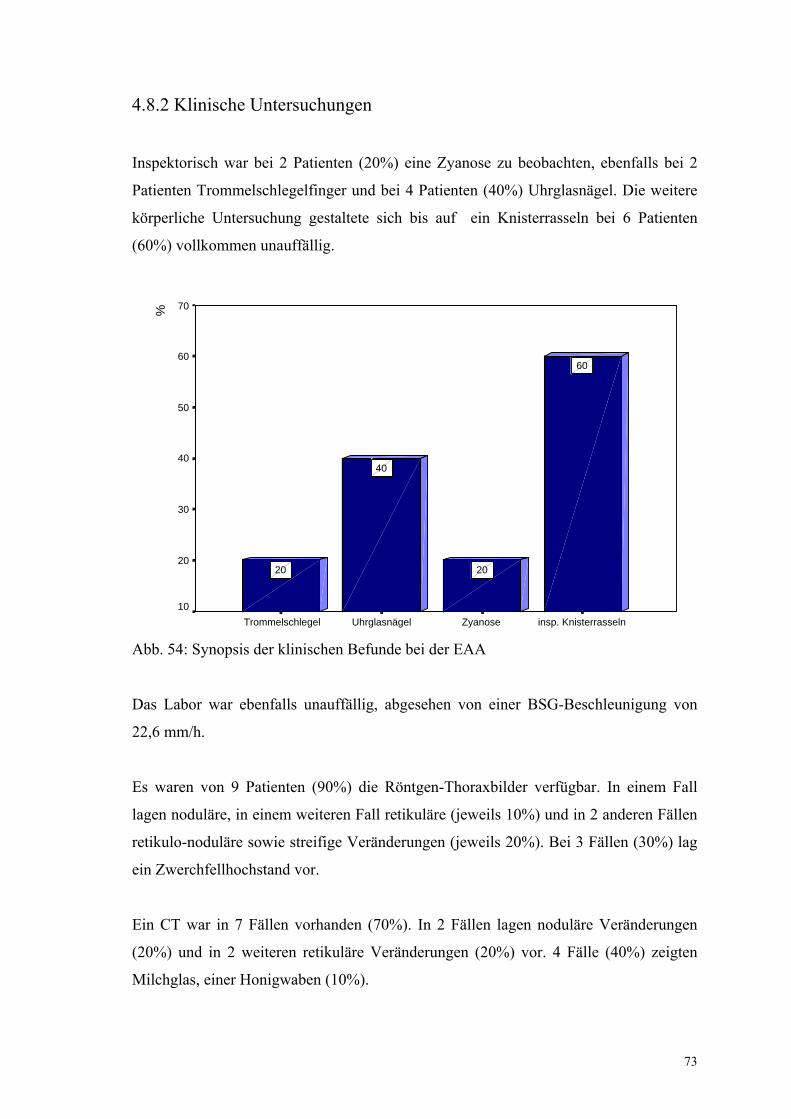
73
4.8.2 Klinische Untersuchungen
Inspektorisch war bei 2 Patienten (20%) eine Zyanose zu beobachten, ebenfalls bei 2
Patienten Trommelschlegelfinger und bei 4 Patienten (40%) Uhrglasnägel. Die weitere
körperliche Untersuchung gestaltete sich bis auf ein Knisterrasseln bei 6 Patienten
(60%) vollkommen unauffällig.
Abb. 54: Synopsis der klinischen Befunde bei der EAA
Das Labor war ebenfalls unauffällig, abgesehen von einer BSG-Beschleunigung von
22,6 mm/h.
Es waren von 9 Patienten (90%) die Röntgen-Thoraxbilder verfügbar. In einem Fall
lagen noduläre, in einem weiteren Fall retikuläre (jeweils 10%) und in 2 anderen Fällen
retikulo-noduläre sowie streifige Veränderungen (jeweils 20%). Bei 3 Fällen (30%) lag
ein Zwerchfellhochstand vor.
Ein CT war in 7 Fällen vorhanden (70%). In 2 Fällen lagen noduläre Veränderungen
(20%) und in 2 weiteren retikuläre Veränderungen (20%) vor. 4 Fälle (40%) zeigten
Milchglas, einer Honigwaben (10%).
insp. KnisterrasselnZyanoseUhrglasnägelTrommelschlegel
% 70
60
50
40
30
20
10
60
20
40
20

74
Eine BAL war bei 9 Patienten verfügbar. Die Ergebnisse waren: 62,6% Lymphozyten,
30,7% Makrophagen, 5,5% Neutrophile, 2% Eosinophile und 0,4% Mastzellen.
Abb. 55: BAL-Zelldifferenzierung bei EAA (Angaben in %)
Folgende Messwerte waren bei der LUFU festzustellen: IVC: 57,4% (vom Soll), TLC:
70,7% (vom Soll), RV/TLC: 47,86% (absoluter Wert), FEV1%: 84,12% (absoluter
Wert), RAW: 102,45% (vom Soll).
Die BGA war bis auf wenige „Ausreißer“ unauffällig. Der BE war einmal unter- und
einmal oberhalb der Norm, der pO2 dreimal unterhalb der Norm.
30,862,6
Mastzellen
Eosinophile
Neutrophile
Makrophagen
Lymphozyten
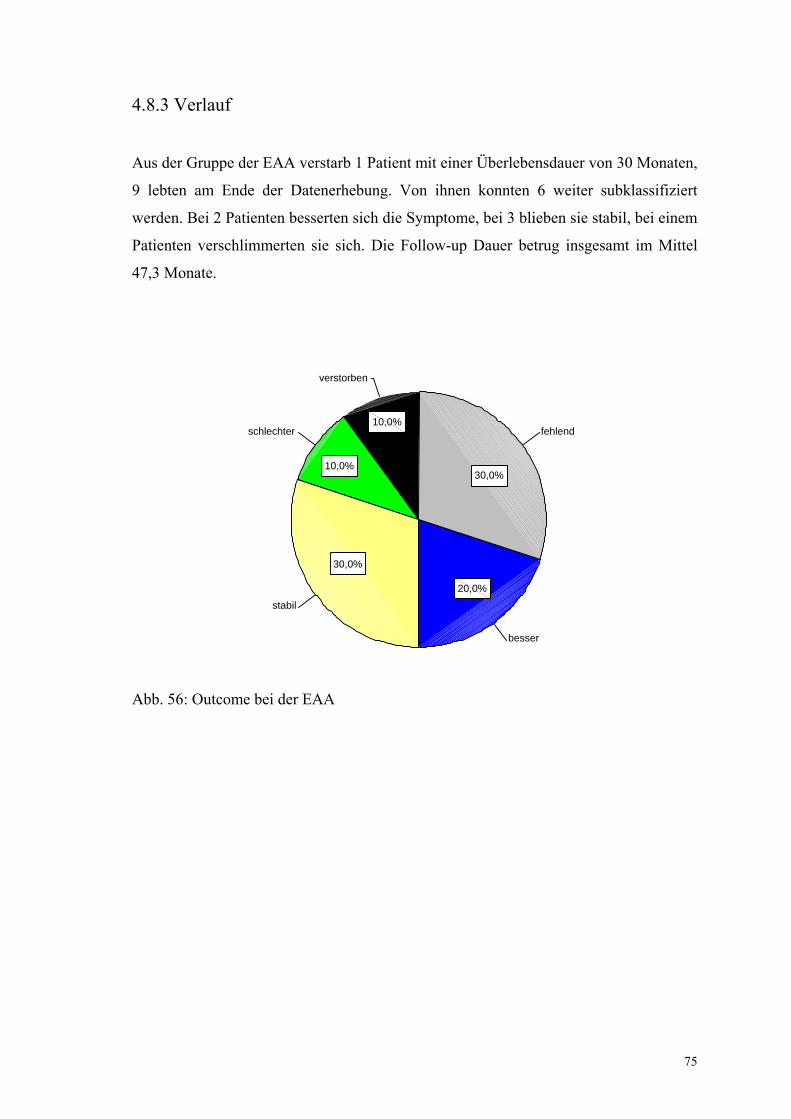
75
4.8.3 Verlauf
Aus der Gruppe der EAA verstarb 1 Patient mit einer Überlebensdauer von 30 Monaten,
9 lebten am Ende der Datenerhebung. Von ihnen konnten 6 weiter subklassifiziert
werden. Bei 2 Patienten besserten sich die Symptome, bei 3 blieben sie stabil, bei einem
Patienten verschlimmerten sie sich. Die Follow-up Dauer betrug insgesamt im Mittel
47,3 Monate.
Abb. 56: Outcome bei der EAA
10,0%
10,0%
30,0%
20,0%
30,0%
verstorben
schlechter
stabil
besser
fehlend
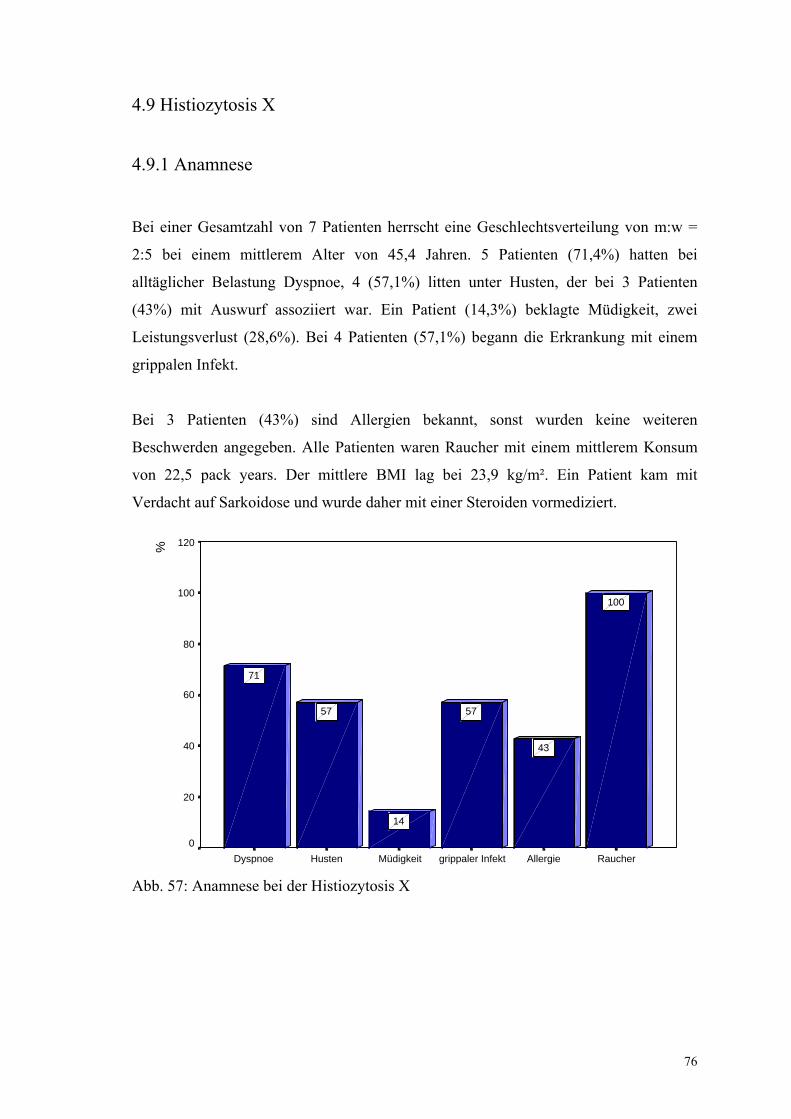
76
4.9 Histiozytosis X
4.9.1 Anamnese
Bei einer Gesamtzahl von 7 Patienten herrscht eine Geschlechtsverteilung von m:w =
2:5 bei einem mittlerem Alter von 45,4 Jahren. 5 Patienten (71,4%) hatten bei
alltäglicher Belastung Dyspnoe, 4 (57,1%) litten unter Husten, der bei 3 Patienten
(43%) mit Auswurf assoziiert war. Ein Patient (14,3%) beklagte Müdigkeit, zwei
Leistungsverlust (28,6%). Bei 4 Patienten (57,1%) begann die Erkrankung mit einem
grippalen Infekt.
Bei 3 Patienten (43%) sind Allergien bekannt, sonst wurden keine weiteren
Beschwerden angegeben. Alle Patienten waren Raucher mit einem mittlerem Konsum
von 22,5 pack years. Der mittlere BMI lag bei 23,9 kg/m². Ein Patient kam mit
Verdacht auf Sarkoidose und wurde daher mit einer Steroiden vormediziert.
Abb. 57: Anamnese bei der Histiozytosis X
RaucherAllergiegrippaler InfektMüdigkeitHustenDyspnoe
% 120
100
80
60
40
20
0
100
43
57
14
57
71

77
4.9.2 Klinische Untersuchungen
Hypoxiezeichen waren nur bei einem Patienten zu entdecken, und zwar
Trommelschlegelfinger und Uhrglasnägel, aber keine Zyanose. Bei dem Patienten mit
der Steroidvormedikation wurde eine Steroidakne gesehen. Ein Patient zeigte bei der
Perkussion eine eingeschränkte Lungenbeweglichkeit, ein Patient wies bei der
Auskultation Knisterrasseln auf und ein weiterer feinblasige Rasselgeräusche.
Ansonsten waren bei der körperlichen Untersuchung keine Besonderheiten zu finden.
Im Blutbild war eine erhöhte BSG von 30,3 mm/h, Leukozyten von 10.200/μl und
grenzwertige Thrombozyten von 349.500/μl zu sehen.
Röntgenbilder waren bei allen Patienten vorhanden. Bei 2 Patienten lagen noduläre
(28,6%), bei einem retikuläre (14,3%) und bei einem weiteren streifige (14,3%)
Veränderungen vor.
Ein CT wurde von 6 Patienten (85,7%) angefertigt, das bei 3 Patienten noduläre
(42,9%), bei 2 Patienten retikuläre (28,6%) und bei einem streifige (14,3%)
Veränderungen zeigte. Bei einem Patienten wurde Milchglas gefunden (14,3%)
Die BAL war bei 5 Patienten mit folgendem Resultat durchgeführt worden: 4,9%
Lymphozyten, 84% Makrophagen, 9% Neutrophile, 1,7% Eosinophile und 0,5%
Mastzellen.
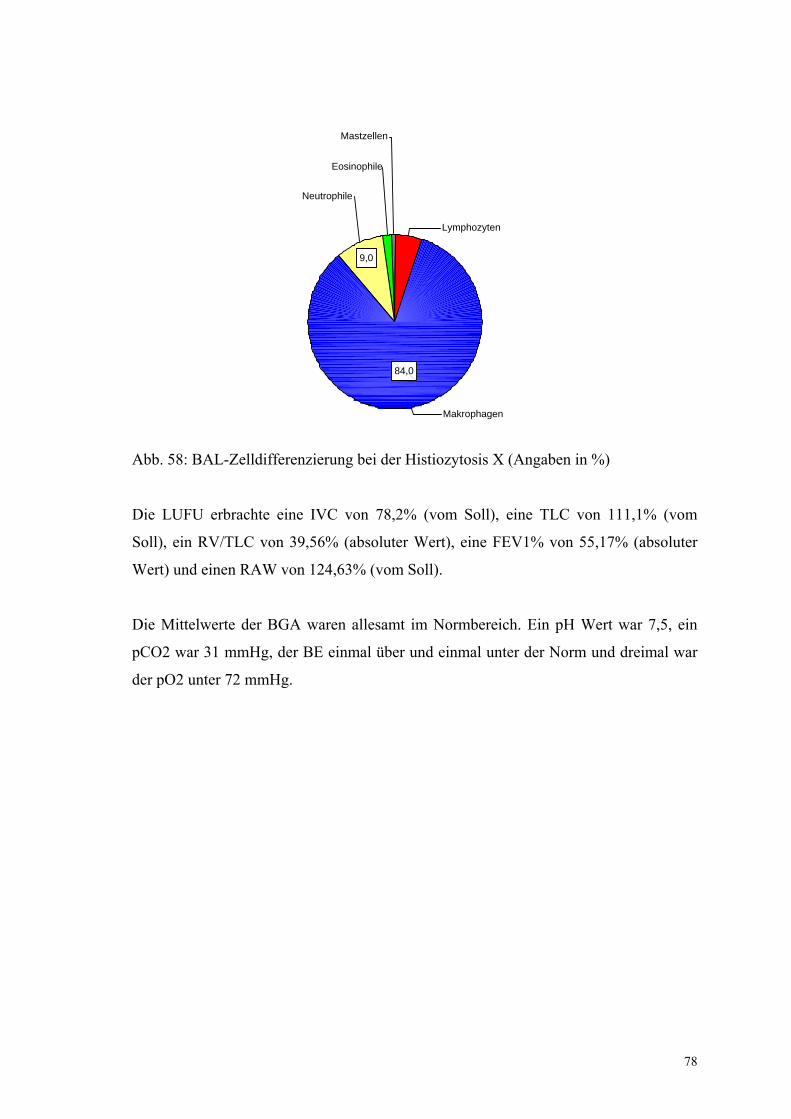
78
Abb. 58: BAL-Zelldifferenzierung bei der Histiozytosis X (Angaben in %)
Die LUFU erbrachte eine IVC von 78,2% (vom Soll), eine TLC von 111,1% (vom
Soll), ein RV/TLC von 39,56% (absoluter Wert), eine FEV1% von 55,17% (absoluter
Wert) und einen RAW von 124,63% (vom Soll).
Die Mittelwerte der BGA waren allesamt im Normbereich. Ein pH Wert war 7,5, ein
pCO2 war 31 mmHg, der BE einmal über und einmal unter der Norm und dreimal war
der pO2 unter 72 mmHg.
9,0
84,0
Mastzellen
Eosinophile
Neutrophile
Makrophagen
Lymphozyten
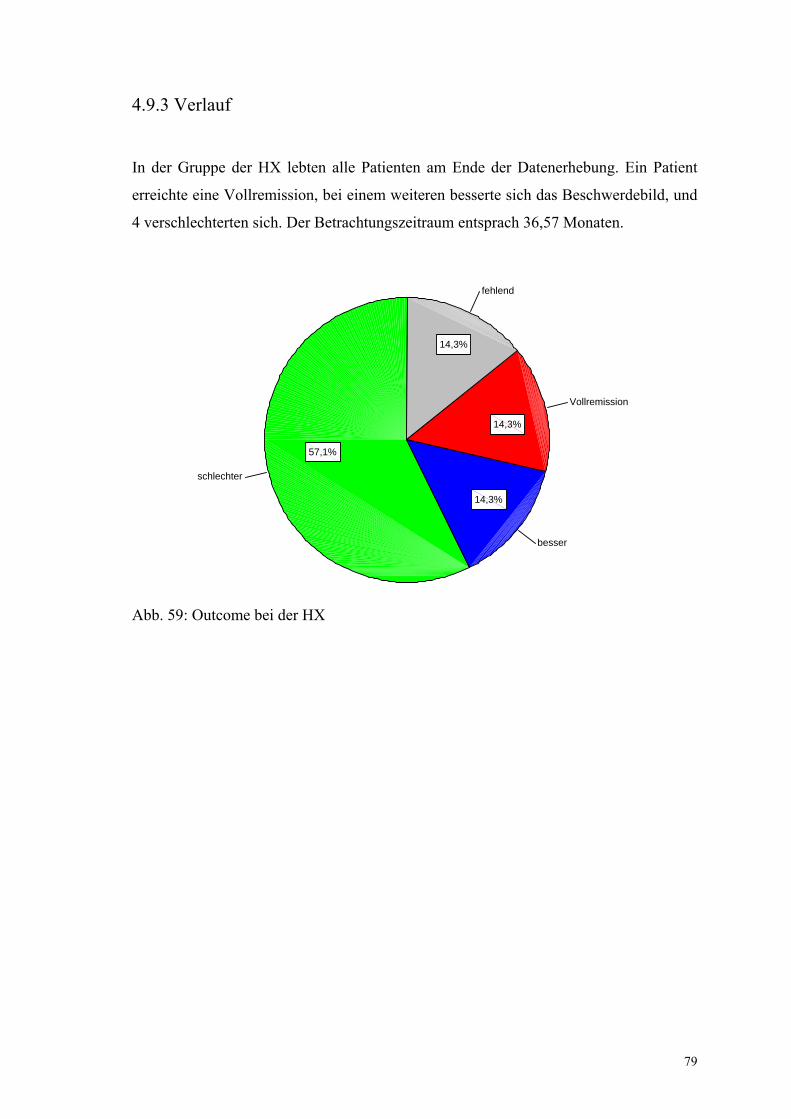
79
4.9.3 Verlauf
In der Gruppe der HX lebten alle Patienten am Ende der Datenerhebung. Ein Patient
erreichte eine Vollremission, bei einem weiteren besserte sich das Beschwerdebild, und
4 verschlechterten sich. Der Betrachtungszeitraum entsprach 36,57 Monaten.
Abb. 59: Outcome bei der HX
57,1%
14,3%
14,3%
14,3%
schlechter
besser
Vollremission
fehlend
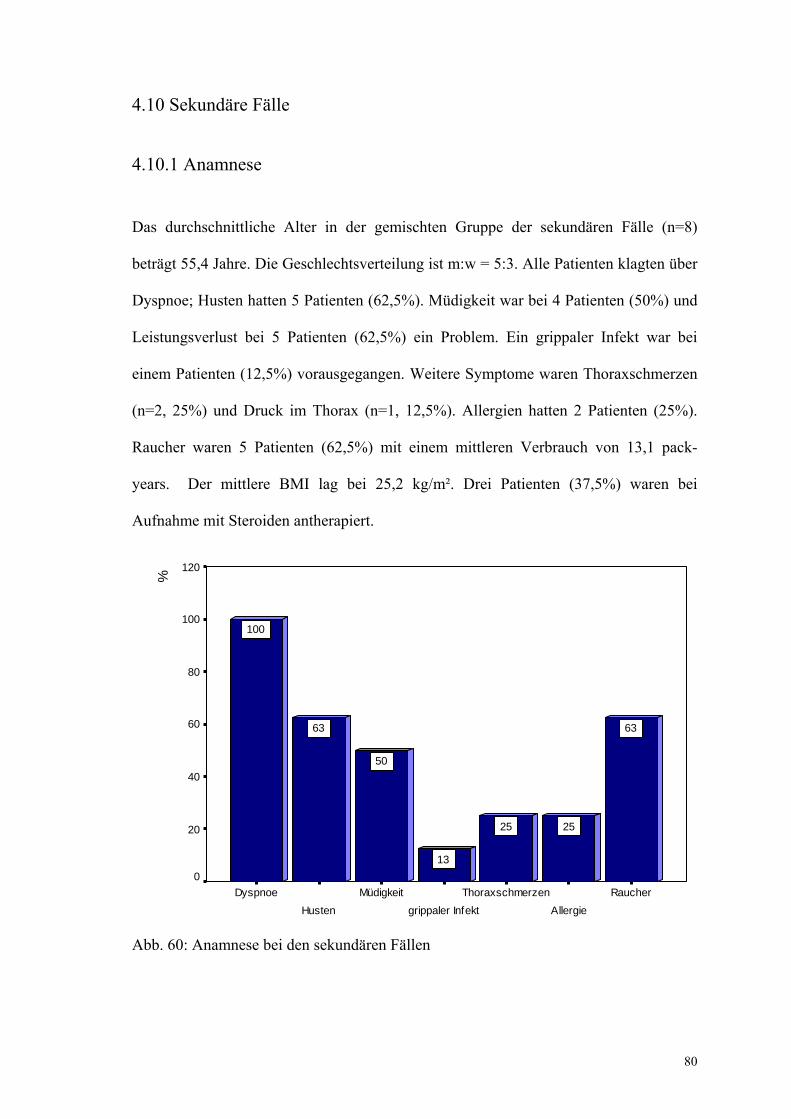
80
4.10 Sekundäre Fälle
4.10.1 Anamnese
Das durchschnittliche Alter in der gemischten Gruppe der sekundären Fälle (n=8)
beträgt 55,4 Jahre. Die Geschlechtsverteilung ist m:w = 5:3. Alle Patienten klagten über
Dyspnoe; Husten hatten 5 Patienten (62,5%). Müdigkeit war bei 4 Patienten (50%) und
Leistungsverlust bei 5 Patienten (62,5%) ein Problem. Ein grippaler Infekt war bei
einem Patienten (12,5%) vorausgegangen. Weitere Symptome waren Thoraxschmerzen
(n=2, 25%) und Druck im Thorax (n=1, 12,5%). Allergien hatten 2 Patienten (25%).
Raucher waren 5 Patienten (62,5%) mit einem mittleren Verbrauch von 13,1 pack-
years. Der mittlere BMI lag bei 25,2 kg/m². Drei Patienten (37,5%) waren bei
Aufnahme mit Steroiden antherapiert.
RaucherAllergie
Thoraxschmerzengrippaler Infekt
MüdigkeitHusten
Dyspnoe
% 120
100
80
60
40
20
0
63
2525
13
50
63
100
Abb. 60: Anamnese bei den sekundären Fällen

81
4.10.2 Klinische Untersuchungen
Als Hypoxiezeichen trat lediglich bei einem Patienten (12,5%) eine Zyanose auf. Zwei
Patienten zeigten ein door-stop-Phänomen (25%). Die Perkussion wies bei einem
Patienten (12,5%) Schenkelschall, die Auskultation bei 3 Patienten (37,5%)
inspiratorisches Knisterrasseln auf.
Die Blutsenkungsgeschwindigkeit war mit 26 mm/h erhöht, die Leukozyten mit
11.100/µl und das LDH mit 251 U/l ebenfalls.
Die Röntgenbilder ließen bei allen Patienten pathologische Veränderungen erkennen,
die retikulär (25%, n=2), retikulo-nodulär (12,5%, n=1) oder streifig (25%, n=2) waren.
Die CT’s zeigten außerdem noch in 2 Fällen (25%) milchglasartige Veränderungen und
in einem Fall auch honigwabige Veränderungen (12,5%).
Eine BAL wurde in 6 Fällen durchgeführt und ergab folgende Werte: Lymphozyten
29,3%, Makrophagen 48,0%, Neutrophile 14,7%, Eosinophile 15,7%, und Mastzellen
0,4%.
15,7
14,7
48,0
29,3
Mastzellen
Eosinophile
Neutrophile
Makrophagen
Lymphozyten
Abb. 61: BAL-Zelldifferenzierung bei den sekundären Fällen (Angaben in %)
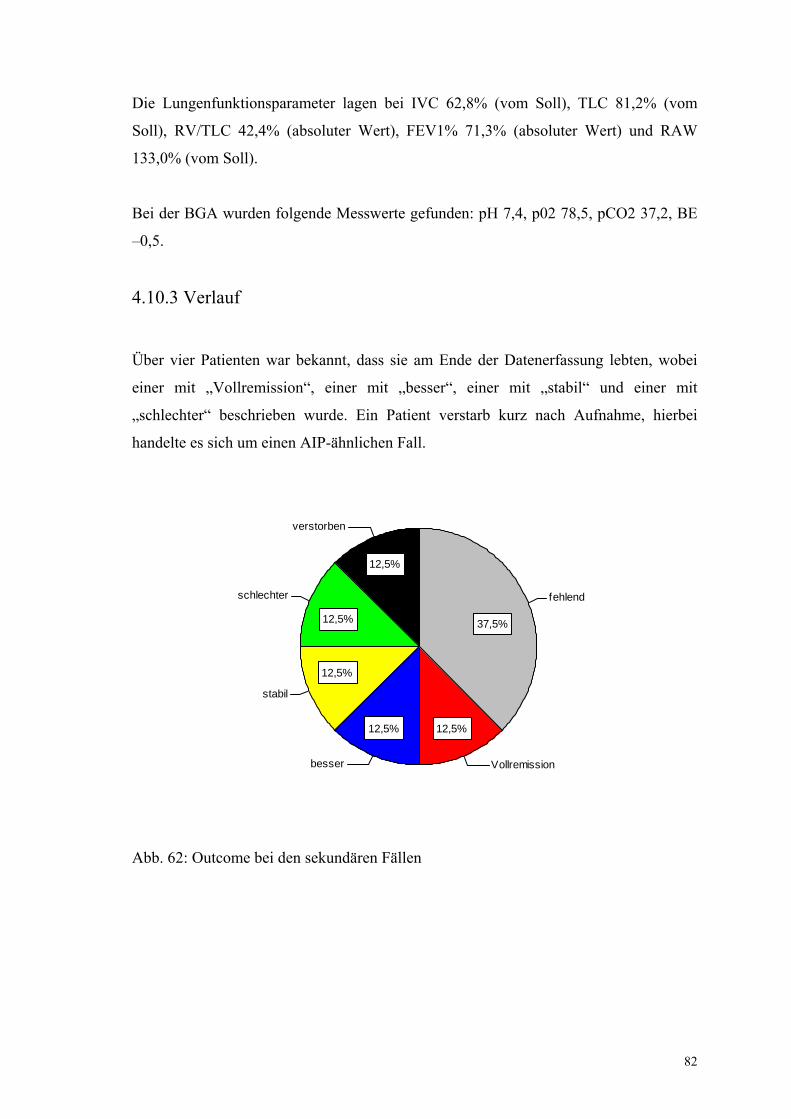
82
Die Lungenfunktionsparameter lagen bei IVC 62,8% (vom Soll), TLC 81,2% (vom
Soll), RV/TLC 42,4% (absoluter Wert), FEV1% 71,3% (absoluter Wert) und RAW
133,0% (vom Soll).
Bei der BGA wurden folgende Messwerte gefunden: pH 7,4, p02 78,5, pCO2 37,2, BE
–0,5.
4.10.3 Verlauf
Über vier Patienten war bekannt, dass sie am Ende der Datenerfassung lebten, wobei
einer mit „Vollremission“, einer mit „besser“, einer mit „stabil“ und einer mit
„schlechter“ beschrieben wurde. Ein Patient verstarb kurz nach Aufnahme, hierbei
handelte es sich um einen AIP-ähnlichen Fall.
12,5%
12,5%
12,5%
12,5% 12,5%
37,5%
verstorben
schlechter
stabil
besser Vollremission
fehlend
Abb. 62: Outcome bei den sekundären Fällen
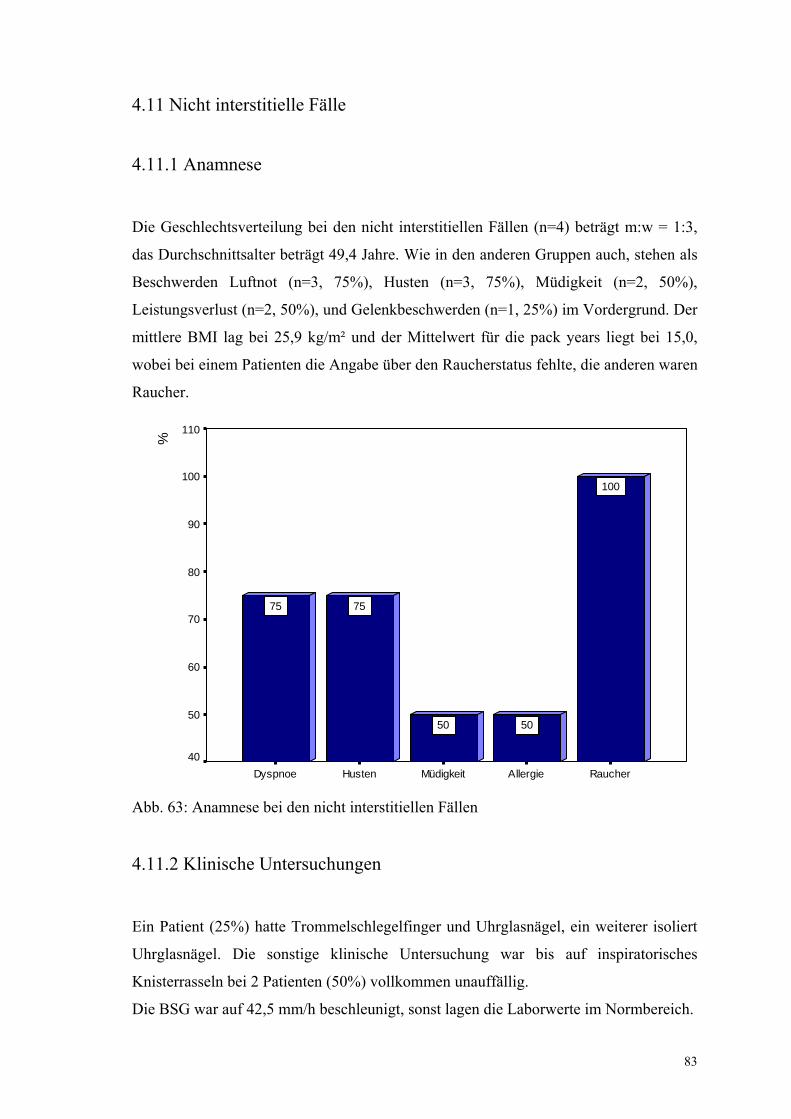
83
4.11 Nicht interstitielle Fälle
4.11.1 Anamnese
Die Geschlechtsverteilung bei den nicht interstitiellen Fällen (n=4) beträgt m:w = 1:3,
das Durchschnittsalter beträgt 49,4 Jahre. Wie in den anderen Gruppen auch, stehen als
Beschwerden Luftnot (n=3, 75%), Husten (n=3, 75%), Müdigkeit (n=2, 50%),
Leistungsverlust (n=2, 50%), und Gelenkbeschwerden (n=1, 25%) im Vordergrund. Der
mittlere BMI lag bei 25,9 kg/m² und der Mittelwert für die pack years liegt bei 15,0,
wobei bei einem Patienten die Angabe über den Raucherstatus fehlte, die anderen waren
Raucher.
RaucherAllergieMüdigkeitHustenDyspnoe
% 110
100
90
80
70
60
50
40
100
5050
7575
Abb. 63: Anamnese bei den nicht interstitiellen Fällen
4.11.2 Klinische Untersuchungen
Ein Patient (25%) hatte Trommelschlegelfinger und Uhrglasnägel, ein weiterer isoliert
Uhrglasnägel. Die sonstige klinische Untersuchung war bis auf inspiratorisches
Knisterrasseln bei 2 Patienten (50%) vollkommen unauffällig.
Die BSG war auf 42,5 mm/h beschleunigt, sonst lagen die Laborwerte im Normbereich.

84
Röntgenbefunde lagen bei 3 (75%) Patienten vor. Sie zeigten bei einem Patienten
streifige Veränderungen.
Eine BAL wurde bei 1 (25%) Patienten durchgeführt. Die Zellverteilung ergab 3%
Lymphozyten, 5% Makrophagen, 92% Neutrophile und 0% Eosinophile.
Lungenfunktionsuntersuchungen wurden bei allen Patienten durchgeführt. Sie ergaben:
IVC 79,1% (vom Soll), TLC 119,2% (vom Soll), RV/TLC 55,5% (absoluter Wert),
FEV1% 58,3% (absoluter Wert) und RAW 123,9% (vom Soll).
Die BGA lag mit allen Parametern im Normbereich.
4.11.3 Verlauf
Hier war von keinem Patienten der weitere Verlauf bekannt.

85
4.12 Kasuistik: Rheumatoide Arthritis mit AIP-ähnlicher Histologie
Im Kollektiv war ein Fall mit den typischen histologischen Merkmalen einer AIP, dem
die Sicherheitsstufe „sicher“ zugewiesen wurde.
Es handelte sich um eine 60-jährige Hausfrau, die sich mit einer antibiotika-resistenten
Dyspnoe zunächst in der Ambulanz vorstellte und drei Wochen später stationär
aufgenommen wurde. Die Dyspnoe war erstmals zwei Monate zuvor aufgetreten und
hatte sich nun bis zur Ruhedyspnoe, die auch mit 2 l Sauerstoffzufuhr anhielt,
gesteigert. Die Patientin klagte weder über Husten noch über Auswurf. Es war aber ein
Gewichtsverlust von 11 kg, starke Müdigkeit und Leistungsverlust auffällig.
An Vorerkrankungen war eine Tuberkulose 50 Jahre zuvor, eine Struma nodosa, eine
Linksherzinsuffizienz sowie eine rheumatoide Arthritis bekannt, die erstmals 9 Monate
zuvor diagnostiziert worden war und wegen der die Patientin auch Goldpräparate
eingenommen hatte. Die aktuelle Medikation bestand in einem Nitrat, einem
Schleifendiuretikum, einem H2-Blocker, einem Neuroleptikum, einem Laxans, Jod und
Kalzium.
Außerdem bestand klinisch eine Lippenzyanose, Trommelschlegelfinger,
Unterschenkelödeme sowie ein Knisterrasseln in der Auskultation.
In den routinemäßigen Blutuntersuchungen waren die Erythrozyten mit 5,3 Mill./μl
grenzwertig, die Leukozyten mit14.100/μl erhöht und das LDH mit 566 U/l stark
erhöht. Die Körpertemperatur lag bei 38°C.
Das Thorax-Röntgenbild zeigte mikronoduläre und retikuläre fleckige Infiltrate im
linken Unterfeld. Im CT waren ausgedehnte Fibrosierungen und leichte milchglasartige
Verschattungen zu sehen. Ambulant wurde keine BAL durchgeführt, da die
Sauerstoffsättigung auch unter 8 l O2 nur 86% betrug. Die BAL wurde zwar während
des stationären Aufenthaltes nachgeholt, die Auswertung war aber leider nicht möglich.
Die stationäre Aufnahme erfolgte 21 Tage nach der Vorstellung in der Ambulanz. Nun
wurde eine offene Lungenbiopsie durchgeführt. Die Patientin verstarb weitere 8 Tage
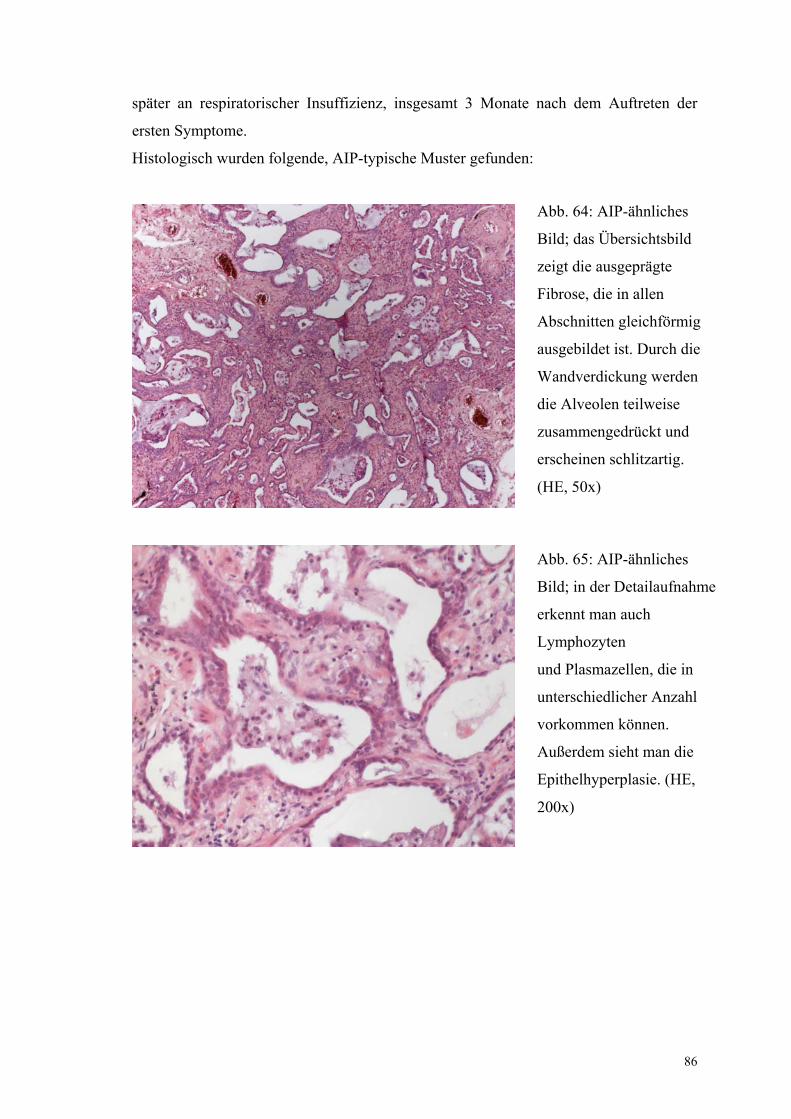
86
später an respiratorischer Insuffizienz, insgesamt 3 Monate nach dem Auftreten der
ersten Symptome.
Histologisch wurden folgende, AIP-typische Muster gefunden:
Abb. 65: AIP-ähnliches
Bild; in der Detailaufnahme
erkennt man auch
Lymphozyten
und Plasmazellen, die in
unterschiedlicher Anzahl
vorkommen können.
Außerdem sieht man die
Epithelhyperplasie. (HE,
200x)
Abb. 64: AIP-ähnliches
Bild; das Übersichtsbild
zeigt die ausgeprägte
Fibrose, die in allen
Abschnitten gleichförmig
ausgebildet ist. Durch die
Wandverdickung werden
die Alveolen teilweise
zusammengedrückt und
erscheinen schlitzartig.
(HE, 50x)
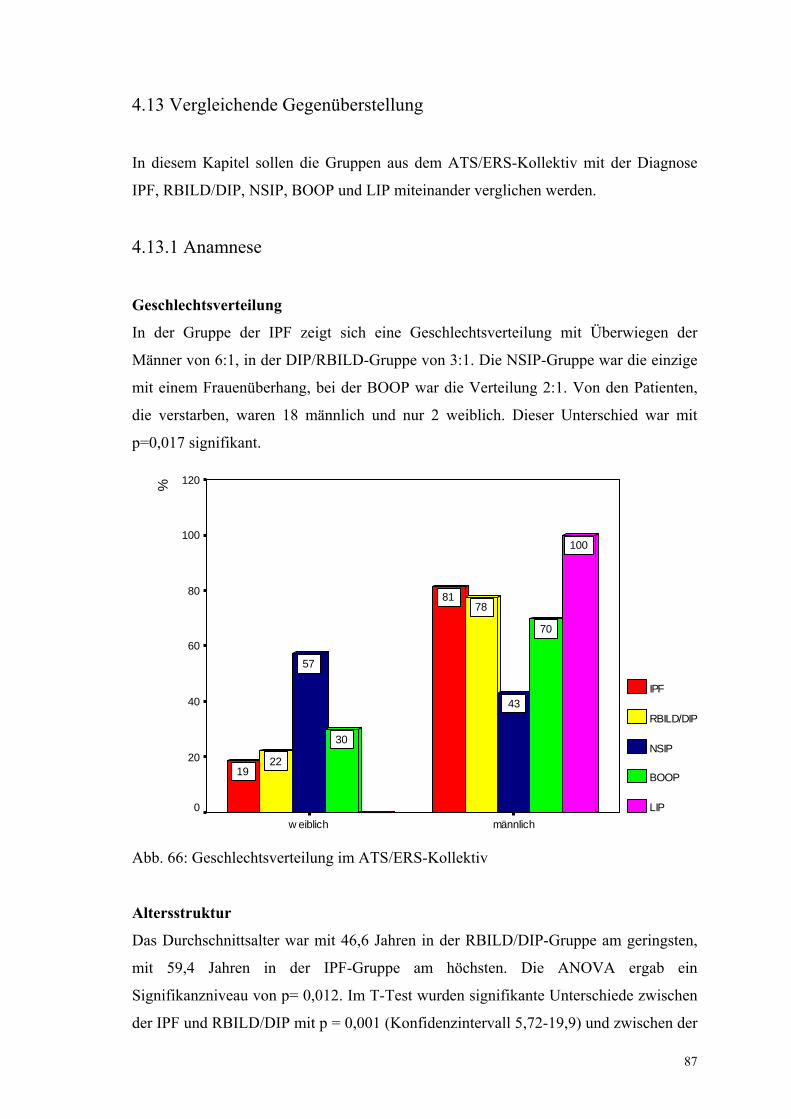
87
4.13 Vergleichende Gegenüberstellung
In diesem Kapitel sollen die Gruppen aus dem ATS/ERS-Kollektiv mit der Diagnose
IPF, RBILD/DIP, NSIP, BOOP und LIP miteinander verglichen werden.
4.13.1 Anamnese
Geschlechtsverteilung
In der Gruppe der IPF zeigt sich eine Geschlechtsverteilung mit Überwiegen der
Männer von 6:1, in der DIP/RBILD-Gruppe von 3:1. Die NSIP-Gruppe war die einzige
mit einem Frauenüberhang, bei der BOOP war die Verteilung 2:1. Von den Patienten,
die verstarben, waren 18 männlich und nur 2 weiblich. Dieser Unterschied war mit
p=0,017 signifikant.
männlichw eiblich
% 120
100
80
60
40
20
0
IPF
RBILD/DIP
NSIP
BOOP
LIP
100
70
30
43
57
78
22
81
19
Abb. 66: Geschlechtsverteilung im ATS/ERS-Kollektiv
Altersstruktur
Das Durchschnittsalter war mit 46,6 Jahren in der RBILD/DIP-Gruppe am geringsten,
mit 59,4 Jahren in der IPF-Gruppe am höchsten. Die ANOVA ergab ein
Signifikanzniveau von p= 0,012. Im T-Test wurden signifikante Unterschiede zwischen
der IPF und RBILD/DIP mit p = 0,001 (Konfidenzintervall 5,72-19,9) und zwischen der
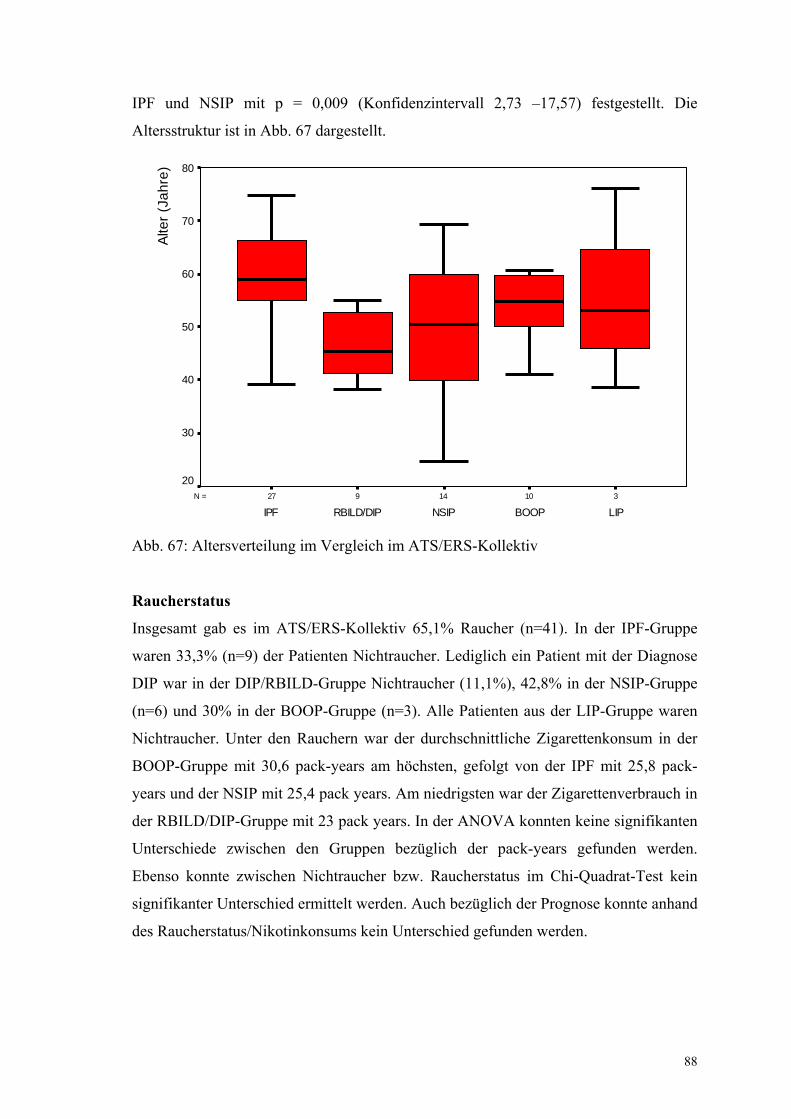
88
IPF und NSIP mit p = 0,009 (Konfidenzintervall 2,73 –17,57) festgestellt. Die
Altersstruktur ist in Abb. 67 dargestellt.
31014927N =
LIPBOOPNSIPRBILD/DIPIPF
Alte
r (Ja
hre) 80
70
60
50
40
30
20
Abb. 67: Altersverteilung im Vergleich im ATS/ERS-Kollektiv
Raucherstatus
Insgesamt gab es im ATS/ERS-Kollektiv 65,1% Raucher (n=41). In der IPF-Gruppe
waren 33,3% (n=9) der Patienten Nichtraucher. Lediglich ein Patient mit der Diagnose
DIP war in der DIP/RBILD-Gruppe Nichtraucher (11,1%), 42,8% in der NSIP-Gruppe
(n=6) und 30% in der BOOP-Gruppe (n=3). Alle Patienten aus der LIP-Gruppe waren
Nichtraucher. Unter den Rauchern war der durchschnittliche Zigarettenkonsum in der
BOOP-Gruppe mit 30,6 pack-years am höchsten, gefolgt von der IPF mit 25,8 pack-
years und der NSIP mit 25,4 pack years. Am niedrigsten war der Zigarettenverbrauch in
der RBILD/DIP-Gruppe mit 23 pack years. In der ANOVA konnten keine signifikanten
Unterschiede zwischen den Gruppen bezüglich der pack-years gefunden werden.
Ebenso konnte zwischen Nichtraucher bzw. Raucherstatus im Chi-Quadrat-Test kein
signifikanter Unterschied ermittelt werden. Auch bezüglich der Prognose konnte anhand
des Raucherstatus/Nikotinkonsums kein Unterschied gefunden werden.
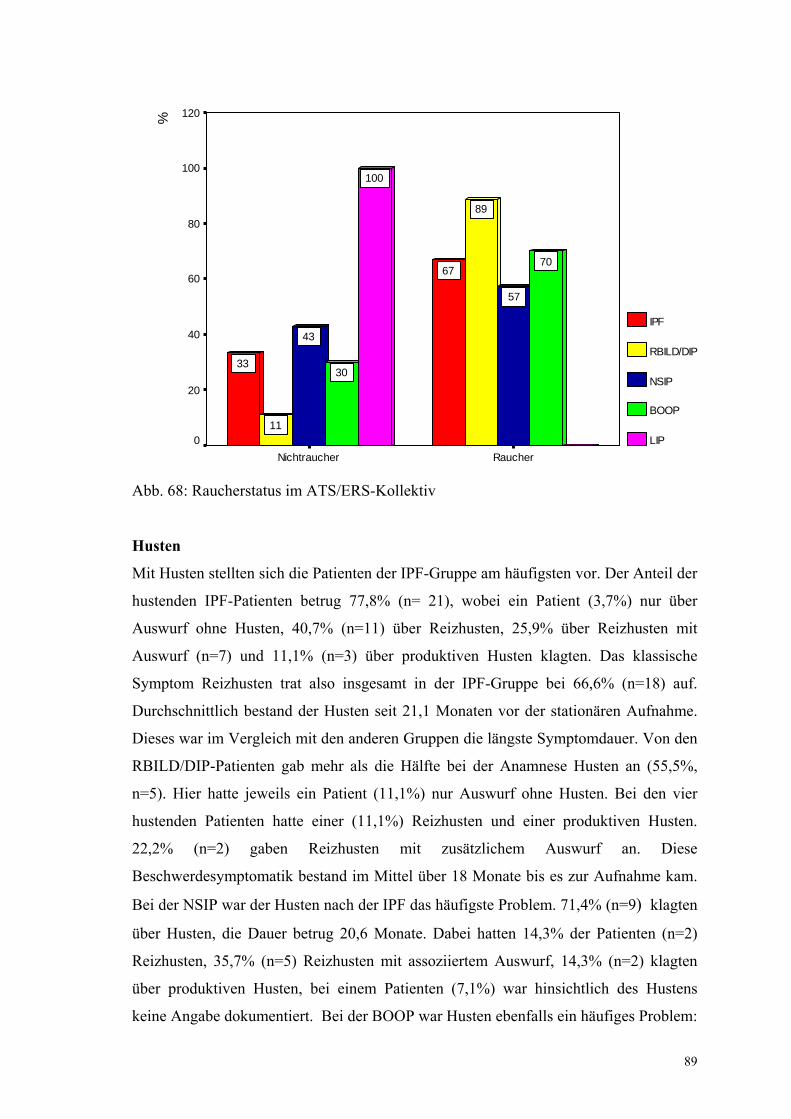
89
RaucherNichtraucher
% 120
100
80
60
40
20
0
IPF
RBILD/DIP
NSIP
BOOP
LIP
100
70
30
57
43
89
11
67
33
Abb. 68: Raucherstatus im ATS/ERS-Kollektiv
Husten
Mit Husten stellten sich die Patienten der IPF-Gruppe am häufigsten vor. Der Anteil der
hustenden IPF-Patienten betrug 77,8% (n= 21), wobei ein Patient (3,7%) nur über
Auswurf ohne Husten, 40,7% (n=11) über Reizhusten, 25,9% über Reizhusten mit
Auswurf (n=7) und 11,1% (n=3) über produktiven Husten klagten. Das klassische
Symptom Reizhusten trat also insgesamt in der IPF-Gruppe bei 66,6% (n=18) auf.
Durchschnittlich bestand der Husten seit 21,1 Monaten vor der stationären Aufnahme.
Dieses war im Vergleich mit den anderen Gruppen die längste Symptomdauer. Von den
RBILD/DIP-Patienten gab mehr als die Hälfte bei der Anamnese Husten an (55,5%,
n=5). Hier hatte jeweils ein Patient (11,1%) nur Auswurf ohne Husten. Bei den vier
hustenden Patienten hatte einer (11,1%) Reizhusten und einer produktiven Husten.
22,2% (n=2) gaben Reizhusten mit zusätzlichem Auswurf an. Diese
Beschwerdesymptomatik bestand im Mittel über 18 Monate bis es zur Aufnahme kam.
Bei der NSIP war der Husten nach der IPF das häufigste Problem. 71,4% (n=9) klagten
über Husten, die Dauer betrug 20,6 Monate. Dabei hatten 14,3% der Patienten (n=2)
Reizhusten, 35,7% (n=5) Reizhusten mit assoziiertem Auswurf, 14,3% (n=2) klagten
über produktiven Husten, bei einem Patienten (7,1%) war hinsichtlich des Hustens
keine Angabe dokumentiert. Bei der BOOP war Husten ebenfalls ein häufiges Problem:
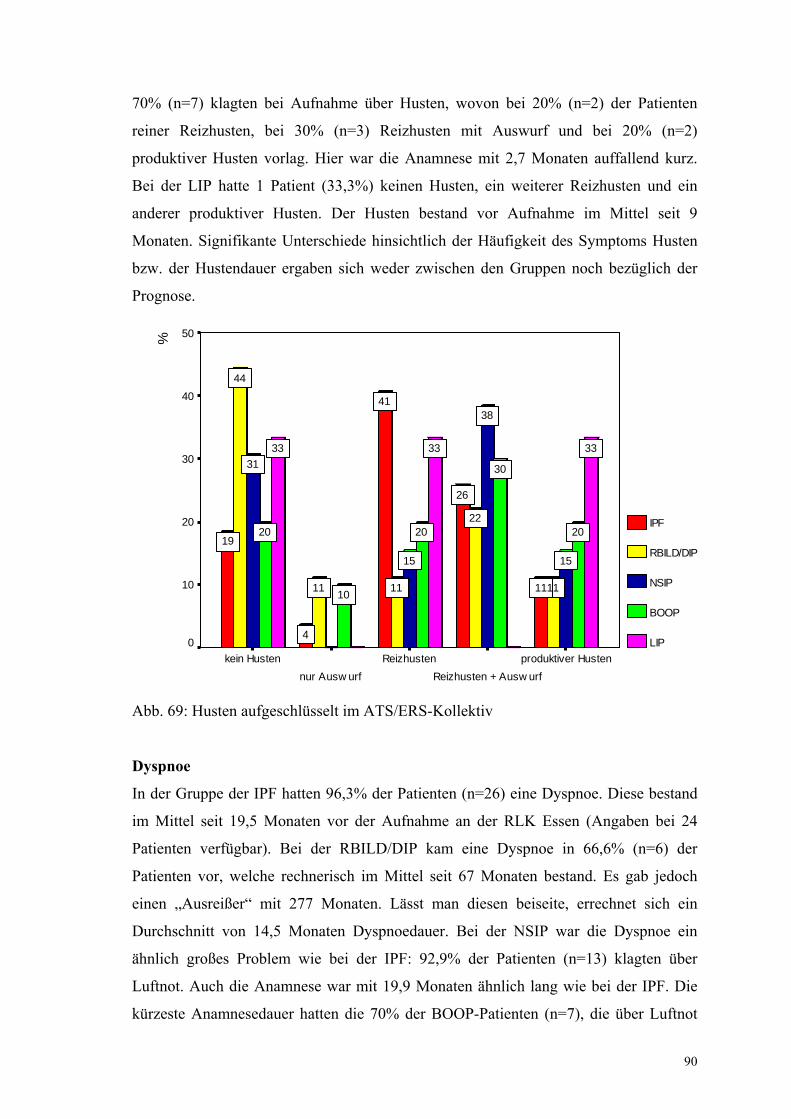
90
70% (n=7) klagten bei Aufnahme über Husten, wovon bei 20% (n=2) der Patienten
reiner Reizhusten, bei 30% (n=3) Reizhusten mit Auswurf und bei 20% (n=2)
produktiver Husten vorlag. Hier war die Anamnese mit 2,7 Monaten auffallend kurz.
Bei der LIP hatte 1 Patient (33,3%) keinen Husten, ein weiterer Reizhusten und ein
anderer produktiver Husten. Der Husten bestand vor Aufnahme im Mittel seit 9
Monaten. Signifikante Unterschiede hinsichtlich der Häufigkeit des Symptoms Husten
bzw. der Hustendauer ergaben sich weder zwischen den Gruppen noch bezüglich der
Prognose.
produktiver HustenReizhusten + Ausw urf
Reizhustennur Ausw urf
kein Husten
% 50
40
30
20
10
0
IPF
RBILD/DIP
NSIP
BOOP
LIP
333333
20
30
20
10
20
15
38
15
31
11
22
1111
44
11
26
41
4
19
Abb. 69: Husten aufgeschlüsselt im ATS/ERS-Kollektiv
Dyspnoe
In der Gruppe der IPF hatten 96,3% der Patienten (n=26) eine Dyspnoe. Diese bestand
im Mittel seit 19,5 Monaten vor der Aufnahme an der RLK Essen (Angaben bei 24
Patienten verfügbar). Bei der RBILD/DIP kam eine Dyspnoe in 66,6% (n=6) der
Patienten vor, welche rechnerisch im Mittel seit 67 Monaten bestand. Es gab jedoch
einen „Ausreißer“ mit 277 Monaten. Lässt man diesen beiseite, errechnet sich ein
Durchschnitt von 14,5 Monaten Dyspnoedauer. Bei der NSIP war die Dyspnoe ein
ähnlich großes Problem wie bei der IPF: 92,9% der Patienten (n=13) klagten über
Luftnot. Auch die Anamnese war mit 19,9 Monaten ähnlich lang wie bei der IPF. Die
kürzeste Anamnesedauer hatten die 70% der BOOP-Patienten (n=7), die über Luftnot
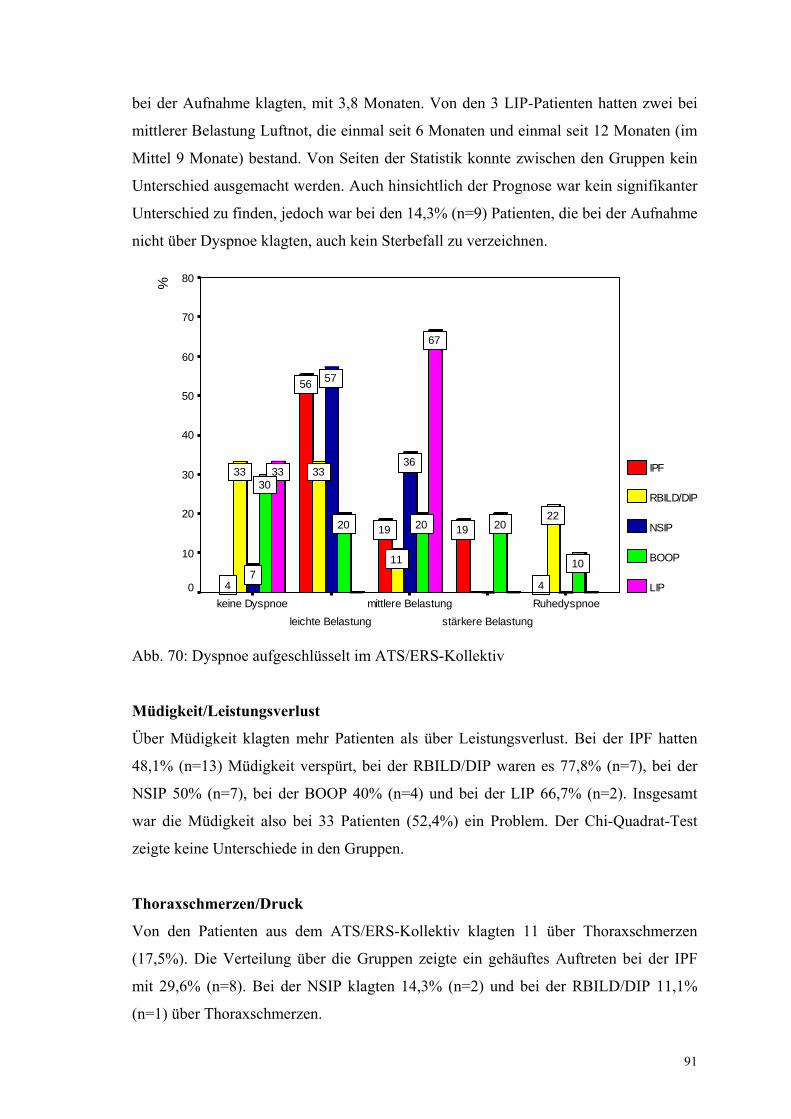
91
bei der Aufnahme klagten, mit 3,8 Monaten. Von den 3 LIP-Patienten hatten zwei bei
mittlerer Belastung Luftnot, die einmal seit 6 Monaten und einmal seit 12 Monaten (im
Mittel 9 Monate) bestand. Von Seiten der Statistik konnte zwischen den Gruppen kein
Unterschied ausgemacht werden. Auch hinsichtlich der Prognose war kein signifikanter
Unterschied zu finden, jedoch war bei den 14,3% (n=9) Patienten, die bei der Aufnahme
nicht über Dyspnoe klagten, auch kein Sterbefall zu verzeichnen.
Ruhedyspnoestärkere Belastung
mittlere Belastungleichte Belastung
keine Dyspnoe
% 80
70
60
50
40
30
20
10
0
IPF
RBILD/DIP
NSIP
BOOP
LIP
67
33
10
202020
30
36
57
7
22
11
3333
4
1919
56
4
Abb. 70: Dyspnoe aufgeschlüsselt im ATS/ERS-Kollektiv
Müdigkeit/Leistungsverlust
Über Müdigkeit klagten mehr Patienten als über Leistungsverlust. Bei der IPF hatten
48,1% (n=13) Müdigkeit verspürt, bei der RBILD/DIP waren es 77,8% (n=7), bei der
NSIP 50% (n=7), bei der BOOP 40% (n=4) und bei der LIP 66,7% (n=2). Insgesamt
war die Müdigkeit also bei 33 Patienten (52,4%) ein Problem. Der Chi-Quadrat-Test
zeigte keine Unterschiede in den Gruppen.
Thoraxschmerzen/Druck
Von den Patienten aus dem ATS/ERS-Kollektiv klagten 11 über Thoraxschmerzen
(17,5%). Die Verteilung über die Gruppen zeigte ein gehäuftes Auftreten bei der IPF
mit 29,6% (n=8). Bei der NSIP klagten 14,3% (n=2) und bei der RBILD/DIP 11,1%
(n=1) über Thoraxschmerzen.

92
Weder bei der BOOP noch bei der LIP kamen diese Beschwerden vor. Die
Unterschiede waren nicht signifikant.
Gelenke
Gelenkbeschwerden wurden bei der Anamnese von 19% der Patienten (n=12)
angegeben. Aus den Gruppen IPF, RBILD/DIP, NSIP und BOOP hatten jeweils 3
Patienten Gelenkbeschwerden. Anteilsmäßig traten die Gelenkbeschwerden am
häufigsten bei der RBILD/DIP auf mit 33,3%, gefolgt von der BOOP mit 30%, dann der
NSIP mit 21,4% und zuletzt der IPF mit 11,1%.
Allergie
Insgesamt gab es 22 Allergiker (34,9%). Davon waren 11 in der IPF-Gruppe (40,7%), 6
in der NSIP-Gruppe (42,9%), 3 in der BOOP-Gruppe (30%) und 2 in der RBILD/DIP-
Gruppe (22,2%). Es konnte im Chi-Quadrat-Test kein signifikanter Unterschied
ermittelt werden.
BMI
Die Werte für den Body-Mass-Index lagen zwischen 22,51 bei der LIP und 28,15 kg/m²
bei der IPF. Dazwischen lagen die RBILD/DIP mit 27,7, NSIP mit 26,0 und die BOOP
mit 25,7 kg/m². Damit waren im Durchschnitt, abgesehen von der LIP, alle Patienten
übergewichtig, jedoch noch nicht im Bereich der Adipositas (>30kg/m²). Für die
Prognose war der BMI kein prädiktiver Faktor.
Vogelhaltung
Im ATS/ERS Kollektiv waren 4 Patienten (6,4%) (1 IPF-, 1 NSIP-, 2 RBILD-
Patienten) zum Zeitpunkt der Diagnose gegenüber Vögeln exponiert. Bei allen zeigte
die BAL keine Lymphozytose und die Präzipitine waren negativ, so dass eine EAA
auszuschließen war.
Pneumonien
Ein Viertel der Patienten hatte zuvor bereits einmal eine Pneumonie durchgemacht
(n=16). Sie waren bei der Hälfte der BOOP-Patienten aufgetreten (n=5). Bei der NSIP
waren es 42,9% (n=6), bei der RBILD/DIP 22,2% (n=2) und bei der IPF 11,1% (n=3).
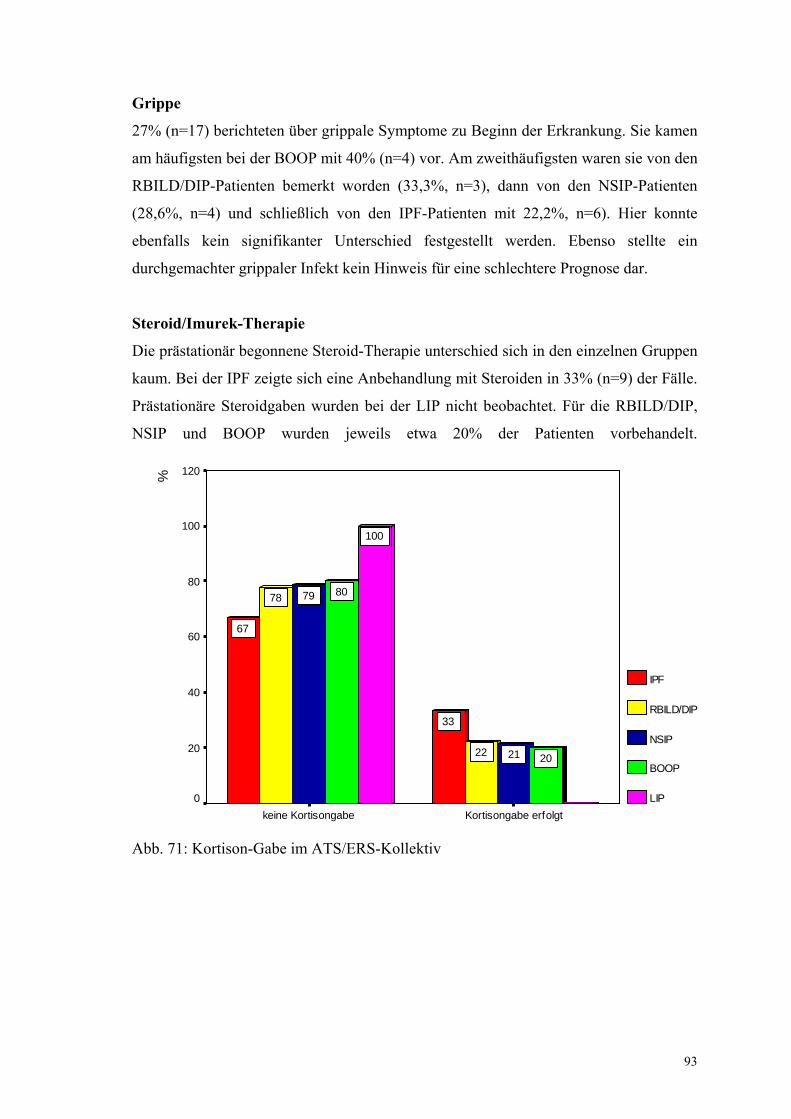
93
Grippe
27% (n=17) berichteten über grippale Symptome zu Beginn der Erkrankung. Sie kamen
am häufigsten bei der BOOP mit 40% (n=4) vor. Am zweithäufigsten waren sie von den
RBILD/DIP-Patienten bemerkt worden (33,3%, n=3), dann von den NSIP-Patienten
(28,6%, n=4) und schließlich von den IPF-Patienten mit 22,2%, n=6). Hier konnte
ebenfalls kein signifikanter Unterschied festgestellt werden. Ebenso stellte ein
durchgemachter grippaler Infekt kein Hinweis für eine schlechtere Prognose dar.
Steroid/Imurek-Therapie
Die prästationär begonnene Steroid-Therapie unterschied sich in den einzelnen Gruppen
kaum. Bei der IPF zeigte sich eine Anbehandlung mit Steroiden in 33% (n=9) der Fälle.
Prästationäre Steroidgaben wurden bei der LIP nicht beobachtet. Für die RBILD/DIP,
NSIP und BOOP wurden jeweils etwa 20% der Patienten vorbehandelt.
Kortisongabe erfolgtkeine Kortisongabe
% 120
100
80
60
40
20
0
IPF
RBILD/DIP
NSIP
BOOP
LIP
100
20
80
21
79
22
78
33
67
Abb. 71: Kortison-Gabe im ATS/ERS-Kollektiv
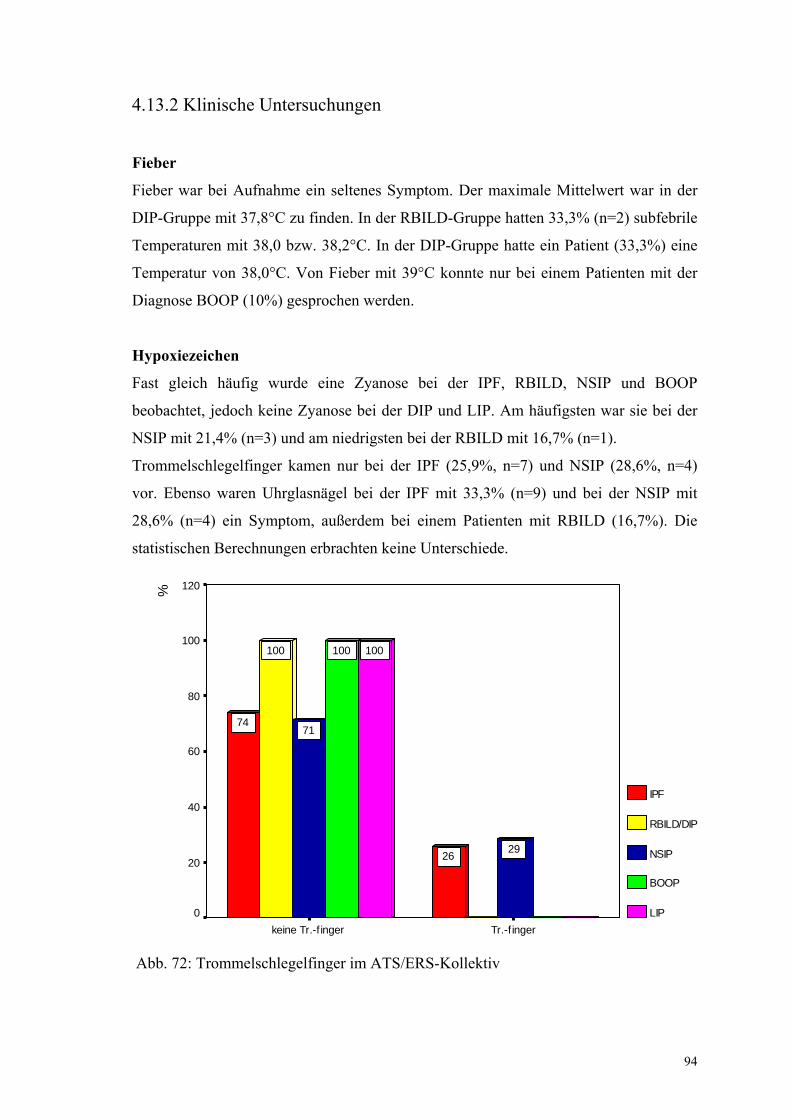
94
4.13.2 Klinische Untersuchungen
Fieber
Fieber war bei Aufnahme ein seltenes Symptom. Der maximale Mittelwert war in der
DIP-Gruppe mit 37,8°C zu finden. In der RBILD-Gruppe hatten 33,3% (n=2) subfebrile
Temperaturen mit 38,0 bzw. 38,2°C. In der DIP-Gruppe hatte ein Patient (33,3%) eine
Temperatur von 38,0°C. Von Fieber mit 39°C konnte nur bei einem Patienten mit der
Diagnose BOOP (10%) gesprochen werden.
Hypoxiezeichen
Fast gleich häufig wurde eine Zyanose bei der IPF, RBILD, NSIP und BOOP
beobachtet, jedoch keine Zyanose bei der DIP und LIP. Am häufigsten war sie bei der
NSIP mit 21,4% (n=3) und am niedrigsten bei der RBILD mit 16,7% (n=1).
Trommelschlegelfinger kamen nur bei der IPF (25,9%, n=7) und NSIP (28,6%, n=4)
vor. Ebenso waren Uhrglasnägel bei der IPF mit 33,3% (n=9) und bei der NSIP mit
28,6% (n=4) ein Symptom, außerdem bei einem Patienten mit RBILD (16,7%). Die
statistischen Berechnungen erbrachten keine Unterschiede.
Tr.-f ingerkeine Tr.-f inger
% 120
100
80
60
40
20
0
IPF
RBILD/DIP
NSIP
BOOP
LIP
100100
29
71
100
26
74
Abb. 72: Trommelschlegelfinger im ATS/ERS-Kollektiv
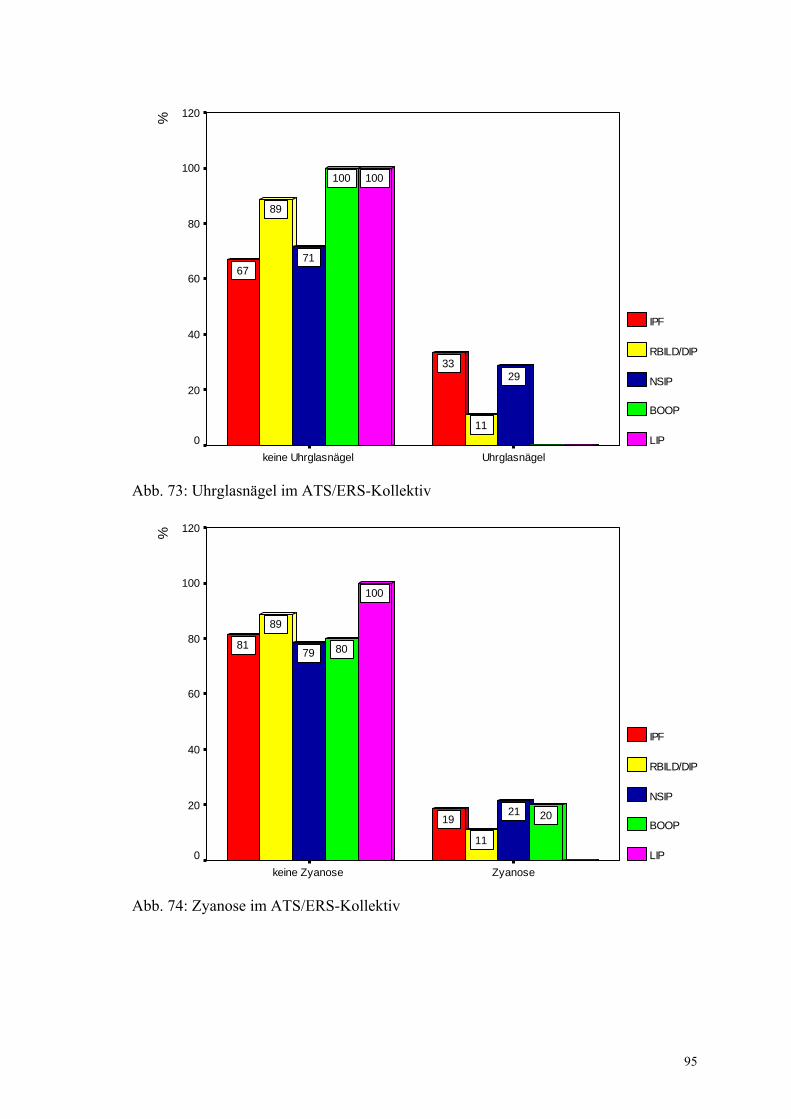
95
Uhrglasnägelkeine Uhrglasnägel
% 120
100
80
60
40
20
0
IPF
RBILD/DIP
NSIP
BOOP
LIP
100100
29
71
11
89
33
67
Abb. 73: Uhrglasnägel im ATS/ERS-Kollektiv
Zyanosekeine Zyanose
% 120
100
80
60
40
20
0
IPF
RBILD/DIP
NSIP
BOOP
LIP
100
20
80
21
79
11
89
19
81
Abb. 74: Zyanose im ATS/ERS-Kollektiv

96
Inspiratorisches Knisterrasseln
Ein häufig auftretendes Symptom ist das inspiratorische Knisterrasseln. Bei der IPF war
es immer vorhanden (100%, n=27). Bei der RBILD war es bei einem Patienten (16,7%)
auskultierbar, bei der DIP ebenfalls bei einem Patienten (33,3%), bei der NSIP konnte
bei 64,3% (n=9) ein Knistergeräusch gehört werden, bei der BOOP in der Hälfte der
Fälle und bei der LIP bei zwei Patienten (66,7%). Auch hier wurden keine signifikanten
Unterschiede gefunden.
Knisterrasselnkein Knistern
% 120
100
80
60
40
20
0
IPF
RBILD/DIP
NSIP
BOOP
LIP
67
33
5050
64
36
22
78
100
Abb. 75: inspiratorisches Knisterrasseln im ATS/ERS-Kollektiv
Laborwerte
Im Blutbild waren bei den Erythrozyten, dem Hämoglobin, dem Hämatokrit und den
Leukozyten alle Mittelwerte im jeweiligen Normbereich. Die Thrombozyten waren bei
der LIP im Mittel auf 484.000/µl erhöht, in den anderen Gruppen im Normbereich. Hier
ergab sich auch in der ANOVA ein signifikanter Unterschied mit p = 0,01.
Routinemäßig wurde außerdem die LDH bestimmt. Hier zeigte sich nur bei der IPF eine
leichte Erhöhung auf 247,5 U/l, wobei der Wert bei 33,3% (n=9) bis maximal 373 U/l
auffällig war.

97
Andere Laboruntersuchungen wurden nach Anordnung durchgeführt: Das CRP war bei
den erhobenen Werten im Mittel leicht erhöht: es lag bei der IPF bei 1,66, bei der
RBILD bei 1,34, bei der DIP bei 5,48 (wobei einer von 2 Werten auf 10,4 erhöht war),
bei der BOOP bei 2,63 und bei der LIP war das CRP nicht bestimmt worden.
Die CEA-Werte waren normwertig bis leicht erhöht. Der maximale Mittelwert war bei
der NSIP mit 5,9 U/l zu finden. Leichte Erhöhungen über den Normwert von 5 U/l
waren außerdem bei der RBILD und der BOOP zu finden. Bei der DIP wurde der CEA-
Wert nicht bestimmt.
Rheumafaktoren wurden insgesamt 19 Mal ausgewertet, bei der IPF waren sie stets
negativ, ebenso bei der DIP und der BOOP. Lediglich bei der NSIP waren von 8
bestimmten Werten die Rheumafaktoren 2 Mal positiv.
Lungenfunktionsanalyse
Für die IVC wurden die höchsten Werte bei der LIP mit 76,6% vom Soll gesehen, der
niedrigste Wert war bei der NSIP mit 58,0% vom Soll zu finden. Betrachtet man die
TLC, findet sich der höchste Wert ebenfalls für die LIP mit fast normalen Werten von
92,86% vom Soll, der niedrigste Wert liegt bei 66,6% für die IPF. Die NSIP hat die
besten Werte bezüglich des Atemwegswiderstandes (98,6%), die schlechtesten liegen
bei der LIP mit 72,2% vor. Pathologische Werte mit über 40% fanden sich mit
Ausnahme der IPF (35,2%) in allen Gruppen, hier hatte die NSIP mit 49,8% den
höchsten Quotienten. Bei der FEV1% war ebenfalls nur ein Wert, nämlich der der LIP
mit 69% normwertig, der am weitesten abweichende Wert war bei der BOOP mit 80,3%
auszumachen.
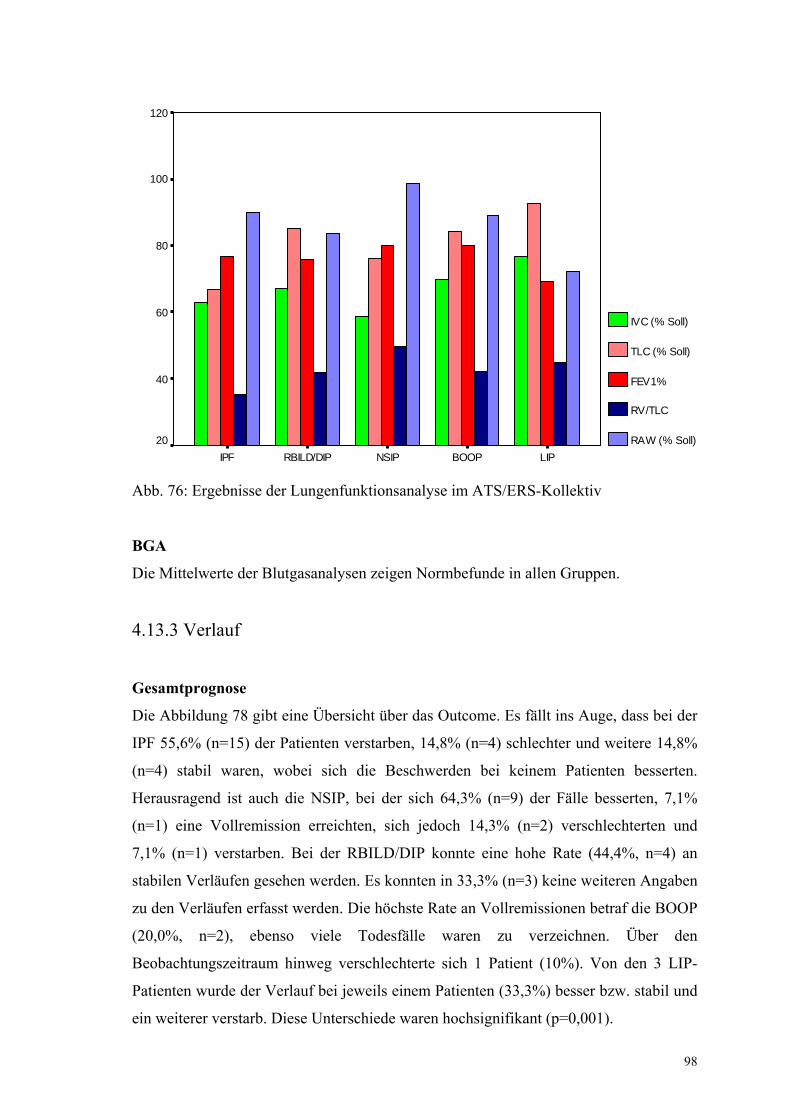
98
LIPBOOPNSIPRBILD/DIPIPF
120
100
80
60
40
20
IVC (% Soll)
TLC (% Soll)
FEV1%
RV/TLC
RAW (% Soll)
Abb. 76: Ergebnisse der Lungenfunktionsanalyse im ATS/ERS-Kollektiv
BGA
Die Mittelwerte der Blutgasanalysen zeigen Normbefunde in allen Gruppen.
4.13.3 Verlauf
Gesamtprognose
Die Abbildung 78 gibt eine Übersicht über das Outcome. Es fällt ins Auge, dass bei der
IPF 55,6% (n=15) der Patienten verstarben, 14,8% (n=4) schlechter und weitere 14,8%
(n=4) stabil waren, wobei sich die Beschwerden bei keinem Patienten besserten.
Herausragend ist auch die NSIP, bei der sich 64,3% (n=9) der Fälle besserten, 7,1%
(n=1) eine Vollremission erreichten, sich jedoch 14,3% (n=2) verschlechterten und
7,1% (n=1) verstarben. Bei der RBILD/DIP konnte eine hohe Rate (44,4%, n=4) an
stabilen Verläufen gesehen werden. Es konnten in 33,3% (n=3) keine weiteren Angaben
zu den Verläufen erfasst werden. Die höchste Rate an Vollremissionen betraf die BOOP
(20,0%, n=2), ebenso viele Todesfälle waren zu verzeichnen. Über den
Beobachtungszeitraum hinweg verschlechterte sich 1 Patient (10%). Von den 3 LIP-
Patienten wurde der Verlauf bei jeweils einem Patienten (33,3%) besser bzw. stabil und
ein weiterer verstarb. Diese Unterschiede waren hochsignifikant (p=0,001).
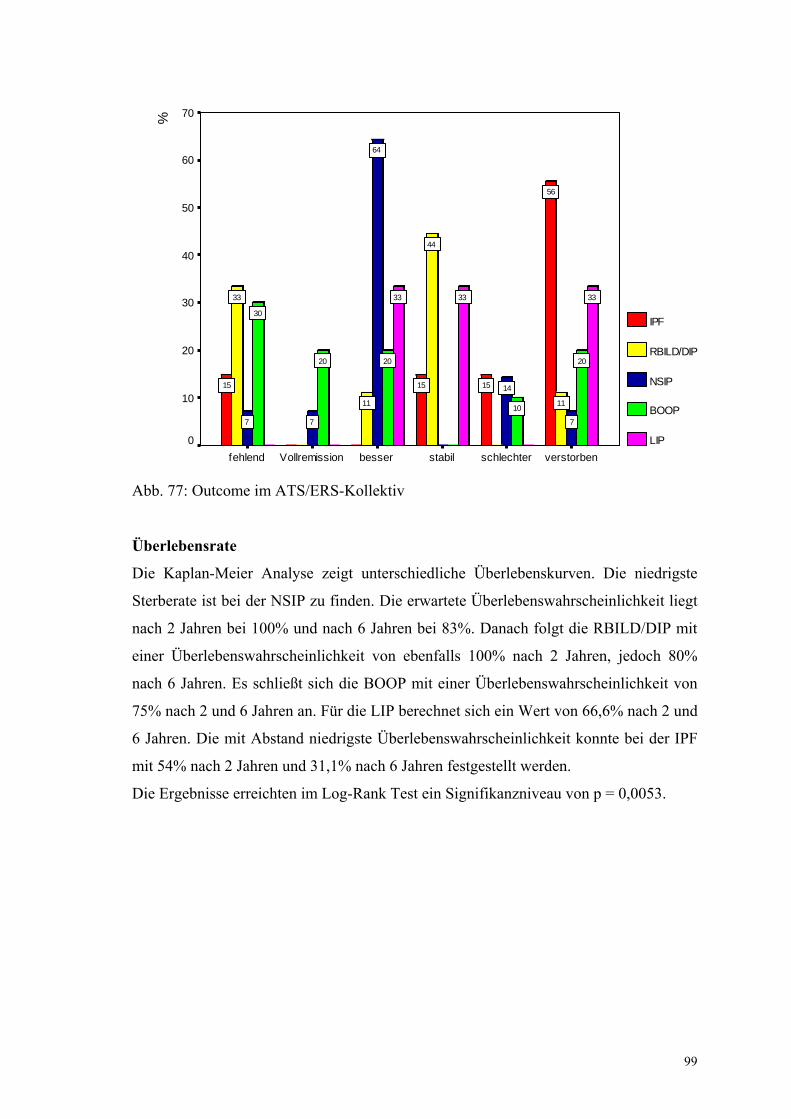
99
verstorbenschlechterstabilbesserVollremissionfehlend
% 70
60
50
40
30
20
10
0
IPF
RBILD/DIP
NSIP
BOOP
LIP
333333
20
10
2020
30
7
14
64
77
11
44
11
33
56
151515
Abb. 77: Outcome im ATS/ERS-Kollektiv
Überlebensrate
Die Kaplan-Meier Analyse zeigt unterschiedliche Überlebenskurven. Die niedrigste
Sterberate ist bei der NSIP zu finden. Die erwartete Überlebenswahrscheinlichkeit liegt
nach 2 Jahren bei 100% und nach 6 Jahren bei 83%. Danach folgt die RBILD/DIP mit
einer Überlebenswahrscheinlichkeit von ebenfalls 100% nach 2 Jahren, jedoch 80%
nach 6 Jahren. Es schließt sich die BOOP mit einer Überlebenswahrscheinlichkeit von
75% nach 2 und 6 Jahren an. Für die LIP berechnet sich ein Wert von 66,6% nach 2 und
6 Jahren. Die mit Abstand niedrigste Überlebenswahrscheinlichkeit konnte bei der IPF
mit 54% nach 2 Jahren und 31,1% nach 6 Jahren festgestellt werden.
Die Ergebnisse erreichten im Log-Rank Test ein Signifikanzniveau von p = 0,0053.
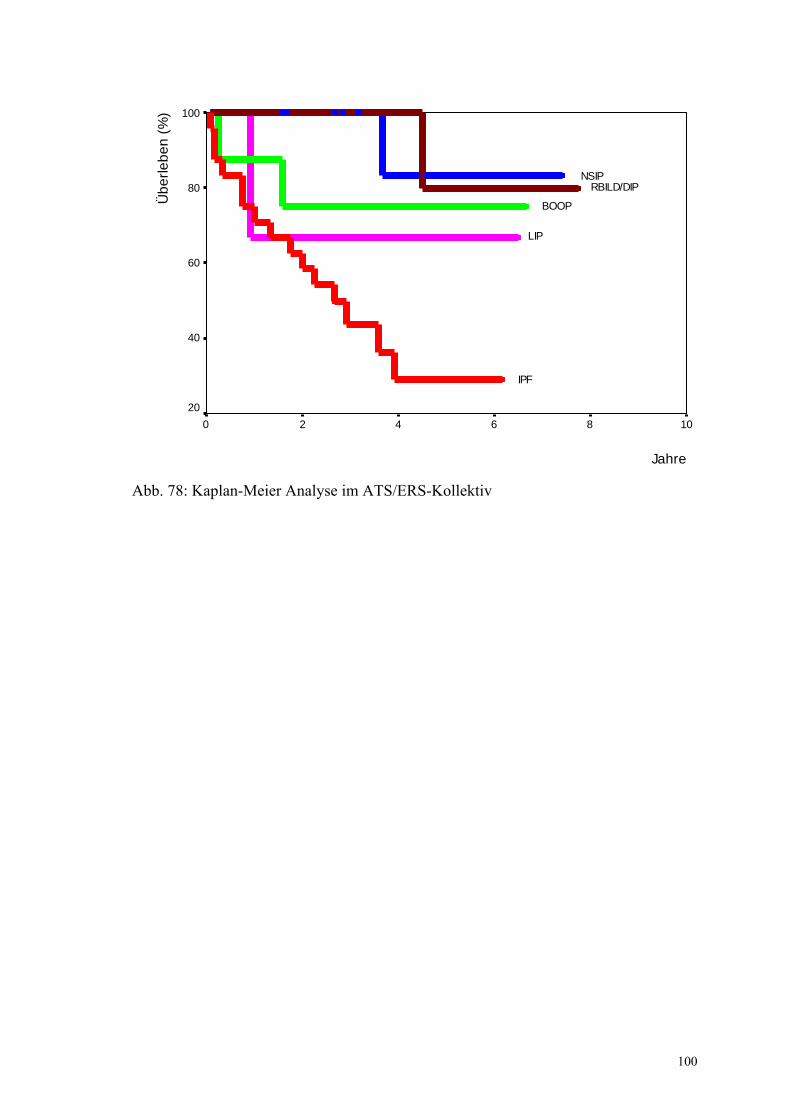
100
Jahre
1086420
Übe
rlebe
n (%
) 100
80
60
40
20
NSIPRBILD/DIP
BOOP
LIP
IPF
Abb. 78: Kaplan-Meier Analyse im ATS/ERS-Kollektiv

101
4.14 Status in BAL-Subgruppen
Die BAL-Zelldifferenzierung zeigt Unterschiede der einzelnen Zelltypen in den IPF-
Untergruppen. Die Makrophagen waren am höchsten in der DIP/RBILD-Gruppe mit
76% und am niedrigsten in der BOOP-Gruppe mit 44%. Dazwischen lag die IPF-
Gruppe mit einem Makrophagen-Anteil von 66% und die NSIP-Gruppe mit 49%.
Die Lymphozyten waren in allen Gruppen erhöht, der höchste Lymphozyten-Anteil
wurde bei der NSIP-Gruppe mit 34% und BOOP mit 41% gesehen, gefolgt von der IPF-
Gruppe mit 19% und der DIP/RBILD-Gruppe mit 14%. Neutrophile waren in der IPF-
Gruppe mit 9 ebenso in der NSIP-Gruppe mit 12% erhöht.
Mit 5% waren die Eosinophilen als Fibrosemarker bei allen Gruppen in gleicher Weise
erhöht.
Die Mastzellen lagen bei der IPF bei 0,5%, in absteigender Reihenfolge schließen sich
NSIP, BOOP und DIP/RBILD an.
In der ANOVA konnten keine signifikanten Unterschiede ausgemacht werden, bei den
Makrophagen und Lymphozyten wurde ein ausreichendes Signifikanzniveau jedoch nur
knapp verfehlt.
Tab. 11: BAL Daten in den Untergruppen
IPF DIP/RBILD NSIP BOOP p
n=22 n=8 n=14 n=9
Makrophagen (%±SD) 66±19 76±20 49±31 44±35 0,051
Lymphozyten (%±SD) 19±14 14±19 34±29 41±36 0,059
Neutrophile (%±SD) 9±6 7±9 12±14 8±15 0,865
Eosinophile (%±SD) 5±12 5±12 5±7 5±12 0,981
Mastzellen (%±SD) 0,5±0,4 0,1±0,2 0,4±0,5 0,3±0,4 0,286
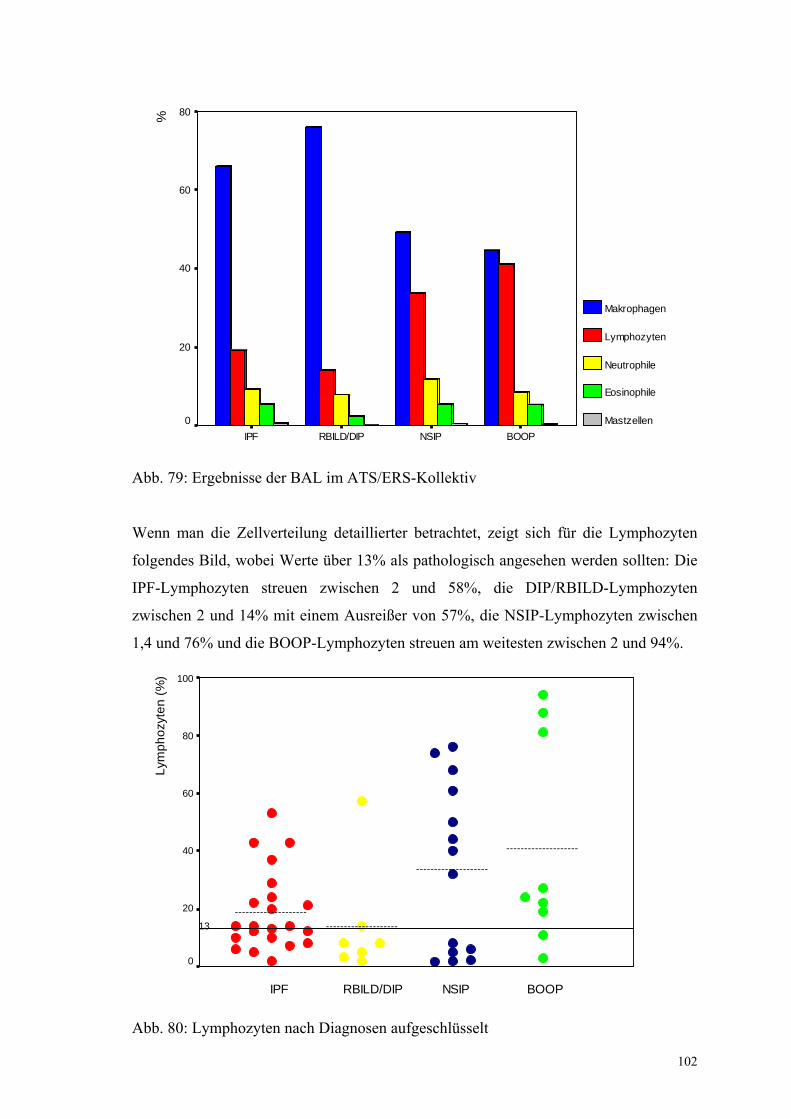
102
BOOPNSIPRBILD/DIPIPF
% 80
60
40
20
0
Makrophagen
Lymphozyten
Neutrophile
Eosinophile
Mastzellen
Abb. 79: Ergebnisse der BAL im ATS/ERS-Kollektiv
Wenn man die Zellverteilung detaillierter betrachtet, zeigt sich für die Lymphozyten
folgendes Bild, wobei Werte über 13% als pathologisch angesehen werden sollten: Die
IPF-Lymphozyten streuen zwischen 2 und 58%, die DIP/RBILD-Lymphozyten
zwischen 2 und 14% mit einem Ausreißer von 57%, die NSIP-Lymphozyten zwischen
1,4 und 76% und die BOOP-Lymphozyten streuen am weitesten zwischen 2 und 94%.
IPF RBILD/DIP NSIP BOOP
Lym
phoz
yten
(%) 100
80
60
40
20
0
----------------------------------------
--------------------
--------------------
13
Abb. 80: Lymphozyten nach Diagnosen aufgeschlüsselt
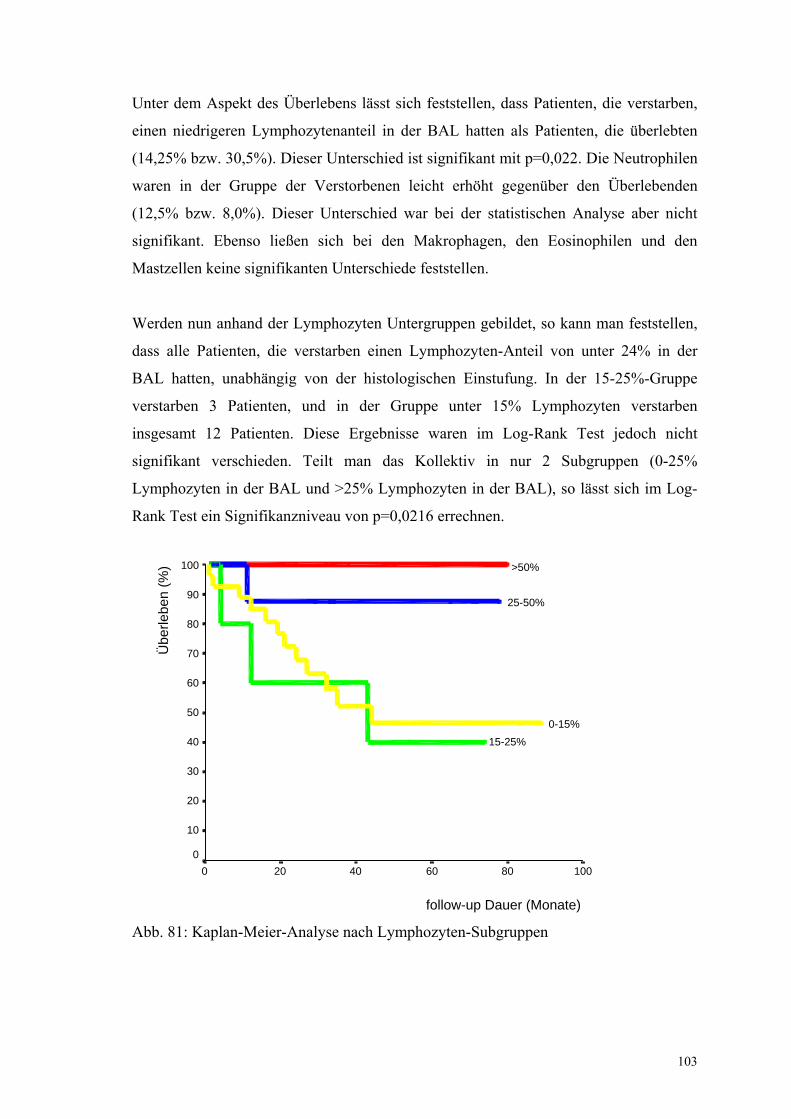
103
Unter dem Aspekt des Überlebens lässt sich feststellen, dass Patienten, die verstarben,
einen niedrigeren Lymphozytenanteil in der BAL hatten als Patienten, die überlebten
(14,25% bzw. 30,5%). Dieser Unterschied ist signifikant mit p=0,022. Die Neutrophilen
waren in der Gruppe der Verstorbenen leicht erhöht gegenüber den Überlebenden
(12,5% bzw. 8,0%). Dieser Unterschied war bei der statistischen Analyse aber nicht
signifikant. Ebenso ließen sich bei den Makrophagen, den Eosinophilen und den
Mastzellen keine signifikanten Unterschiede feststellen.
Werden nun anhand der Lymphozyten Untergruppen gebildet, so kann man feststellen,
dass alle Patienten, die verstarben einen Lymphozyten-Anteil von unter 24% in der
BAL hatten, unabhängig von der histologischen Einstufung. In der 15-25%-Gruppe
verstarben 3 Patienten, und in der Gruppe unter 15% Lymphozyten verstarben
insgesamt 12 Patienten. Diese Ergebnisse waren im Log-Rank Test jedoch nicht
signifikant verschieden. Teilt man das Kollektiv in nur 2 Subgruppen (0-25%
Lymphozyten in der BAL und >25% Lymphozyten in der BAL), so lässt sich im Log-
Rank Test ein Signifikanzniveau von p=0,0216 errechnen.
Abb. 81: Kaplan-Meier-Analyse nach Lymphozyten-Subgruppen
follow-up Dauer (Monate)
100806040200
Übe
rlebe
n (%
) 100
90
80
70
60
50
40
30
20
10
0
>50%
25-50%
0-15%15-25%
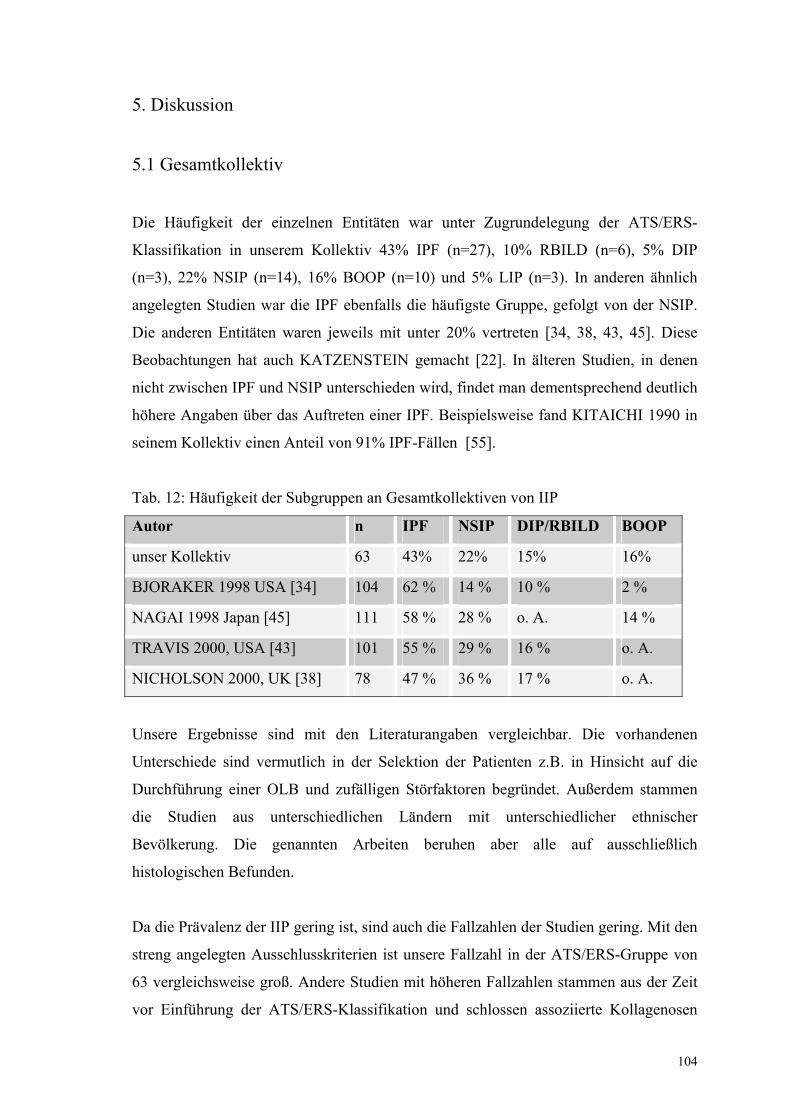
104
5. Diskussion
5.1 Gesamtkollektiv
Die Häufigkeit der einzelnen Entitäten war unter Zugrundelegung der ATS/ERS-
Klassifikation in unserem Kollektiv 43% IPF (n=27), 10% RBILD (n=6), 5% DIP
(n=3), 22% NSIP (n=14), 16% BOOP (n=10) und 5% LIP (n=3). In anderen ähnlich
angelegten Studien war die IPF ebenfalls die häufigste Gruppe, gefolgt von der NSIP.
Die anderen Entitäten waren jeweils mit unter 20% vertreten [34, 38, 43, 45]. Diese
Beobachtungen hat auch KATZENSTEIN gemacht [22]. In älteren Studien, in denen
nicht zwischen IPF und NSIP unterschieden wird, findet man dementsprechend deutlich
höhere Angaben über das Auftreten einer IPF. Beispielsweise fand KITAICHI 1990 in
seinem Kollektiv einen Anteil von 91% IPF-Fällen [55].
Tab. 12: Häufigkeit der Subgruppen an Gesamtkollektiven von IIP
Autor n IPF NSIP DIP/RBILD BOOP
unser Kollektiv 63 43% 22% 15% 16%
BJORAKER 1998 USA [34] 104 62 % 14 % 10 % 2 %
NAGAI 1998 Japan [45] 111 58 % 28 % o. A. 14 %
TRAVIS 2000, USA [43] 101 55 % 29 % 16 % o. A.
NICHOLSON 2000, UK [38] 78 47 % 36 % 17 % o. A.
Unsere Ergebnisse sind mit den Literaturangaben vergleichbar. Die vorhandenen
Unterschiede sind vermutlich in der Selektion der Patienten z.B. in Hinsicht auf die
Durchführung einer OLB und zufälligen Störfaktoren begründet. Außerdem stammen
die Studien aus unterschiedlichen Ländern mit unterschiedlicher ethnischer
Bevölkerung. Die genannten Arbeiten beruhen aber alle auf ausschließlich
histologischen Befunden.
Da die Prävalenz der IIP gering ist, sind auch die Fallzahlen der Studien gering. Mit den
streng angelegten Ausschlusskriterien ist unsere Fallzahl in der ATS/ERS-Gruppe von
63 vergleichsweise groß. Andere Studien mit höheren Fallzahlen stammen aus der Zeit
vor Einführung der ATS/ERS-Klassifikation und schlossen assoziierte Kollagenosen

105
mit ein, oder es wurde nur eine TBB durchgeführt bzw. auf histologische
Untersuchungen ganz verzichtet [14, 31, 56, 57, 58, 59].
Die vier in der Tabelle aufgeführten Studien hatten Fallzahlen zwischen 78 und 111
Patienten und erfüllen alle das Kriterium der offenen Lungenbiopsie. NAGAI et al.
verglichen nicht nur eigene Patienten miteinander [45], auch andere Studien erreichten
hohe Fallzahlen, indem sie Fälle aus verschiedenen Kliniken zusammengetragen haben,
beispielsweise werteten YOUSEM et al. 18 RBILD und 36 DIP-Fälle aus, die an drei
verschiedenen Kliniken diagnostiziert worden waren [27]. Für die anderen drei Studien
[34, 38, 43] stammen die Biopsien aus einem längeren Zeitraum als in unserer Studie,
was zu der höheren Fallzahl geführt hat. BJORAKER et al. erfasste Biopsien zwischen
1976 und 1989, TRAVIS et al. zwischen 1970 und 1992 und NICHOLSON et al.
zwischen 1978 und 1989. Wir beschränkten uns aus praktischen Gründen auf die Zeit
nach 1993, da ab diesem Zeitpunkt durch Einführung der EDV eine computergestützte
Recherche möglich war und sowohl das Biopsiematerial als auch die Krankenakten aus
den Archiven direkt verfügbar waren.
Wir fanden eine Geschlechtsverteilung von annähernd 3:2 (♂:♀) und eine
Altersverteilung zwischen 25 bis 76 Jahren mit einem Durchschnitt von 53 Jahren,
wobei die Frauen im Schnitt 3 Jahre jünger waren als die Männer. Sehr ähnliche
Angaben wurden bereits 1964 von LIVINGSTONE et al. gemacht [60]. Er fand ein
Verhältnis von 5:4 (♂:♀) und ein Alter zwischen 20 bis 78 Jahren bei einem
Durchschnitt von 50,5 Jahren. Gelegentlich wird aber auch ein Frauenüberhang wie bei
GELB et al. von 7:13 (♂:♀) gefunden [61]. Als Hauptsymptome gelten Dyspnoe,
Husten und Allgemeinsymptome wie Müdigkeit, Leistungsverlust, Gelenkbeschwerden,
grippale Infekte und Gewichtsverlust. Trommelschlegelfinger, Knisterrasseln und
Zyanose fallen bei der körperlichen Untersuchung auf. Die Laborbefunde sind in der
Regel normwertig, gelegentlich ist die Blutsenkungsgeschwindigkeit erhöht. Außerdem
treten zumeist restriktive Ventilationsstörungen auf. Radiologisch werden bilaterale
Infiltrate gesehen [6, 16, 60, 62]. Diese Ergebnisse konnten mit unserem Kollektiv
bestätigt werden. Es war aber in unserem Kollektiv ein geschlechtsspezifischer
Unterschied bei der Anamnesedauer auffällig. Diese lag bei den Frauen um mehr als 2
Jahre höher als bei den Männern. Erklärbar ist dieser Unterschied durch eine extrem
lange Anamnesedauer bei drei Frauen. Sie gaben an, die ersten Symptome bis zu 23

106
Jahre vor der Biopsie verspürt zu haben. Lässt man diese „Ausreißer“ bei der
Auswertung weg, gleicht sich die Anamnesedauer denen der Männer an.
Abgesehen von der allgemeinen Problematik, die bei einer retrospektiven Studie
auftritt, ist die Auswertung der radiologischen Befunde besonders schwierig. Hier
wurden die schriftlichen Befunde aus den Krankenakten dargestellt und die Röntgen-
bzw. CT-Bilder nur bei der Plausibilitätsprüfung hinzugezogen. Für die schriftlichen
Befunde gibt es keine Standards oder Befundungsschemata, sie sind in ihrem Umfang
und Präzision stark unterschiedlich, teilweise waren überhaupt keine schriftlichen
Befunde dokumentiert, obwohl Aufnahmen angefertigt wurden.
Während der Follow-Up Dauer von 3 Jahren verstarben 24% der Patienten. Das
entspricht auch dem Anteil der Todesfälle in der Studie von LIVINGSTONE et al. .
Allerdings bestehen deutliche Unterschiede im Outcome zwischen seinem und unserem
Kollektiv. Die prozentualen Anteile der Verläufe, die sich verbesserten lagen bei uns
um 25% höher, die stabilen um 13% niedriger und die Fälle, die sich verschlechterten
bzw. verstarben um 20% niedriger [60]. Eine höhere Mortalitätsrate und geringe Fälle
von Remissionen finden sich auch in anderen Studien [62]. Die vergleichsweise
höheren Mortalitätsraten und niedrigeren Remissionsraten in den Studien lassen sich
dadurch erklären, dass in diesen Studien idiopathische Fälle ausgewertet wurden. In
unserem Gesamtkollektiv sind aber noch relativ viele nicht-IIP-Fälle mit einem Anteil
von insgesamt 31,5% enthalten.
5.2 ATS/ERS Kollektiv
5.2.1 Geschlechts- und Altersverteilung
Anhand in der Literatur vorhandener, in den Tabellen aufgelisteter konkreter Daten,
aber auch anhand zahlreicher anderer Artikel lässt sich feststellen, dass unsere
Ergebnisse bei der Anamnese gut mit den zuvor beschriebenen übereinstimmen.
Es sind mehr Männer als Frauen von einer IPF betroffen. In unserem IPF-Kollektiv
waren mehr als viermal mehr Männer als Frauen zu finden. In der Literatur finden sich
je nach Studie variierende Angaben bezüglich der Geschlechtsverteilung. Bei der IPF

107
wurde aber bisher in keiner Studie ein Frauenüberhang gefunden [34, 35, 36, 37, 38].
Ebenso verhält es sich mit der RBILD: Wir fanden einen fünfmal höheren Anteil an
Männern als an Frauen, wobei in der Literatur teilweise auch ausgeglichene Kollektive
beschrieben werden. Aber auch hier wird von keinem Frauenüberhang berichtet [15, 27,
39, 40, 63], was nicht verwunderlich ist, da es sich bei der RBILD um eine
raucherassoziierte Erkrankung handelt. Die einzige Gruppe mit einem dominierenden
Frauenanteil in unserem Kollektiv war die NSIP (♂:♀=3:4), was ebenfalls in der
Literatur berichtet wird [26, 42]. Allerdings schwanken hier die Angaben und teilweise
wird auch von einem Männerüberschuss in den NSIP-Gruppen berichtet [34, 37, 41], so
dass letztendlich kein Geschlecht eindeutig überwiegt. Bei der BOOP tritt wieder eine
klare Androtropie auf, sowohl bei uns als auch in allen Literaturstellen [45, 46, 47, 48].
Das Durchschnittsalter war in unserem Kollektiv am höchsten in der IPF mit 59,4
Jahren und am niedrigsten in der RBILD-Gruppe mit 44,6 Jahren. Dies wird nicht nur
durch die in den Tabellen aufgeführten Artikel, sondern auch in anderen
Übersichtsartikeln ebenso beschrieben. KATZENSTEIN führt beispielsweise für die
IPF ein Durchschnittsalter von 57 Jahren, für die DIP 42 Jahre, für die RBILD 36 Jahre
und für die NSIP 49 Jahre auf [22].
5.2.2 Klinik
Die ersten klinischen Symptome der IPF sind ein schleichend zunehmender Husten und
Dyspnoe, die schließlich in einer Ruhedyspnoe mündet. Übereinstimmend mit unseren
Ergebnissen werden in der Literatur noch weitere Symptome wie Müdigkeit,
Leistungsverlust, grippale Infekte zu Beginn der Erkrankung, Thoraxschmerzen,
Gelenkbeschwerden, Gewichtsverlust und ein so genanntes door-stop-Phänomen
beschrieben. Je nach Kollektiv sind dabei die jeweils beobachteten Häufigkeiten stark
unterschiedlich. Diese Unterschiede können einerseits zufällig sein und vom jeweiligen
Kollektiv abhängen, andererseits können retrospektiv natürlich nur Daten ausgewertet
werden, die auch erfasst wurden. Die Ausführlichkeit und Dokumentation war in den
Krankenakten stark verschieden; das gilt sowohl für die Anamnesen als auch die
körperlichen Untersuchungen. Als Beispiel hierfür sind Nagelveränderungen wie
Trommelschlegelfinger und Uhrglasnägel in der Tabelle mit aufgeführt. Wir fanden bei
33,3% der IPF-Patienten Uhrglasnägel und bei 26% Trommelschlegelfinger. DANIIL et

108
al. konnten diese Veränderungen bei fast allen Patienten feststellen, BJORAKER et al.
hingegen nur bei 19%.
Tab. 13: Übersicht der anamnestischen und klinischen Angaben bei der IPF
Der RBILD wurde zunächst als Zufallsbefund keine besondere klinische Bedeutung
zugeschrieben [39]. Über die Inzidenz gibt es keine Angaben [15, 40].
Man sollte bei jüngeren Rauchern mit milder Symptomatik, auffälligem Röntgenbild
und restriktiver Lungenfunktionsuntersuchung an eine RBILD denken. Die Symptome
sind aber so unspezifisch, dass für die Diagnose eine OLB notwendig ist [39]. Wie die
Tabelle zeigt, ist die RBILD quasi ausschließlich bei Rauchern zu finden. In der Studie
von MOON et al. war jedoch ein Nichtraucher von einer RBILD betroffen. Dieser
Patient war beruflich Lötdämpfen ausgesetzt, die vermutlich ursächlich für die RBILD
waren [63]. Dieser Fall kann daher nicht als idiopathisch angesehen werden. Für die
RBILD sind die anamnestischen und klinischen Daten unserer Studie den
Literaturangaben in der Tabelle 14 gegenübergestellt. Unsere Ergebnisse, auch die nicht
tabellarisch aufgeführten, weichen nicht von den Literaturangaben ab.
unse
r K
olle
ktiv
2003
D
CA
RR
ING
TON
1978
USA
[35]
MA
TUSO
19
96
Japa
n [3
6]
BJO
RA
KER
1998
USA
[34]
DA
NII
L 19
99
UK
[37]
NIC
HO
LSO
N
2000
UK
[38]
n 27 53 30 64 15 37
♂:♀ 4,4:1 1,7:1 2,5:1 1:1 4:1 8,3:1
Alter (Jahre) 59,4 51 60 65 56 57,2
Dyspnoe (%) 96,3 o. A. 88,6 89 100 o. A.
Husten (%) 77,7 o. A. 100 71 60 o. A.
Symptomdauer (J) 1,7 2,5 1,1 o. A. 1,5 1,3
Raucher (%) 66,7 71 60 54 80 78
Nagelveränd. (%) 33,3 o. A. o. A. 19 93,3 o. A.
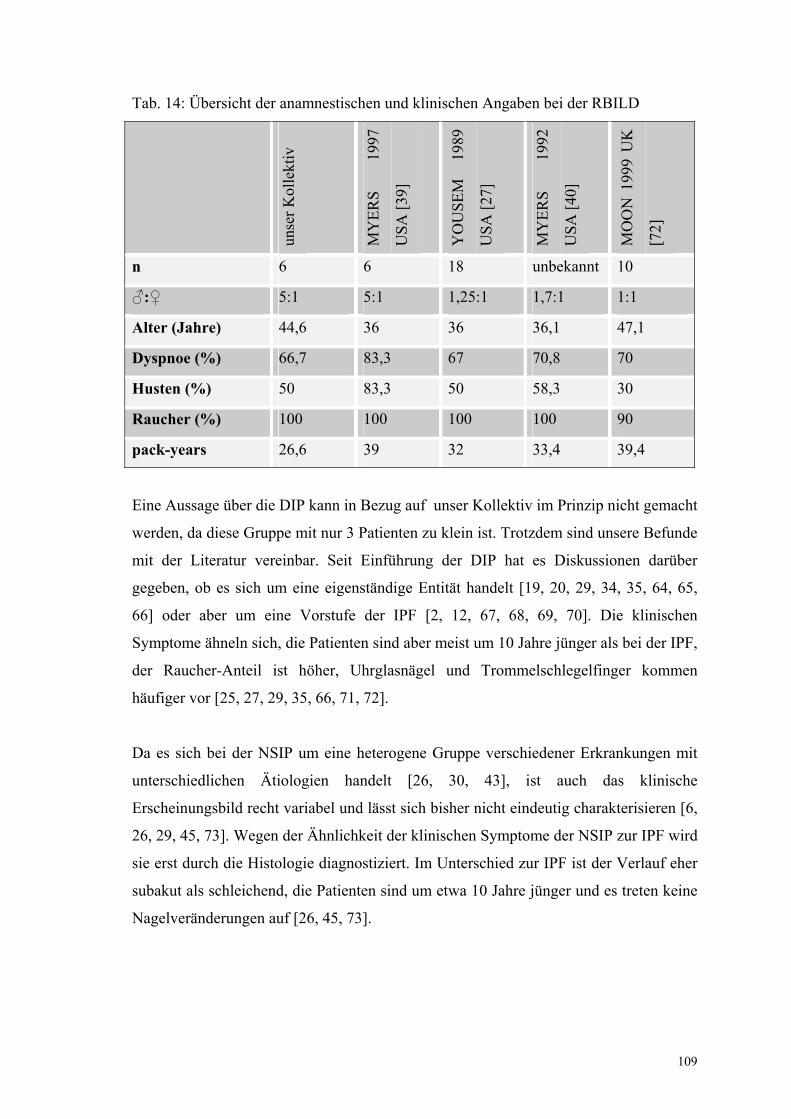
109
Tab. 14: Übersicht der anamnestischen und klinischen Angaben bei der RBILD
unse
r Kol
lekt
iv
MY
ERS
1997
USA
[39]
YO
USE
M
1989
USA
[27]
MY
ERS
1992
USA
[40]
MO
ON
199
9 U
K
[72]
n 6 6 18 unbekannt 10
♂:♀ 5:1 5:1 1,25:1 1,7:1 1:1
Alter (Jahre) 44,6 36 36 36,1 47,1
Dyspnoe (%) 66,7 83,3 67 70,8 70
Husten (%) 50 83,3 50 58,3 30
Raucher (%) 100 100 100 100 90
pack-years 26,6 39 32 33,4 39,4
Eine Aussage über die DIP kann in Bezug auf unser Kollektiv im Prinzip nicht gemacht
werden, da diese Gruppe mit nur 3 Patienten zu klein ist. Trotzdem sind unsere Befunde
mit der Literatur vereinbar. Seit Einführung der DIP hat es Diskussionen darüber
gegeben, ob es sich um eine eigenständige Entität handelt [19, 20, 29, 34, 35, 64, 65,
66] oder aber um eine Vorstufe der IPF [2, 12, 67, 68, 69, 70]. Die klinischen
Symptome ähneln sich, die Patienten sind aber meist um 10 Jahre jünger als bei der IPF,
der Raucher-Anteil ist höher, Uhrglasnägel und Trommelschlegelfinger kommen
häufiger vor [25, 27, 29, 35, 66, 71, 72].
Da es sich bei der NSIP um eine heterogene Gruppe verschiedener Erkrankungen mit
unterschiedlichen Ätiologien handelt [26, 30, 43], ist auch das klinische
Erscheinungsbild recht variabel und lässt sich bisher nicht eindeutig charakterisieren [6,
26, 29, 45, 73]. Wegen der Ähnlichkeit der klinischen Symptome der NSIP zur IPF wird
sie erst durch die Histologie diagnostiziert. Im Unterschied zur IPF ist der Verlauf eher
subakut als schleichend, die Patienten sind um etwa 10 Jahre jünger und es treten keine
Nagelveränderungen auf [26, 45, 73].
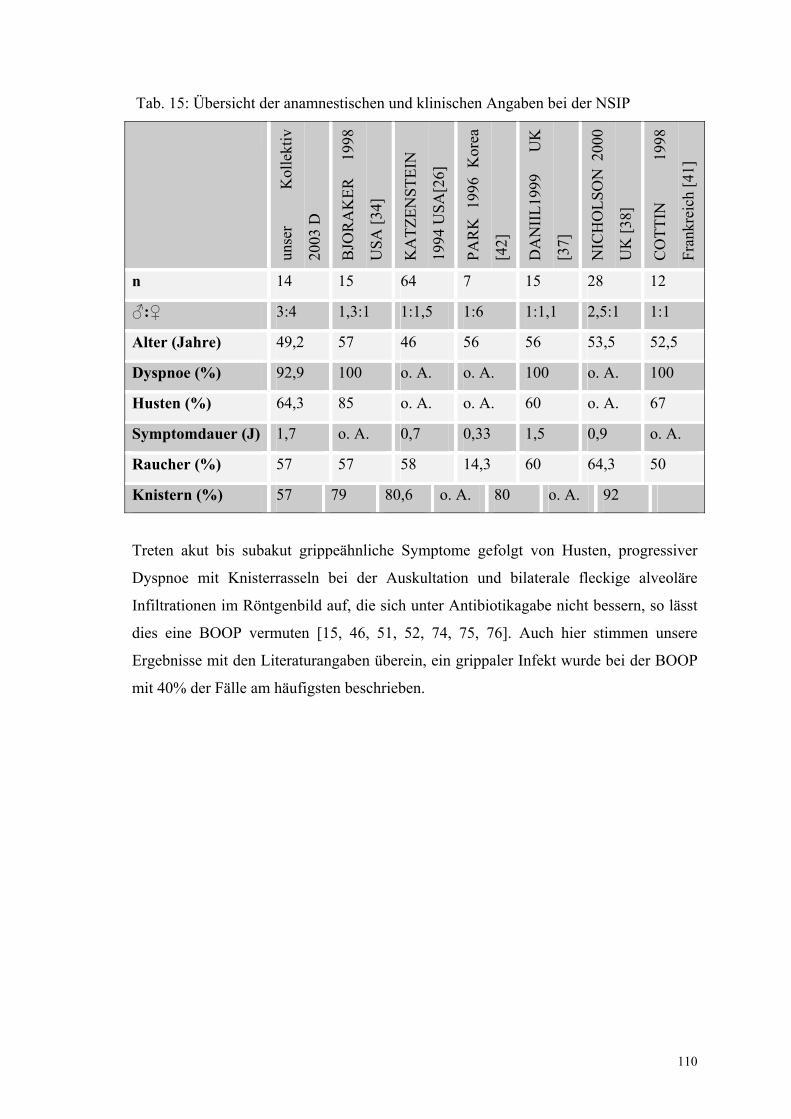
110
Tab. 15: Übersicht der anamnestischen und klinischen Angaben bei der NSIP
unse
r K
olle
ktiv
2003
D
BJO
RA
KER
19
98
USA
[34]
KA
TZEN
STEI
N
1994
USA
[26]
PAR
K 1
996
Kor
ea
[42]
DA
NII
L199
9 U
K
[37]
NIC
HO
LSO
N 2
000
UK
[38]
CO
TTIN
19
98
Fran
krei
ch [4
1]
n 14 15 64 7 15 28 12
♂:♀ 3:4 1,3:1 1:1,5 1:6 1:1,1 2,5:1 1:1
Alter (Jahre) 49,2 57 46 56 56 53,5 52,5
Dyspnoe (%) 92,9 100 o. A. o. A. 100 o. A. 100
Husten (%) 64,3 85 o. A. o. A. 60 o. A. 67
Symptomdauer (J) 1,7 o. A. 0,7 0,33 1,5 0,9 o. A.
Raucher (%) 57 57 58 14,3 60 64,3 50
Knistern (%) 57 79 80,6 o. A. 80 o. A. 92
Treten akut bis subakut grippeähnliche Symptome gefolgt von Husten, progressiver
Dyspnoe mit Knisterrasseln bei der Auskultation und bilaterale fleckige alveoläre
Infiltrationen im Röntgenbild auf, die sich unter Antibiotikagabe nicht bessern, so lässt
dies eine BOOP vermuten [15, 46, 51, 52, 74, 75, 76]. Auch hier stimmen unsere
Ergebnisse mit den Literaturangaben überein, ein grippaler Infekt wurde bei der BOOP
mit 40% der Fälle am häufigsten beschrieben.
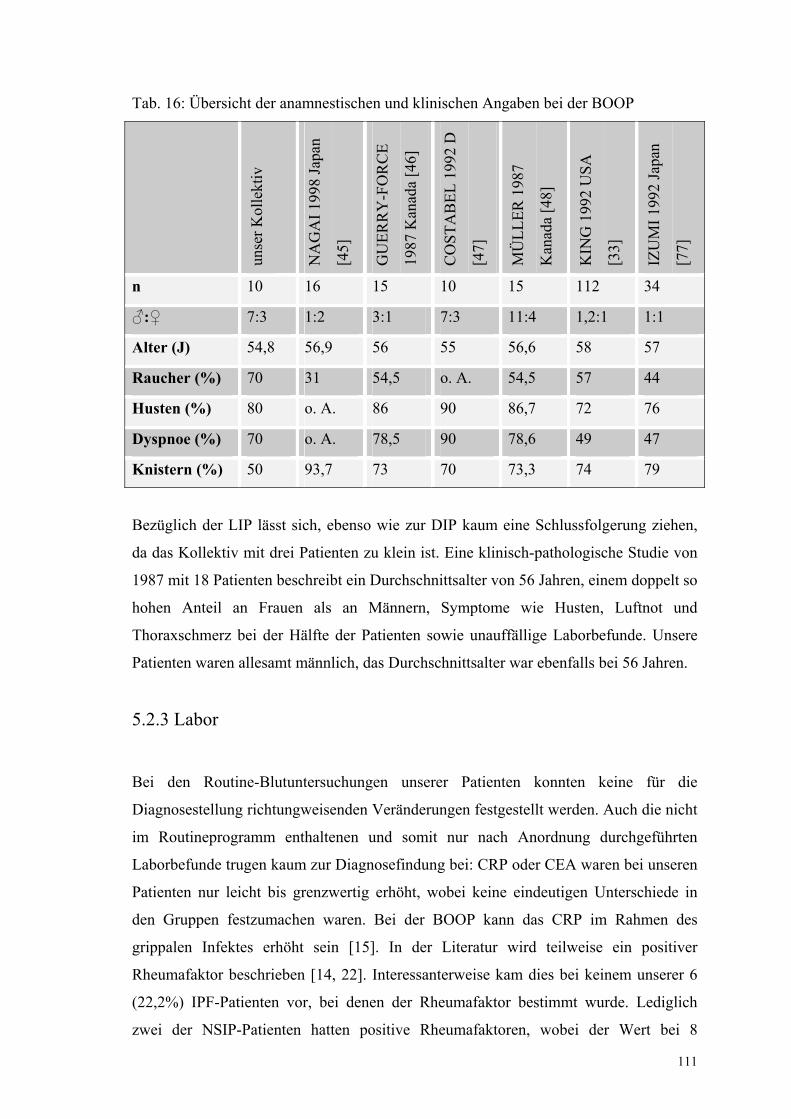
111
Tab. 16: Übersicht der anamnestischen und klinischen Angaben bei der BOOP
unse
r Kol
lekt
iv
NA
GA
I 199
8 Ja
pan
[45]
GU
ERR
Y-F
OR
CE
1987
Kan
ada
[46]
CO
STA
BEL
199
2 D
[47]
MÜ
LLER
198
7
Kan
ada
[48]
KIN
G 1
992
USA
[33]
IZU
MI 1
992
Japa
n
[77]
n 10 16 15 10 15 112 34
♂:♀ 7:3 1:2 3:1 7:3 11:4 1,2:1 1:1
Alter (J) 54,8 56,9 56 55 56,6 58 57
Raucher (%) 70 31 54,5 o. A. 54,5 57 44
Husten (%) 80 o. A. 86 90 86,7 72 76
Dyspnoe (%) 70 o. A. 78,5 90 78,6 49 47
Knistern (%) 50 93,7 73 70 73,3 74 79
Bezüglich der LIP lässt sich, ebenso wie zur DIP kaum eine Schlussfolgerung ziehen,
da das Kollektiv mit drei Patienten zu klein ist. Eine klinisch-pathologische Studie von
1987 mit 18 Patienten beschreibt ein Durchschnittsalter von 56 Jahren, einem doppelt so
hohen Anteil an Frauen als an Männern, Symptome wie Husten, Luftnot und
Thoraxschmerz bei der Hälfte der Patienten sowie unauffällige Laborbefunde. Unsere
Patienten waren allesamt männlich, das Durchschnittsalter war ebenfalls bei 56 Jahren.
5.2.3 Labor
Bei den Routine-Blutuntersuchungen unserer Patienten konnten keine für die
Diagnosestellung richtungweisenden Veränderungen festgestellt werden. Auch die nicht
im Routineprogramm enthaltenen und somit nur nach Anordnung durchgeführten
Laborbefunde trugen kaum zur Diagnosefindung bei: CRP oder CEA waren bei unseren
Patienten nur leicht bis grenzwertig erhöht, wobei keine eindeutigen Unterschiede in
den Gruppen festzumachen waren. Bei der BOOP kann das CRP im Rahmen des
grippalen Infektes erhöht sein [15]. In der Literatur wird teilweise ein positiver
Rheumafaktor beschrieben [14, 22]. Interessanterweise kam dies bei keinem unserer 6
(22,2%) IPF-Patienten vor, bei denen der Rheumafaktor bestimmt wurde. Lediglich
zwei der NSIP-Patienten hatten positive Rheumafaktoren, wobei der Wert bei 8

112
Patienten (57,1%) bestimmt wurde. Das könnte auf die strengen Ausschlusskriterien
zurückzuführen sein, die wir angelegt hatten. Patienten mit Systemerkrankungen
schieden als nicht idiopathisch aus unserem Kollektiv aus, in anderen Studien wurden
sie jedoch noch eingeschlossen [14].
5.2.4 BAL Da die Ergebnisse der BAL nicht pathognomonisch sind und eine normale BAL eine IIP
bzw. IPF nicht ausschließt, wird ihr Stellenwert teilweise kontrovers diskutiert [78]. Bei
der Auswertung der Daten der BAL muss man sich vor Augen halten, dass dieses
Diagnose-Verfahren zwar wegweisende Befunde liefern, jedoch keinesfalls eine
Lungenbiopsie ersetzen kann. Dennoch kann die BAL bei der Indikationsstellung zur
Lungenbiopsie helfen. Wichtig ist es, mit der BAL andere Erkrankungen wie eine
Sarkoidose oder exogen-allergische Alveolitis auszuschließen. Eine sichere Korrelation
zur jeweiligen Untergruppe bzw. zum Krankheitsverlauf konnte bisher nicht
nachgewiesen werden [6, 45, 79, 80]. Vorteilhaft an der BAL ist, dass mit ihr ein
größeres Lungenareal untersucht wird als bei einer offenen Lungenbiopsie, sie
wiederholt durchführbar ist und kaum Risiken birgt. Sie liefert repräsentatives
Zellmaterial und Flüssigkeit [5, 81, 82, 83, 84]. Anhand des Zellmaterials kann in der
BAL die entzündliche Komponente der Erkrankung beurteilt werden, um Rückschlüsse
auf das Stadium, das Ansprechen auf eine Therapie und die Prognose abzuschätzen [3,
15, 81]. Wiederholte Lavagen können bei der Bewertung des Therapieansprechens bzw.
einer Dosisanpassung hilfreich sein [57]. WATTERS et al.[82] stellen in einer
Untersuchung über die IPF eine positive Korrelation zwischen der Histologie und BAL
fest: erhöhte Lymphozyten korrelieren positiv mit interstitieller Entzündung und negativ
mit honigwabigem Umbau, wohingegen erhöhte Makrophagen positiv mit
honigwabigem Umbau und Hypertrophie der glatten Muskelzellen korrelieren. Einer
Erhöhung der Eosinophilen könnte ein honigwabiger Umbau zugrunde liegen, eine
eindeutige Korrelation ergab sich hier nicht. In einem weiteren Schritt stellt er den
gefundenen Zellen die Klinik gegenüber. Bei Patienten mit Lymphozytose war die
Symptomdauer kürzer als bei anderen und nach 6 Monaten Therapie war bei allen eine
Besserung eingetreten. Bei Eosinophilie von über 5 % in der BAL war die Erkrankung
bereits in einem weiter fortgeschrittenen Stadium. Eine Neutrophilie ist keiner
histopathologischen Veränderung zugeordnet und auch nicht als nützlicher Marker für
die Prognose angesehen [82, 83]. Zahlreiche andere Autoren fanden im Unterschied

113
dazu eine Assoziation zwischen Neutrophilie und schlechterer Prognose. Ebenso sind
erhöhte Eosinophile ein Zeichen für ein schlechtes Therapieansprechen [2, 5, 36, 57, 83,
85, 86, 87]. Eine Lymphozytose ist hingegen ein Marker für ein gutes
Steroidansprechen und eine gute Prognose [36, 57, 83, 85]. SCHWARZ et al. sehen
jedoch keinen Zusammenhang zwischen der Zellverteilung in der BAL und dem
Überleben [58]. ROBINSON hat in der BAL-Flüssigkeit die von Typ II-Zellen
produzierten Phospholipide untersucht und festgestellt, dass eine Erhöhung der
Phospholipide sowohl histopathologisch mit einer fortgeschrittenen Fibrose als auch
klinisch mit einem schlechten Steroidansprechen einhergehen [88]. Allerdings ergibt
sich weder aus dieser Studie mit geringer Fallzahl noch aus anderen Untersuchungen
über die löslichen Substanzen der Spülflüssigkeit ein Verhaltensratschlag für das
klinische Vorgehen [15].
Unsere Ergebnisse bei der BAL waren für die IPF mit den Literaturangaben
vergleichbar. Bei der RBILD ist die BAL zumeist unauffällig, verglichen mit
„gesunden“ Rauchern findet man lediglich eine höhere Zellzahl [15]. Auch dies kann
mit unseren Ergebnissen bestätigt werden. Als Unterschied zwischen der NSIP im
Vergleich zur IPF wird allgemein ein erniedrigter CD4/CD8-Quotient bei bestehender
Lymphozytose in der BAL angesehen [26, 45, 73]. Wir konnten diese Aussage nicht
bestätigen. Der CD4/CD8-Quotient lag bei der IPF bei 1,3 und bei der NSIP bei 2,0. Bei
der BOOP hat die BAL eine besondere Wertigkeit, da sie bei typischen Befunden wie
einer Lymphozytose von über 20%, 2 bis 25% Eosinophile, ein CD4/CD8-Quotient von
unter 1, ein normaler CD 57-Wert und eine sterile Spülflüssigkeit in der BAL und
außerdem typischen radiologischen Veränderungen einen Therapiebeginn ohne
vorherige Lungenbiopsie rechtfertigen kann [15, 47, 89]. Die Erhöhung des
Lymphozytenanteils und der erniedrigte CD4/CD8-Quotient gilt als
Unterscheidungskriterium zwischen BOOP und UIP [15].
Ebenso wie bei der NSIP ist in unserem Kollektiv aber der CD4/CD8-Quotient bei der
BOOP wider Erwarten mit 2,0 deutlich erhöht. Die durchschnittlich erhöhte
Lymphozytenzahl stimmt mit den Literaturangaben überein. Die CD4/CD8-Quotienten
sind relativ gleichmäßig verteilt, es gibt weder bei der NSIP noch bei der BOOP
extreme Ausreißer, die die hohen Werte erklären würden. Um die Diskrepanz zur
Literatur mit sicherlich gegebenen unterschiedlichen Laborverfahren oder
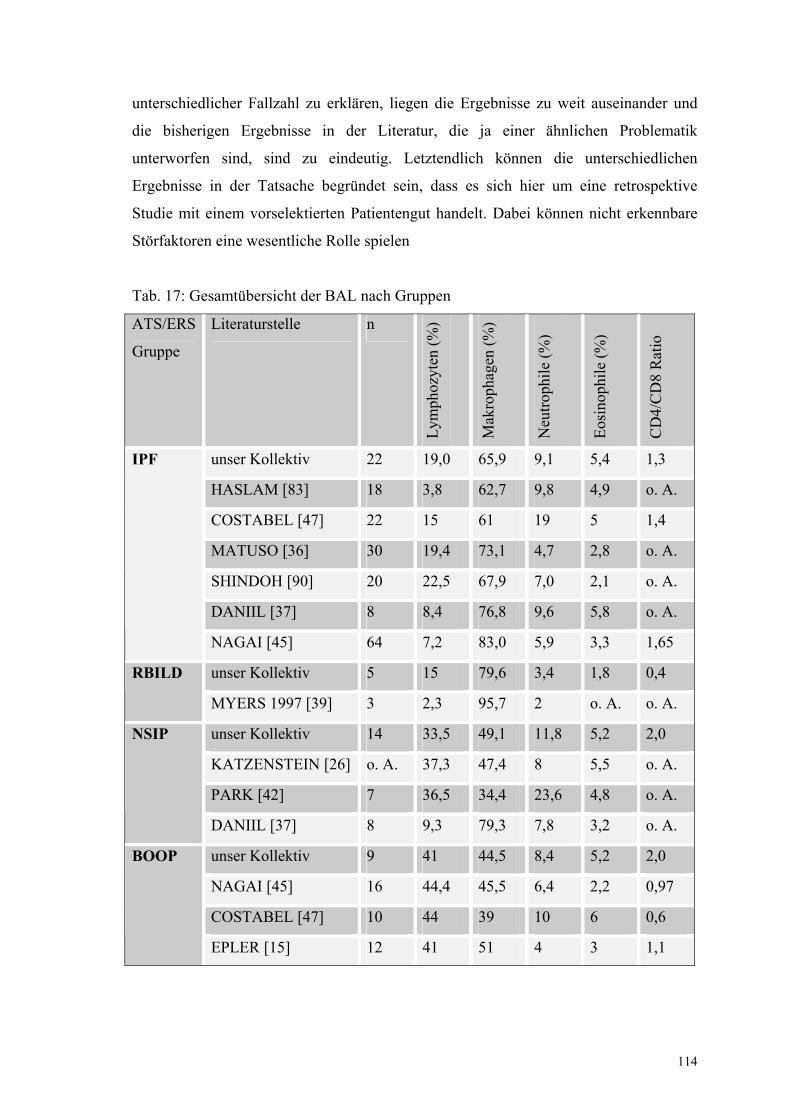
114
unterschiedlicher Fallzahl zu erklären, liegen die Ergebnisse zu weit auseinander und
die bisherigen Ergebnisse in der Literatur, die ja einer ähnlichen Problematik
unterworfen sind, sind zu eindeutig. Letztendlich können die unterschiedlichen
Ergebnisse in der Tatsache begründet sein, dass es sich hier um eine retrospektive
Studie mit einem vorselektierten Patientengut handelt. Dabei können nicht erkennbare
Störfaktoren eine wesentliche Rolle spielen
Tab. 17: Gesamtübersicht der BAL nach Gruppen
ATS/ERS
Gruppe
Literaturstelle n
Lym
phoz
yten
(%)
Mak
roph
agen
(%)
Neu
troph
ile (%
)
Eosi
noph
ile (%
)
CD
4/C
D8
Rat
io
unser Kollektiv 22 19,0 65,9 9,1 5,4 1,3
HASLAM [83] 18 3,8 62,7 9,8 4,9 o. A.
COSTABEL [47] 22 15 61 19 5 1,4
MATUSO [36] 30 19,4 73,1 4,7 2,8 o. A.
SHINDOH [90] 20 22,5 67,9 7,0 2,1 o. A.
DANIIL [37] 8 8,4 76,8 9,6 5,8 o. A.
IPF
NAGAI [45] 64 7,2 83,0 5,9 3,3 1,65
unser Kollektiv 5 15 79,6 3,4 1,8 0,4 RBILD
MYERS 1997 [39] 3 2,3 95,7 2 o. A. o. A.
unser Kollektiv 14 33,5 49,1 11,8 5,2 2,0
KATZENSTEIN [26] o. A. 37,3 47,4 8 5,5 o. A.
PARK [42] 7 36,5 34,4 23,6 4,8 o. A.
NSIP
DANIIL [37] 8 9,3 79,3 7,8 3,2 o. A.
unser Kollektiv 9 41 44,5 8,4 5,2 2,0
NAGAI [45] 16 44,4 45,5 6,4 2,2 0,97
COSTABEL [47] 10 44 39 10 6 0,6
BOOP
EPLER [15] 12 41 51 4 3 1,1

115
5.2.5 Lungenfunktionsanalyse
Eine Lungenfunktionsuntersuchung erfasst den Funktionszustand der Lunge. Sie kann
zur Verlaufskontrolle und zur Beurteilung des Therapieeffektes wiederholt werden [80].
Pathologische Lungenfunktionsuntersuchungen sind oft ein frühes Zeichen für das
Vorhandensein einer interstitiellen Lungenerkrankung [80, 90, 91, 92, 93]. Auch für die
Lungenfunktionsuntersuchung zeigt sich, dass unsere Ergebnisse im Bereich dessen
liegen, was zuvor in der Literatur angegeben wurde.
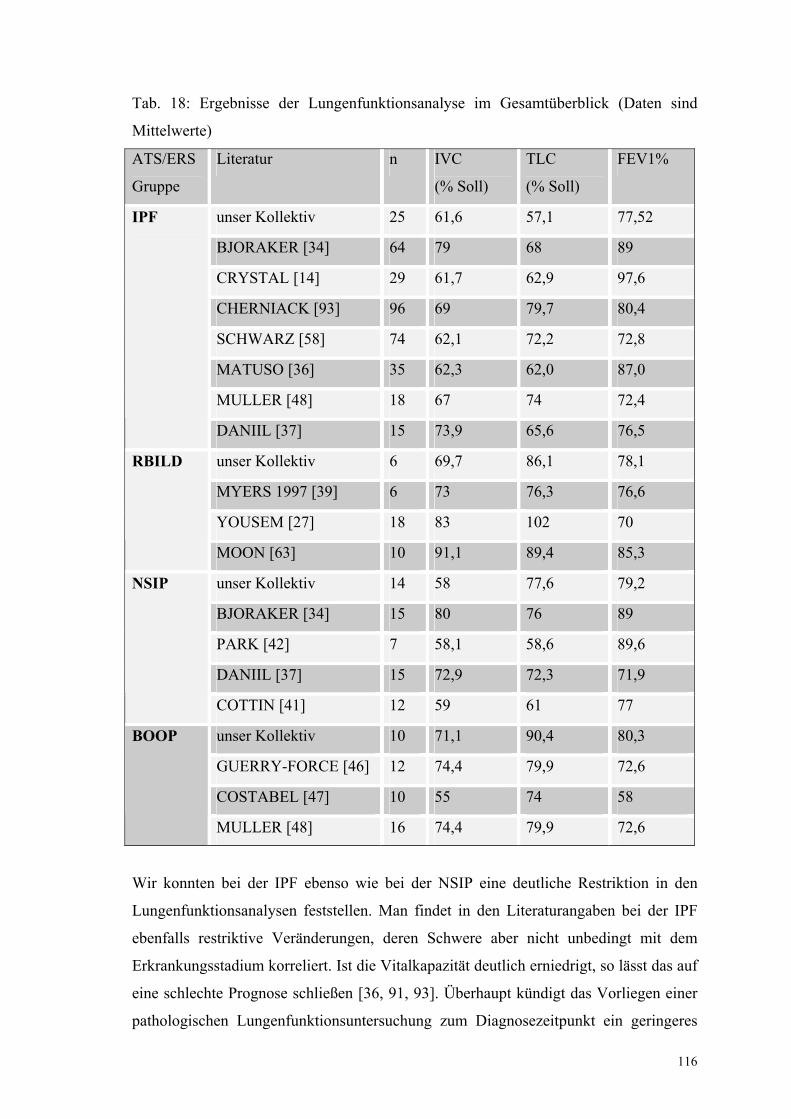
116
Tab. 18: Ergebnisse der Lungenfunktionsanalyse im Gesamtüberblick (Daten sind
Mittelwerte)
ATS/ERS
Gruppe
Literatur n IVC
(% Soll)
TLC
(% Soll)
FEV1%
unser Kollektiv 25 61,6 57,1 77,52
BJORAKER [34] 64 79 68 89
CRYSTAL [14] 29 61,7 62,9 97,6
CHERNIACK [93] 96 69 79,7 80,4
SCHWARZ [58] 74 62,1 72,2 72,8
MATUSO [36] 35 62,3 62,0 87,0
MULLER [48] 18 67 74 72,4
IPF
DANIIL [37] 15 73,9 65,6 76,5
unser Kollektiv 6 69,7 86,1 78,1
MYERS 1997 [39] 6 73 76,3 76,6
YOUSEM [27] 18 83 102 70
RBILD
MOON [63] 10 91,1 89,4 85,3
unser Kollektiv 14 58 77,6 79,2
BJORAKER [34] 15 80 76 89
PARK [42] 7 58,1 58,6 89,6
DANIIL [37] 15 72,9 72,3 71,9
NSIP
COTTIN [41] 12 59 61 77
unser Kollektiv 10 71,1 90,4 80,3
GUERRY-FORCE [46] 12 74,4 79,9 72,6
COSTABEL [47] 10 55 74 58
BOOP
MULLER [48] 16 74,4 79,9 72,6
Wir konnten bei der IPF ebenso wie bei der NSIP eine deutliche Restriktion in den
Lungenfunktionsanalysen feststellen. Man findet in den Literaturangaben bei der IPF
ebenfalls restriktive Veränderungen, deren Schwere aber nicht unbedingt mit dem
Erkrankungsstadium korreliert. Ist die Vitalkapazität deutlich erniedrigt, so lässt das auf
eine schlechte Prognose schließen [36, 91, 93]. Überhaupt kündigt das Vorliegen einer
pathologischen Lungenfunktionsuntersuchung zum Diagnosezeitpunkt ein geringeres

117
Überleben an [58], eine Reduktion der Lungenvolumina ist bereits frühzeitig bemerkbar
[80]. Bleiben die Untersuchungen über 1 Jahr stabil oder verbessern sich sogar, so ist
das ein Zeichen für therapeutisches Ansprechen und längeres Überleben [94]. Dennoch
ist eine normale Lungenfunktionsuntersuchung bei einem Patienten mit IPF nicht
ungewöhnlich [95]. Eine Abgrenzung der IPF von der NSIP ist allein anhand der
Lungenfunktionsuntersuchungen nicht möglich.
In unserem Kollektiv wurde eine leichte Verminderung der Vitalkapazität bei der
RBILD gesehen. In der Literatur werden allenfalls milde restriktive Veränderungen in
den Lungenfunktionsuntersuchungen angegeben [39].
Bei der BOOP fanden wir ebenfalls leichte restriktive Veränderungen, was den
Literaturangaben entspricht [46, 48, 76]. Man könnte auch eine Obstruktion bei der
BOOP vermuten, da die betroffenen Bronchiolen durch das Granulationsgewebe aber
eher ausgefüllt statt nur verengt werden, fehlt normalerweise eine Obstruktion [46, 48].
Bei Rauchern können aber gelegentlich obstruktive Komponenten hinzukommen [74,
89].
5.2.6 Prognose
Die Prognose war in unserem Kollektiv bei der IPF am schlechtesten. Keiner erreichte
eine Besserung der Symptomatik und über die Hälfte der Patienten verstarben. Die
Überlebenswahrscheinlichkeit in der Kaplan-Meier-Analyse lag nach 2 Jahren bei 54%
und nach 6 Jahren bei nur 31,1%. Zahlreiche Studien bestätigen die schlechte Prognose
der IPF; die Mortalitätsrate wird zwischen 59 bis 70% angegeben. Die mittlere
Überlebensdauer liegt zwischen 3 bis 6 Jahren [2, 14, 22, 34, 35, 70, 85, 96]. Wird eine
niedrigere Mortalitätsrate gefunden, so sollte man die korrekte Diagnosestellung der IPF
anzweifeln. Gerade in älteren Studien mit gemischten Kollektiven findet man längere
mediane Überlebenszeiten [22, 97].
In unserem Kollektiv konnte beobachtet werden, dass die Patienten, die verstarben,
einen signifikant niedrigeren Lymphozytenanteil in der BAL hatten als die
Überlebenden. Außerdem waren die Neutrophilen in der Gruppe der Verstorbenen
höher als in der Gruppe der Überlebenden.

118
Als prognostisch günstige Kriterien werden in der Literatur umgekehrt niedrige
Neutrophile und hohe Lymphozyten in der BAL, eine Vitalkapazität über 60%,
schnelles Therapieansprechen und kurze Anamnesedauer angesehen [13, 36, 70, 72, 95,
98]. Jüngere Patienten mit kurzem Krankheitsverlauf und wenig radiologischen
Veränderungen hatten durchschnittlich ein längeres Überleben [72]. Ob ein Patient auf
die Therapie ansprechen wird oder nicht, ist im Einzelfall nicht vorherzusagen. Günstig
scheint aber eine kurze Erkrankungsdauer von unter 1 Jahr, erhöhte Lymphozyten in der
BAL bei niedrigen Neutrophilen und Eosinophilen sowie ein histologisch eher
entzündliches Bild zu sein [13, 35, 52, 70, 99, 100]. Prognostisch ungünstige Faktoren
sind höheres Alter, männliches Geschlecht, reduzierte Lungenvolumina und
Verschlechterung der Lungenfunktionswerte [59]. Mit unserem Kollektiv können wir
als eindeutig ungünstige Faktoren nur ein männliches Geschlecht bestätigen. Von den
20 Patienten, die verstarben, waren 18 männlich. Tendenziell hatten auch Raucher und
Patienten mit höherem Lebensalter eine schlechtere Prognose; diese Unterschiede waren
aber nicht signifikant. Bezüglich der Dyspnoe lässt sich nicht die Aussage machen, dass
Patienten mit höhergradiger Dyspnoe eine höhere Mortalität hatten, aber alle Patienten,
die bei Aufnahme keine Dyspnoe hatten, leben. Auch erniedrigte Lungenvolumina
erwiesen sich in unserem Kollektiv nicht als Prognosemarker.
In einigen Studien über prognostische Faktoren wurde hingegen einer erniedrigten
Vitalkapazität, einem erniedrigtem totalen Lungenvolumen und erhöhtem FEV1% ein
niedrigeres Überleben zugeschrieben [13, 58, 70, 72]. Wird nach einem Jahr ein
unveränderter oder sogar verbesserter Befund bei der Lungenfunktion erhoben, so
bedeutet dies in der Regel einen therapeutischen Erfolg und ein längeres Überleben
[94]. In den Verlaufskurven der Lungenfunktionsdaten bei unseren Patienten kann man
einen Abfall der Vitalkapazität und totalen Lungenkapazität bis zum Zeitraum von 1,5
Jahren beobachten, danach werden fast normale Werte erreicht. Allerdings wurden dann
nur noch einzelne Patienten beobachtet, da die Patienten mit den schlechteren Befunden
zwischenzeitlich verstorben sind oder für eine Kontroll-Lungenfunktionsuntersuchung
zu schwer krank waren, so dass diese Zahlen nur grob orientierend betrachtet werden
dürfen.

119
Die Prognose in den anderen Gruppen war durchweg deutlich besser als bei der IPF. Es
kam bei allen Gruppen meistens zu Verläufen mit klinischer Besserung oder
Vollremission oder zumindest einem stabilen Krankheitsstadium. Die Mortalitätsrate
war bei der RBILD am geringsten, hier trat kein Todesfall auf, gefolgt von der NSIP,
BOOP, DIP und LIP in steigender Reihenfolge. Die scheinbar hohe Mortalität bei der
LIP mit 33% ist in der niedrigen Fallzahl (n=3) begründet, gleiches gilt für die DIP, wo
ein Patient 4,5 Jahre, nachdem die Diagnose DIP gestellt wurde, an einem Bronchial-
Karzinom verstarb, welches erst einige Monate zuvor entdeckt wurde. Hier sind die DIP
und das Bronchialkarzinom am ehesten als unabhängige Erkrankungen anzusehen. Die
eigentliche Todesursache war nicht bekannt. In der Literatur wurden bisher keine
Todesfälle bei der RBILD beschrieben, die Prognose ist durchweg gut und
normalerweise reicht als alleinige Therapie ein Rauchverzicht aus. Dies unterstreicht die
Notwendigkeit einer korrekten Diagnose mit Abgrenzung zu anderen IIP, auch von der
DIP, um den RBILD-Patienten eine unnötige, mit Nebenwirkungen behaftete Therapie
zu ersparen [27, 39, 40, 101, 102], obwohl häufig davon ausgegangen wird, dass es sich
bei der DIP möglicherweise um das Spätstadium einer RBILD handeln könnte [22, 27,
39, 40, 43].
Von den Patienten aus der NSIP-Gruppe verstarb nur einer nach 44 Monaten, die
meisten Patienten erreichten eine Besserung der Symptome. Dies findet in der Literatur
Bestätigung [34, 41, 42, 45, 73]. Wird die NSIP weiter histologisch nach zellulärem
oder fibrotischem Typ aufgeteilt, so findet man eine deutlich schlechtere Prognose bei
der fibrotischen Variante [26, 37, 41].
Die BOOP hat in der Literatur ebenso wie in unserem Kollektiv eine gute Prognose [15,
52, 74], nur 20% unserer Patienten verstarben. Nicht alle Patienten brauchen zur
Therapie Steroide, da auch Spontanremissionen auftreten können. Wird aber eine
Therapie notwendig, so ist die Gefahr eines Rückfalls bei zu kurzer Therapiedauer
(unter 3 Monate) bei einem Drittel der Patienten gegeben [15, 52].

120
5.2.7 Histologie
Im Gegensatz zu anderen Studien [13, 59, 70, 72, 96, 103, 104, 105] wurden hier
ausschließlich Fälle mit offenen Lungenbiopsien eingeschlossen, da nur an einem
ausreichend großen Stück Lungengewebe eine histologische Befundung mit maximaler
Aussagekraft möglich ist. Die OLB ist die optimale Methode, um Lungengewebe zu
gewinnen [106], sie hat überschaubare Risiken [107, 108]. Im Gegensatz zu
Erkrankungen mit spezifischen histologischen Veränderungen ist der Probenfehler
wegen der geringen Probengröße bei der transbronchialen Biopsie bei den IIP zu groß
[10, 91, 99]. Die OLB gilt als das probate Diagnoseinstrument [99, 109, 110].
Mittlerweile liegen einige gut histologisch abgesicherte Studien vor [14, 19, 26, 34, 35,
37, 38, 43, 46, 48, 51, 93].
Die Einstufung in die Sicherheitskriterien „sicher“, „am ehesten“ und „möglich“
veranschaulicht die Schärfe, mit der die einzelnen Gruppen histologisch getrennt
werden können. Bei der NSIP handelt es sich um ein breiter gefächertes histologisches
Spektrum, bei dem einerseits so genannte zelluläre bzw. fibrotische Varianten mit sehr
unterschiedlich histologischen Bildern auftreten können. Hier wurden in 43% der
zelluläre, in 28,5% der gemischte und in 28,5% der fibrotische Subtyp gefunden.
Keine der Entitäten ist per se idiopathisch, allen können bestimmte Umstände oder
andere Krankheiten zugrunde liegen, die die histologischen Veränderungen
verursachen. Wir haben gesehen, dass noch bei rund einem Drittel der Patienten im
Gesamtkollektiv eine Ätiologie gefunden werden konnte, zu einem Großteil bei
Histologien, die mit „sicher“ bewertet wurden. Deswegen ist es so wichtig, bei der
Anamnese penibel nach ätiologischen Faktoren zu fragen. Insbesondere kommen DIP-
ähnliche oder BOOP-ähnliche Veränderungen häufig vor. Sie können in der Nähe
anderer Läsionen auftreten [22, 111] oder sich als Reaktion auf bestimmte Einflüsse
ausbilden. Eine so genannte DIP-like Reaktion tritt beispielsweise bei Infektionen,
Kontakt mit Stäuben, Medikamenten und sekundär bei rheumatischen oder
neoplastischen Veränderungen der Lunge auf [19, 62, 67, 111].

121
Bei der BOOP können Ursachen ebenfalls in Infektionen, Medikamentenreaktionen,
rheumatischen Veränderungen oder Nierenversagen, Wegener’schen Granulomatose,
Kollagenosen, HX, Sarkoidose und Zustand nach Bestrahlung liegen [15, 76, 112].
Zunehmend werden in der Literatur histologische Problemfälle behandelt:
beispielsweise unterstreichen NICHOLSON et al. , dass nicht so sehr auf eine exakte
Unterscheidung zwischen der fibrosierenden NSIP und UIP wegen des ähnlichen
Verlaufes ankommt [38, 113]. FLAHERTY et al. beschreiben das Problem, dass nicht
selten innerhalb einer Lunge an unterschiedlichen Biopsieorten zwei verschiedene
histologische Diagnosen gestellt werden können [114].
Da mittlerweile die IIP auch klinisch gut voneinander unterschieden werden können,
wird in Zukunft nicht jede Diagnose histologisch abgesichert werden, beispielsweise
wenn klinisch (also auch im HRCT) keine Zweifel an der Diagnose einer IPF bestehen
und der Patient über 60 Jahre alt ist [6, 38, 57, 115]. Nur für die IPF gibt es bisher
ausreichend sichere klinische / computertomographische Kriterien.
5.3 Ausblick
Mit der vorgelegten Arbeit wurde durch die Anlage einer großen Datenbank der
Grundstein für weitere Untersuchungen gelegt. Es ist wünschenswert, die gesammelten
Fälle weiterhin zu beobachten, um so die Follow-Up Dauer von 3 Jahren zu verlängern.
Ebenso wäre es wünschenswert, die Datenbank weiter zu pflegen, um größere
Fallzahlen zu erreichen. Retrospektive Studien wie diese sind jedoch prinzipiell für eine
Vielzahl von Störfaktoren anfällig, daher wäre das Optimum eine prospektive Multi-
Centerstudie mit möglichst großer Fallzahl.
Wie bereits geschildert, ist eine Auswertung der radiologischen Befunde im Rahmen
dieser Arbeit nicht möglich. Deswegen sollte eine Revision und Auswertung aller
Röntgendokumente stattfinden, um das Gesamtbild abzurunden.
Das lange Zeit gültige Konzept zur Pathogenese der IIP ging von primär chronisch-
entzündlichen Prozessen in den Alveolen und im Interstitium aus [9, 14, 35, 85, 87, 116,
117, 118, 119, 120, 121]. Neueste Studien widerlegen dieses Konzept und schuldigen
eine gestörte Reparation an [122, 123]. Ein starkes Argument gegen die primäre

122
Entzündungstheorie ist die Tatsache, dass anti-inflammatorische Therapie mit Steroiden
nur selten zum erhofften Erfolg führt [59, 124, 125, 126, 127, 128, 129, 130].
Zytokine und Wachstumsfaktoren sind die Mediatoren der Wundheilung. Bei der IPF
gewinnen profibrotische Faktoren wie der Transforming Growth Factor ß die Oberhand.
Es wäre interessant, solche Zytokine und Wachstumsfaktoren in der BAL-
Spülflüssigkeit zu bestimmen. Sie könnten einerseits für Voraussagen über die Prognose
und andererseits als pharmakologischer Angriffspunkt für neue Therapien genutzt
werden.
Ebenso könnte vor dem Hintergrund des neuen Konzeptes zur Pathogenese versucht
werden, histologische Veränderungen zu finden, die als Prognosemarker dienen können,
insbesondere auch unter Hinzuziehung immunhistochemischer Verfahren.

123
6. Zusammenfassung
Im Jahr 2002 wurden die bisherigen Klassifikationen der idiopathischen interstitiellen
Pneumonien (IIP) von LIEBOW und KATZENSTEIN durch die ATS/ERS
Arbeitsgruppe als interdisziplinärer Konsensus revidiert. Die neue Klassifikation
umfasst die klinischen Diagnosen idiopathische pulmonale Fibrose (IPF),
respiratorische Bronchiolitis mit interstitieller Lungenerkrankung (RBILD),
desquamative interstitielle Pneumonie (DIP), nicht-spezifische interstitielle Pneumonie
(NSIP), cryptogene organisierende Pneumonie (COP), akute interstitielle Pneumonie
(AIP) und lymphozytäre interstitielle Pneumonie (LIP) und analog dazu die
histologischen Diagnosen gewöhnliche interstitielle Pneumonie (UIP), respiratorische
Bronchiolitis (RB), desquamative interstitielle Pneumonie (DIP), nicht-spezifische
interstitielle Pneumonie (NSIP), Bronchiolitis obliterans mit organisierender Pneumonie
(BOOP), diffuser Alveolarschaden (DAD) und die lymphozytäre interstitielle
Pneumonie (LIP).
Für diese Arbeit wurden aus dem Einsendegut der Ruhrlandklinik Essen-Heidhausen im
Archiv der Abteilung für Allgemeine und Spezielle Pathologie der Ruhr-Universität
Bochum insgesamt 103 Fälle von 1993 bis 2000 herausgesucht, bei denen eine
interstitielle Lungenerkrankung diagnostiziert worden war. Die Proben wurden nach der
ATS/ERS-Klassifikation neu eingeteilt und die klinischen Daten anhand der
Krankenakten und Nachfragen bei den behandelnden niedergelassenen Ärzten erhoben.
Wir fanden 63 Fälle mit idiopathischer interstitieller Pneumonie nach der ATS/ERS-
Klassifikation. Diese konnten klinisch und histologisch klar voneinander abgegrenzt
und wie folgt eingeteilt werden: 27 Fälle mit IPF (43%), 6 mit RBILD (10%), 3 mit DIP
(5%), 14 mit NSIP (22%), 10 mit BOOP (16%) und 3 mit LIP (5%). Als ungünstige
Prognosemarker fanden wir ein männliches Geschlecht (p=0,017), höheres Alter
(p=0,012) und niedrige Lymphozytenzahl in der BAL (p=0,022), wobei die IPF die
Entität mit der schlechtesten Prognose ist.
In einer umfassenden deutschen Studie konnten erstmals die jüngsten Ergebnisse der
internationalen Literatur bestätigt werden. Unter Berücksichtigung neuer Pathogenese-
Konzepte werden in Zukunft Medikamente verfügbar sein, die die bisher übliche, aber
nur gering wirksame anti-inflammatorische Therapie ersetzen werden. Hierfür ist eine
exakte Klassifikation der IIP von entscheidender Bedeutung.

124
7. Literaturverzeichnis
1) Coultas, D. B., Zumwalt, R. E., Black, W. C., Sobonya, R. E.: Epidemiology of
interstitial lung diseases. Am. J. Respir. Crit. Care Med. 150(4), 967-72 (1994)
2) Crystal, R. G., Gadek, J. E., Victor, J. F., Fulmer, J. D., Line, B. R., Hunninghake, G.
W.: Interstitial lung disease: Current concepts of pathogenesis, staging and therapy. Am.
J. Medicine 70, 542-568 (1981)
3) Crystal, R. G., Bitterman, P. B., Rennard, S. I., Hance, A. J., Keogh, B. A.:
Interstitial lung diseases of unknown cause. N. Engl. J. Med. 310(3), 154-66 (1984)
4) Weissler, J. C.: Idiopathic pulmonary fibrosis: cellular and molecular pathogenesis.
Am. J. Med. Sci. 297(2),91-104 (1989)
5) King, T. E. et al.: ATS/ERS Statement: Idiopathic pulmonary fibrosis: diagnosis and
treatment. Am. J. Respir. Crit. Care Med. 161, 646-664 (2000)
6) Travis, W. D. et al.: ATS/ERS international multidisciplinary consensus
classification of idiopathic intersitial pneumonias. Am. J. Respir. Crit. Care Med. 165,
277-304 (2002)
7) Homolka, J.: Idiopathic pulmonary fibrosis: a historical review. CMAJ 137(11),
1003-5 (1987)
8) Hamman, L., Rich, A. R.: Acute diffuse interstitial fibrosis of the lungs. Bull. John
Hopkins Hospital 74, 177-212 (1944)
9) Liebow, A. A.: Definition and classification of interstitial pneumonias in human
pathology. Prog. Resp. Res.8, 1-33 (1975)
10) Schlick, W.: Current issues in the assessment of interstitial lung disease. Monaldi.
Arch. Chest Dis. 48(3), 237-44 (1993)

125
11) Cegla, U. H.: Die idiopathisch fibrosierende Alveolitis. Thieme Verlag Stuttgart
(1977)
12) Scadding, J. G., Hinson, K. F. W.: Diffuse fibrosing alveolitis . Thorax 22, 291-304
(1967)
13) Stack, B. H. R., Choo-Kang, Y. F., Heard, B. E.: Prognosis of cryptogenic fibrosing
alveolitis. Thorax 27, 535-542 (1972)
14) Crystal, R. G., Fulmer, J. D., Roberts, W., Moss, M., Line, B. R., Reynolds, H. Y.:
Clinical, histological, radiographic, physiologic, scintigraphic, cytologic and
biochemical aspects of idiopathic pulmonary fibrosis. Ann. Intern. Med. 85, 769-788
(1976)
15) Epler, G. R.: Diseases of the bronchioles. Raven Press New York 1994 (1994)
16) Kitaichi, M.: Pathologic features and the classification of interstitial pneumonias of
unknown etology. Bull. Chest Dis. Res. Inst. Kyoto Univ. 23(1-2), 1-18 (1990)
17) Gabrielli, S., Stanflin, N., Melli, G.: Hypothesis of classification of pulmonary
fibrosis. Pathologica 82 (1078), 119-23 (1990)
18) Hogg, J. C.: Benjamin Felson lecture. Chronic interstitial lung disease of unknown
cause: a new classification based on pathogenesis. AJR 156(2), 225-33 (1991)
19) Katzenstein A. L.: Idiopathic interstitial pneumonia:
Classification and diagnosis. Churg, A.: The lung. Current concepts, monographs in
pathology 36 Williams and Wilkins, Baltimore (1993)
20) Liebow, A. A., Steer, A., Billingsley, J. G.: Desquamative interstitial pneumonia.
Am. J. Med. 39, 369-404 (1965)
21) Fromm, G. B., Dunn, L. J., Harris, J. C.: Desquamative interstitial pneumonitis.
Chest 77, 552-554 (1980)

126
22) Katzenstein, A. L., Myers, J. L.: IPF: clinical relevance. Am. J. Respir. Crit. Care
Med.. 157,1301-1315 (1998)
23) Katzenstein, A. L., Myers, J. L., Mazur, M. T.: Acute interstitial pneumonia. Am. J.
Surgical Pathology 10(4), 256-267 (1986)
24) Ohori, N. P., Sciurba, F. C., Owens, G. R., Hodgson, M. J., Yousem, S. A.: Giant-
cell interstitial pneumonia and hard-metal pneumoconiosis. Am. J. Surg. Pathol.
13(7), 581-587 (1989)
25) Katzenstein and Askin: Surgical pathology of non-neoplastic lung disease. ISBN
072165755-9
26) Katzenstein, A. L., Fiorelli, R. F.: Nonspecific interstitial pneumonia/fibrosis.
Histologic features and clinical significance. Am. J. Surg. Pathol. 18(2),136-147 (1994)
27) Yousem, S. A., Colby, T. V., Gaensler, E. A.: Respiratory bronchiolitis-associated
interstitial lung disease and its relationship to desquamative interstitial pneumonia.
Mayo. Clin. Proc. 64(11), 1373-80 (1989)
28) Primack, S. L., Hartmann, T. E., Ikezoe, J., Akira, M., Askatani, M., Müller, N. L.:
Acute interstitial pneumonia: radiographic and CT findings in nine patients. Radiology
188, 817-820 (1993)
29) Costabel, U.: Die neue Klassifikation der interstitiellen Pneumonien. Pneumologie
54, 447-453 (2000)
30) Katzenstein, A. L., Myers, J. L.: Nonspecific interstitial pneumonia and the other
idiopathic interstitial pneumonias: classification and diagnostic criteria. Am. J. Surg.
Pathol. 24 (1), 1-3 (2000)
31) Tubbs, R. R., Benjamin, S. P., Reich, N. E., McCormack, L. J., VanOrdstand, H. S.:
Desquamative interstitial pneumonitis. Chest, 72(8), 159-165 (1977)

127
32) Fishman, H.: UIP, DIP and all that. N. Engl. J. Med. 298(15), 843-845 (1978)
33) King, T. E.: Cryptogenic organizing pneumonitis: the North American experience.
Chest 102, 8S-13S (1992)
34) Bjoraker, J. A., Ryu, J. H., Edwin, M. K., Myers, J. L., Tazelaar, H. D., Schroeder,
D. R., Offord, K. P.: Prognostic significance of histopathologic subsets in idiopathic
pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 157(1), 199-203 (1998)
35) Carrington, C. B., Gaensler, E. A., Couto, R. E., Fitzgerald, M. X., Gupta, R. G.:
Natural history and treated course of usual and desquamative interstitial pneumonia. N.
Engl. J. Med. 28, 13 801-809 (1978)
36) Matsuo, K., Tada, S., Shibayama, T., Ueno, Y., Miyake, T., Takehara, H., Kataoka,
M., Harada, M., Kimura, I.: Factors affecting prognosis of idiopathic interstitial
pneumonia. Acta. Med. Okayama 50(1),37-46 (1996)
37) Daniil, Z. D., Gilchrist, F., C., Nicholson, A. G., Hansell, D., M., Harris, J., Colby,
T. V., DuBois, R. M.: A histologic pattern of nonspecific interstitial pneumonia is
associated with a better prognosis than usual interstitial pneumonia in patients with
cryptogenic fibrosing alveolitis. Am. J. Respir. Crit. Care Med. 160, 899-905 (1999)
38) Nicholson, A. G., Colby, T. V., DuBois, R. M., Hansell, D. M., Wells, A. U.:
Prognostic significance of the histologic pattern of interstitial pneumonia in patients
presenting with the clinical entitiy of cryptogenic fibrosing alveolitis. Am. J. Respir.
Crit. Care Med. 162, 2213-2217 (2000)
39) Myers, J. L., Veal, C. F. Jr, Shin, M. S., Katzenstein, A. L.: Respiratory
bronchiolitis causing interstitial lung disease. Am. Rev. Respir. Dis. 135(4), 880-4
(1987)
40) Myers, J. L.: Respiratory bronchiolitis with interstitial lung disease. Sem. Respir.
Med. 13, 134- 139 (1992)

128
41) Cottin, V., Donsbeck, A.-V., Revel, D., Loire, R., Cordier, J-F.: Idiopathic
interstitial pneumonia: individualization of a clinicopathologic entity in a series of 12
patients. Am. J. Respir. Crit. Care Med. 158, 1286-1293 (1998)
42) Park, C. S., Jeon, J. W., Park, S. W., Lim, G. I., Jeong, S. H., Uh, S. T., Park, J. S.,
Choi, D. L., Jin, S. Y., Kang, C. H.: NSIP: clinical manifestations, histologic and
radiologic features. Korean. J. Intern. Med. 11(2), 122-132 (1996)
43) Travis, W. D., Matsui, K., Moss, J., Ferrans, V. : Idiopathic nonspecific interstitial
pneumonia: prognostic significance of cellular and fibrosing patterns. Am. J. Surg.
Pathol. 24(1), 19-23 (2000)
44) Kim, T. S., Lee, K. S., Chung, M. P., Han, M., Park, J. S., Hwang, J. H., Kwon, O.
J., Rhee, C. H.: Nonspecific interstitial pneumonia with fibrosis: high resolution CT and
pathologic findings. AJR 171, 1645-1650 (1998)
45) Nagai, S., Kitaichi, M., Itoh, H., Nishimura, K., Izumi, T., Colby, T. V.: Idiopathic
nonspecific interstitial pneumonia/fibrosis: comparison with idiopathic pulmonary
fibrosis and bronchiolitis obliterans with interstitial pneumonia. Eur. Respir. J. 12,1010-
1019 (1998)
46) Guerry-Force, M. L., Müller, N. L., Wright, J. L., Wiggs, B., Coppin, C., Pare, P.
D., Hogg, J. C.: A comparison of bronchiolitis obliterans with organizing pneumonia,
usual interstitial pneumonia and small airways disease. Am. Rev. Respir. Dis 135(3),
705-12 (1987)
47) Costabel, U., Teschler, H., Guzman, J.: Bronchiolitis obliterans organizing
pneumonia: the cytological and immunocytological profile of bronchoalveolar lavage.
Eur. Respir. J. 5(7), 791-7 (1992)
48) Müller, N. L., Guerry-Force, M. L., Staples, C. A., Wright, J. L., Wiggs, B., Coppin,
C., Paré, P. Hogg, J. C.: Differential diagnosis of bronchiolitis obliterans with
organizing pneumonia and usual interstitial pneumonia: clinical, functional and
radiologic findings. Radiology 162, 151-6 (1987)

129
49) Giessler, H. M., Roos, N., Diederich, S., Thomas, M., Schmid, K.W., Peters, P. E.:
Bronchiolitis obliterans mit organisierender Pneumonie: Klinik, Pathologie und
radiologisches Erscheinungsbild. Radiologe 36(7), 560-6 (1996)
50) Nagai, S., Izumi, T.: Bronchiolitis obliterans with organizing pneumonia. Curr.
Opin. Pulm. Med. 2(5), 419-23 (1996)
51) Katzenstein, A. L., Myers, J. L., Prophet, W.. D., Corley, L. S. 3rd, Shin, M. S.:
Bronchiolitis obliterans and usual interstitial pneumonia. Am. J. Path. 10(6), 373-381
(1986)
52) Costabel, U., Guzman, J., Teschler, H.: Bronchiolitis obliterans with organising
pneumonia: outcome. Thorax 50 (Suppl. 1)59-64 (1995)
53) Koss, M. N., Hochholzer, L., Langloss, J. M., Wehunt, W. D., Lazarus, A. A.:
Lymphoid interstitial pneumonia: clinicopathological and immunopathological findings.
Pathology 19(2), 178-185 (1987)
54) Teirstein, A. S., Rosen, M. J.: Lymphocytic interstitial pneumonia. Clin. Chest Med.
9, 467-71 (1998)
55) Kitaichi, M.: Pathologic features and the classification of interstitial pneumonia of
unknown etology. Bull. Chest. Dis. Res. Inst. Kyoto Univ. 23(1-2), 1-18 (1990)
56) Turner-Warwick, M.: Interstitial lung disease of unknown etology. Chest 100, 232-
233 (1991)
57) Turner-Warwick, M., Haslam, P. L.: The value of serial bronchoalveolar lavages in
assessing the clinical progress of patients with cryptogenic fibrosing alveolitis. Am. Rev.
Respir. Dis. 135(1), 26-34 (1987)
58) Schwarz, D. A., Helmers, R. A., Galvin, J. R., Scott van Fossen, D., Frees, K. L.,
Dayton, C. S., Burmeister, L. F., Hunninghake, G. W.: Determinants of survival in
idiopathic pulmonary fibrosis. Am. J. Respir. Crit .Care Med. 149, 450-4 (1994)

130
59) Douglas, W. W., Ryu, J. H., Schroeder, D. R.: Idiopathic pulmonary fibrosis:
impact of oxygen and colchicine prednisone or no therapy on survival. Am. J. Respir.
Crit. Care Med. 161, 1172-1178 (2000)
60) Livingstone, J. L., Lewis, J. G., Reid, L. Jefferson, K. E.: Diffuse interstitial
pulmonary fibrosis: a clinical, radiological and pathological study based on 45 patients.
Quart. J. Med. 33, 71-103 (1964)
61) Gelb, A. F., Dreisen, R. B., Epstein, J. D., Silverthorne, J. D., Bickel, Y, Fields, M.,
´Border, W. A., Taylor, C. R.: Immune complexes, gallium lung scans and
bronchoalveolar lavage in idiopathic interstitial pneumonitis-fibrosis. Chest 84( 2), 148-
153 (1983)
62) Stack, B. H. R., Grant, I. W. B., Irvine, W. J., Moffat, M. A. J.: Idiopathic diffuse
interstitial lung disease. Am.Rev.Resp.Dis. 92, 939-948 (1965)
63) Moon, J., DuBois, R. M., Colby, T. V., Hansell, D. M., Nicholson, A. G.: Clinical
significance of respiratory bronchiolitis on open lung biopsy and its relationship to
smoking related interstitial lung disease. Thorax 54, 1009-1014 (1999)
64) Lipworth, B., Woodcock, A., Addis, B., Turner-Warwick, M.: Late relapse of
desquamative interstitial pneumonia. Am. Rev. Respir. Dis. 136(5), 1253-5 (1987)
65) Hartmann, T. E., Primack, S. L., Kang, E. Y., Swensen, S. J., Hansell, D. M.,
McGuiness, G., Müller, N. L.: Disease progression in usual interstitial pneumonia
compared with desquamative interstitial pneumonia. Chest, 110, 378-82 (1996)
66) Akira, M., Yamamoto, S., Hara, S., Sakatani, M., Ueda, E.: Serial computed
tomographic evaluation in desquamative interstitial pneumonia. Thorax 52(4), 333-337
(1997)
67) Henderson, D. W.: Morphogenesis and classification of diffuse interstitial lung
diseases. Aust. NZ J. Med. 14, 735-748 (1984)

131
68) McCann, B. G., Brewer, D. B.: A case of desquamative interstitial pneumonia
progressing to honeycomb lung. J. Pathol. 112(4), 199-202 (1973)
69) Patchefsky, A. S. H., Israel, H. L., Hoch, W. S., Gordon, G.: Desquamative
interstitial pneumonia: relationship to interstitial fibrosis. Thorax 28, 680-693 (1973)
70) Turner-Warwick, M., Burrows, B., Johnson, A.: Cryptogenic fibrosing alveolitis:
clinical features and their influence on survival. Thorax 35, 171-180 (1980)
71) Gaensler, E. A., Goff, A. M., Prowse, C. M.: Desquamative interstitial pneumonia.
N. Engl. J. Med. 274, 113-128 (1966)
72) Tukiainen, P., Taskinen, E., Holsti, P., Korhola, O., Valle, M.: Prognosis of
cryptogenic fibrosing alveolitis. Thorax 38, 349-355 (1983)
73) Lee, K. S., Chung, M. P.: Idiopathic interstitial pneumonias: clinical findings,
pathogenesis, pathology and radiologic findings. J. Korean. Med. Sci. 14, 113-27 (1999)
74) Epler, G. R., Colby, T. V., McLoud, T. C., Carrington, C. B., Gaensler, E. A.:
Bronchiolitis obliterans organizing pneumonia. N. Engl. J. Med. 312 (3), 152-8 (1985)
75) Chandler, P. W., Shin, M. S., Friedman, S. E., Myers, J. L., Katzenstein, A. L.:
Radiographic manifestations of bronchiolitis obliterans with organizing pneumonia vs.
usual interstitial pneumonia. AJR 147(5), 899-906 (1986)
76) Cordier, J. F., Loire, R., Brune, J. : Idiopathic bronchiolitis obliterans organizing
pneumonia. Definition of characteristic clinical profiles in a series of 16 patients. Chest
96(5), 999-1004 (1989)
77) Izumi, T.: Bronchiolitis obliterans organizing pneumonia. Chest 102, 712-719
(1992)
78) Raghu, G.: Interstitial lung disease: a diagnostic approach. Am. J. Respir. Crit. Care
Med. 151, 909-914 (1995)

132
79) Costabel, U., Guzman, J.: Bronchoalveolar lavage in interstitial lung disease Curr.
Opin. Pulm. Med. 7(5), 255-61 (2001)
80) Pforte, A.: Diagnostik der idiopathischen Lungenfibrose und anderer interstitieller
Pneumonien. Atemw. Lungenkrh. 29(3), 108-114 (2003)
81) Costabel, U.: Technik und Methodik der bronchoalveolären Lavage bei
interstitiellen Lungenkrankheiten. Schweiz. Med. Wochenschr. 116 (37), 1238-1244
(1986)
82) Watters, L. C., Schwarz, M. I., Cherniack, R. M., Waldron, J. A., Dunn, T. L.,
Stanford, R. E., King, T. E.: Idiopathic pulmonary fibrosis. Am. Rev. Respir. Dis. 135,
696-704 (1987)
83) Haslam, P., Turton, C. W. G., Heard, B., Lukoszek, A., Collins, J. B., Salsbury, A.
J., Turner-Warwick, M.: Bronchoalveolar lavage in pulmonary fibrosis: comparison of
cells obtained with lung biopsy and clinical features. Thorax 35, 9- 18 (1980)
84) Keogh, B. A.: Alveolitis: the key to the interstitial lung disorders. Thorax 37, 1-10
(1982)
85) Cherniack, R. M., Crystal, R. G., Kalica, A. R.: Current concepts in idiopathic
pulmonary fibrosis: a roadmap for the future. Am. Rev. Rspir. Dis. 143, 680-683 (1991)
86) Sheppard, M. N., Harrison, N. K.: Lung injury, inflammatory mediators and
fibroblast activation in fibrosing alveolitis. Thorax 47, 1064- 1074 (1992)
87) Costabel, U.: Atlas der bronchiolären Lavage. Thieme Verlag Stuttgart (1994)
88) Robinson, R.: Idiopathic pulmonary fibrosis: abnormalities in BALF phospholipids.
Am. Rev. Respir. Dis. 137(3), 585-589 (1988)
89) Erzi, T., Kunichezky, S., Eliraz, A., Soroker, D., Halperin, D., Schattner, A.:
Bronchiolitis obliterans: current concepts. Q. J. Med. 87(1), 1-10 (1994)

133
90) Shindoh, Y., Shimura, S., Tomioka, M., Aikawa, T., Sasaki, H., Takishima, T.:
Cellular analysis in bronchoalveolar lavage fluids in infiltrative and fibrotic stages of
idiopathic pulmonary fibrosis. Tohoku. J. Exp. Med. 149(1),47-60 (1986)
91) King, T. E.: Diagnostic advances in idiopathic pulmonary fibrosis Chest 100 (1),
238-41 (1991)
92) Watters, L. C., King, T. E., Schwarz, M. I., Waldron, J. A., Stanford, R. E.,
Cherniack, R. M.: A clinical, radiographic and scoring system for the longitudinal
assessment of patients with idiopathic pulmonary fibrosis. Am. Rev. Respir. Dis. 133(1),
97-103 (1986)
93) Cherniack, R.M., Colby, T. V., Flint, A., Thurlbeck, W. M., Waldron, J. A.,
Ackerson, L., Schwarz, M. I., King, T. E.: Correlation of structure and function in
idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 151, 1180-8 (1995)
94) Hanson, D., Winterbauer, R. H., Kirtland, S. H., Wu, R.: Changes in pulmonary
function test results after 1 year of therapy as predictors of survival in patients with
idiopathic pulmonary fibrosis. Chest 108, 305-310 (1995)
95) Schwarz, M. I.: Idiopathic pulmonary fibrosis (clinical conference). West. J. Med.
149(2), 199-203 (1988)
96) DeRemee, R. A., Harrison, E. G., Andersen, H. A.: The concept of classic
interstitial pneumonitis-fibrosis as a clinicopathologic syndrome. Chest 61:213-220
(1972)
97) Schmidt, M.: Therapie der idiopathischen interstitiellen Lungenkrankheiten. Atemw.
Lungenkrh. 29(3), 115-120 (2003)
98) Haslam, P., Turton, C. W., Lukoszek, A., Salsbury, A. J., Dewar, A., collins, J. V.,
Turner-Warwick, M.: Bronchoalveolar lavage fluid cell counts in cryptogenic fibrosing
alveolitis and their relation to therapy. Thorax 35(5), 328-39 (1980)

134
99) Winterbauer, R.: The treatment of idiopathic pulmonary fibrosis. Chest 100, 233-
235 (1991)
100) Winterbauer, R. H., Hammar, S. P., Hallmann, K. O., Hays, J. E., Pardee, N. E.,
Morgan, E. H., Allen, J. D., Moores, K. D., Bush, W. , Walker, J. H.: Diffuse interstitial
pneumonitis. Am. J. Med. 65, 661-672 (1978)
101) Sadikot , R. T., Johnson, J., Loyd, J. E., Christman, J. W.: Respiratory
bronchiolitis associated with severe dyspnea, exertional hypoxemia and clubbing. Chest
171, 282-285 (2000)
102) Ryu, J. H., Colby, T. V., Hartman, T. E., Vassallo, R.: Smoking-related interstitial
lung diseases: a concise review. Eur. Respir. J. 17(1), 122-132 (2001)
103) Wells, A. U., Cullinan, P., Hansell, D. M., Rubens, M. B., Black, C. M., Newman-
Taylor, A. J., Du Bois, R. M.: Fibrosing alveolitis associated with systemic sclerosis has
a better prognosis than lone cryptogenic alveolitis. Am. J. Respir.Crit. Care Med.
149(6), 1583-1590 (1994)
104) Chiodera, P.: Idiopathic interstitial lung disease: anatomoradiologic pathogenesis.
Rays, 22(1), 127-164 (1997)
105) Johnston, I. D., Prescott, R. J., Chalmers, J. C., Rudd, R. M.: British Thoracic
Society study of cryptogenic fibrosing alveolitis: current presentation and initial
management. Thorax 52(1), 38-44 (1997)
106) Flint, A., Martinez, F. J., Young, M. L., Whyte, R. I., Toews, G. B., Lynch, J. P.
3rd: Influence of sample number and biopsy site on the histologic diagnosis of diffuse
lung disease. Ann. Thorac. Surg. 60(6), 1605-1608 (1995)
107) Walker, W. A., Cole, F. H. Jr., Khandekar, A., Mahfood, S. S., Watson, D. C.:
Does open lung biopsy affect treatment in patients with diffuse pulmonary infiltrates?
J. Thorac. Cardiovasc. Surg. 97(4), 534-40 (1989)

135
108) Venn, G., Goldstaw, P.: Open lung biopsy. Br. J. Hosp. Med. 39(4), 272-3 (1988)
109) Myers, J. L.: NSIP, UIP, and the ABC of idiopathic interstitial pneumonias. Eur.
Respir. J. 12, 1003-1004 (1998)
110) Colby, T. V., Churg, A. C.: Patterns of pulmonary fibrosis. Pathol. Annual
21(part2), 277-309 (1986)
111) Bedrossian, C. W. M, Kuhn, C., Luno, M. A., Conklin, R. H., Byrd, C. R. B.,
Kaplan, P. D.: Desquamative interstitial pneumonia-like reaction accompanying
pulmonary lesions. Chest 72(2), 166-169 (1977)
112) Kitaichi, M.: Differential diagnosis of bronchiolitis obliterans organizing
pneumonia. Chest 102(1 Suppl), 44-49 (1992)
113) Nicholson, A. G., Fulford, L. J. G., Colby, T. V., DuBois, R. M., Hansell, D. M.,
Wells, A. U.: The relationship between individual histologic features and disease
progression in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 166(2),
173-7 (2002)
114) Flaherty, K. R., Travis, W. D., Colby, T. V., Toews, G :, Kazerooni, E. A., Jain,
A., Strawderman, R. L., Flint, A., Lynch, J. P., Martinez, F. J.: Histopathologic
variability in usual and nonspecifiic interstitial pneumonias. Am. J. Repir. Crit. Care
Med. 164(9),1722-7 (2001)
115) DuBois, R. M.: Idiopathic pulmonary fibrosis. Annu. Rev. Med. 44, 441-50 (1993)
117) Katzenstein, A. L.: Pathogenesis of „Fibrosis“ in interstitial pneumonia. Hum.
Pathol. 16, 1015-1024 (1985)
117) Vaillant, P., Menard, O., Vignaud, J. M., Martinet, N., Martinet, Y. : The role of
cytokines in human lung fibrosis. Monaldi. Arch. Chest Dis. 51(2), 145-152 (1996)

136
118) Line, B. R., Fulmer, J. D., Reynolds, H. Y., Roberts, W. C., Jones, A. E., Harris, E.
K., Crystal, R. G.: Gallium-67 citrate scanning in the staging of idiopathic pulmonary
fibrosis: correlation with physiologic and morphologic features. Am. Rev. Respir. Dis.
118, 354-365 (1978)
119) Reiser, K. M., Last, J. A.: Early cellular events in pulmonary fibrosis. Exp. Lung.
Res. 10, 331-55 (1985)
120) Kasper, M., Haroske, G.: Alterations in the alveolar epithelium after injury leading
to pulmonary fibrosis. Histol. Histopathol. 11(2), 463-83 (1996)
121) Rubin, R.: Bronchoalveoläre Lavage. Z. Erkr. Atmungsorgane 164(1), 4-18 (1985)
122) Selman, M, King, T. E., Pardo, A.: Idiopathic pulmonary fibrosis: prevailing and
evolving hypotheses about its pathogenesis and implications for therapy. Ann. Intern.
Med. 134(2), 136-151 (2001)
123) Kolb, M.: Pathogenese der idiopathischen Lungenfibrose. Atemw. Lungenkrh.
29(3), 99- 102 (2003)
124) Raghu, G.: Idiopathic pulmonary fibrosis: a need for treatment with drugs other
than corticosteroids. Mayo Clin. Proc. 72(3), 285-287 (1997)
125) Douglas, W. W., Ryu, J. H., Bjoraker, J. A., Schroeder, D. R., Myers, J. L.,
Tazelaar, H. D., Swensen, S. J., Scanlon, P. D., Peters, S. G., DeRemee, R. A.:
Colchicine versus prednisone as treatment of usual interstitial pneumonia. Mayo. Clin.
Proc. 72, 201-209 (1997)
126) Johnson, M. A., Kwan, S., Snell, N. J. C., Nunn, A. J., Darbyshire, J. H., Turner-
Warwick, M.: Randomized controlled trial comparing prednisolone alone with
cyclophosphamide and low dose prednisolone in combination with cryptogenic
fibrosing alveolitis. Thorax 44, 280-288 (1989)

137
127) Raghu, G., Depaso, W. J., Cain, K., Hammar, S. P., Wetzel, C. E., Dreis, D. F.,
Hutchinson, J. Pardee, N. E., Winterbauer, R. H.: Azathioprine combined with
prednisone in the treatment of idiopathic pulmonary fibrosis. Am. Rev. Respir. Dis.
144(2), 291-296 (1991)
128) Günther, W.: Klassifikation, Diagnostik und Therapie der idiopathischen
interstitiellen Pneumonien. Dtsch. Arztebl. 100(24), 1676-1685 (2003)
129) Moolmann, J. A., Bardin, P. G., Rossouw, D. J., Joubert, J. R.: Cyclosporin as a
treatment for interstitial lung disease of unknown aetiology. Thorax 46, 592-595 (1991)
130) Hunninghake, G. W., Kalica, A. R.: Approaches to the treatment of pulmonary
fibrosis. Am. J. Respir. Crit. Care Med. 151, 915-918 (1995)

138
8. Danksagung
Ich möchte Herrn Prof. Dr. med. K. Morgenroth für die Rahmenbedingungen danken,
die diese Promotionsarbeit ermöglicht haben.
Herrn PD Dr. med. D. Theegarten danke ich für die Überlassung des Themas. Ihm gilt
auch mein ganz besonderer Dank für die ständige zuverlässige Ansprechbarkeit und
fachliche fundierte Unterstützung sowie die ausgesprochen angenehme Atmosphäre bei
der Zusammenarbeit.
Herrn Prof. Dr. med. U. Costabel möchte ich besonders für seine große Hilfe bei der
Erarbeitung des klinischen Teils danken.
Den Mitarbeitern des Archivs der Ruhrlandklinik Essen danke ich für die Einarbeitung
in das Ablagesystem und die freundliche Unterstützung.
Ein besonderer Dank gilt Herrn Birger Kriwet für die motivierenden Worte und die
Beseitigung orthographischer Stolpersteine.

139
9. Curriculum Vitae
Name: Müller
Vorname: Heike Maria
Geburtsdatum: 18.10.1974
Geburtsort: Paderborn
Anschrift: Im Giershagen 37, 34414 Warburg
Telefon: 05642/949510
Konfession: Römisch-katholisch
Familienstand: Ledig, zwei Kinder
Werdegang: 1981-1985 Katholische Grundschule Willebadessen
1985-1991 Realschule Willebadessen
1991-1994 Gymnasium Marianum Warburg
1994-2000 Studium der Humanmedizin an der Ruhr-
Universität Bochum
10/1996-06/1997 Studienaufenthalt an der Université Louis
Pasteur, Straßburg, Frankreich mit
Anerkennung der Studienleistungen als
ersten Abschnitt der ärztlichen Prüfung
04/2000 Zweiter Abschnitt der ärztlichen Prüfung
04/2000-05/2001 Immatrikulation an der Westfälischen
Wilhelms-Universität Münster
05/2001 Dritter Abschnitt der ärztlichen Prüfung
09/2001-02/2003 ÄIP an der St. Vincenz Frauenklinik
Paderborn
seit 03/2003 Assistenzärztin an der St. Vincenz
Frauenklinik Paderborn







