
Bericht der Arbeitsgruppe vom November
2012
Die AG Wirkstoffentwicklung hat im Jahr 2011 ihre Aktivitäten zur Entwicklung neuer
Wirkstoffe im Rahmen akademischer Forschung fortgesetzt. Eine Reihe der bereits vor zwei
Jahren begonnenen Projekte befindet sich derzeit in einem fortgeschrittenen Stadium der
Entwicklung. Gleichzeitig versucht die Arbeitsgruppe, ihre Aktivitäten zur Verbesserung der
Rahmenbedingungen akademischer Wirkstoffentwicklungsprojekte zu intensivieren.
Neben diesen Bemühungen ist es uns auch in diesem Jahr gelungen, ein Symposium zum
Thema „ Akademische Wirkstoffentwicklung“ in Berlin zu organisieren. Wir werden die für
die Onkologie wichtigen Themen des drug development gegen das Onkogen myc sowie neue
Entwicklungen zum Thema Zellteilung als Target in den wissenschaftlichen Mittelpunkt
stellen. Außerdem wollen wir neue Konzepte zur Durchführung von Phase 1 Studien
diskutieren.
Zusammenfassend haben die Bemühungen der AG Wirkstoffentwicklung zur Fortführung von
akademischen Projekten sowie zum Beginn einer großen gemeinsamen Initiative zur
verbesserten Forschungsförderung in diesem Bereich geführt.

Prof. Dr. Walter Fiedler
Universitätsklinik Hamburg-Eppendorf
Hubertus-Wald University Cancer Center
20246 Hamburg
e-mail: [email protected]
Education
1976-1983 Medicals Schools Gent/Belgium and Hamburg/Germany
1983 Medical approbation with the right to exercise the medical profession,
Hamburg, Germany
1983 MD Phesis (Prof.Dr. R. Jaenisch, Heinrich-Pette Institute)
Titlel: Aktivierung von Moloney Leukämievirus in BALB/Mo Mäusen
1996 Habilitation, University Hamburg
Professional Career
1984-1986 Research Fellow, Memorial Sloan Kettering Cancer Center, New York
1986 to date Department of Internal Medicine II (Oncology/Hematology), University
Hospital Hamburg-Eppendorf, Germany
1994 German board certification for Internal Medicine
1998 German board certification for Hematology and Medical Oncology
1999 to date Professor of Internal Medicine (with special emphasis on
Hematology/Oncology)
2008 Head of newly founded Phase I Unit and Phase I Program

Key Publications
1. Fiedler W., Giaccone G., Lasch P., van der Horst I., Brega N., Courtney R.,
Abbattista A., Shalinsky D.R., Bokemeyer C., Boven E.: Phase I Trial of SU14813 in
Patients with Advanced Solid Malignancies. Annals Oncol 22, 195-201, 2011
2. Fiedler W., Tchen N., Bloch J., Fargeot P., Sorio R., Vermorken J.B., Collette L.,
Lacombe D., Twelves C.: A study from the EORTC new drug development group: open
label phase II study of sabarubicin (MEN-10755) in patients with progressive hormone
refractory prostate cancer.. EJC 42: 200-204, 2006
3. Loges S., Heil G., Aykurt M., Schoder V., Butzal M., Fischer U., Gehling U.M.,
Hossfeld D.K.,Fiedler W: Analysis Of Concerted Expression Of Angiogenic Growth
Factors In Acute Myeloid Leukemia: Expression Of Angiopoietin-2 Represents An
Independent Prognostic Factor For Overall Survival. JCO 23: 1109-1117, 2005
4. Fiedler W., Serve H., Döhner H., Schwittay M., Ottmann O.G., O’Farrell A.-M.,
Bello C.L., Allred R., Manning W.C., Cherrington J.M., Louie S.G., Hong W.,
Brega N:M., Massimini G., Scigalla P., Berdel W.E., Hossfeld D.K.: A phase I study
of SU11248 in the treatment of patients with refractory or resistant acute myeloid
leukemia (AML) or not amenable to conventional therapy for the disease. Blood 105:
986-993, 2005
5. Fiedler W.,Mesters R., Tinnefeld H., Loges S., Staib P., Duhrsen U., Flasshove M.,
Ottmann O. G., Jung W., Cavalli F., Kuse R., Thomalla J., Serve H., O´Farrell A.M.,
Jacobs M., Brega N.M., Scigalla P., Hossfeld D.K., Berdel W.E. A phase 2 clinical study
of SU5416 in patients with refractory acute myeloid leukemia. Blood 102:2763-2767,
2003
6. Fiedler W., Mesters R., Tinnefeld H., Loges S., Staib P., Duhrsen U., Flasshove M.,
Ottmann O. G., Jung W., Cavalli F., Kuse R., Thomalla J., Serve H., O´Farrell A.M.,
Jacobs M., Brega N.M., Scigalla P., Hossfeld D.K., Berdel W.E. A phase 2 clinical
study of SU5416 in patients with refractory acute myeloid leukemia. Blood 102:2763-
2767, 2003
7. Gehling U. M., Ergün S., Schumacher U., Wagener C., Pantel K., Otte M., Schuch
G., Schafhausen P., Mende T., Kilic N., Kluge K., Schäfer B., Hossfeld D. K. and W.
Fiedler: In vitro differentiation of endothelial cells from AC133-positive progenitor
cells. Blood 95:3106-3112, 2000
8. Fiedler W., Graeven U., Ergün S., Verago S., Kilic N., Stockschläder M. and D. K.
Hossfeld: Vascular endothelial growth factor, a possible paracrine growth factor in
human acute myeloid leukemia. Blood 6:1870-1875, 1997

Dr. rer. nat. Ronald Frank
Helmholtz-Zentrum für Infektionsforschung, Braunschweig
HZI-Aktivitäten 2011 im Rahmen der AIO AG „Wirkstoffentwicklung“
Zu den laufenden Projekten, die durch ein Screening am HZI oder durch HZI-Substanzen
unterstützt wurden, sei auf die entsprechenden Berichte der Partner verwiesen. Diese sind:
- Inhibitoren der BCL10 Signalkaskade zur Behandlung von Lymphomen (Jürgen Ruland)
- Inhibitoren des STAT3Signalweges zur Behandlung von Pankreaskarzinom (Florian Greten)
- Modulatoren der Nrf2 Expressionzur Krebsprävention und Kombinationschemotherapie
(Arndt Vogel)
- ProteasominhibitorArgyrin zur Behandlung verschiedener Tumore(Nisar Malek, Markus
Kalesse)
- Inhibitoren der Cdc25APhosphatase für die Behandlung von verschiedenen Krebsarten wie
Brustkrebs, Pankreastumoren und dem nicht-kleinzelligen Bronchialkarzinom (Ingrid
Hoffmann / Markus Kalesse)
Die Substanzsammlung am HZI wurde kontinuierlich durch Aufnahme neuer interessanter
Proben von Kollegen aus chemischen Laboren sowie einer Kopie der Sammlung mariner
Naturstoffe des
KiWiZ (Kieler Wirkstoffzentrum am GEOMAR) erweitert. Ein neues S3-Screening-Labor
wird in Kürze in Betrieb genommen, so dass demnächst auch Screens mit pathogenen
Organismen die dieser Sicherheitsstufe unterliegen durchgeführt werden können; das trifft
u.a. auch für die Hepatitiserreger zu (Leberkrebs).
R. Frank berichtete auf dem AIO-Herbstkongress 2011 in Berlin über die Fortschritte beim
Ausbau von Forschungsinfrastrukturen für die Unterstützung von akademischen Projekten der
frühen Wirkstoffforschung (HGF-Verbund Wirkstoffforschung, ChemBioNet und EU-
OPENSCREEN).

Prof. Dr. Ingrid Hoffmann, PhD
Head of the research group
Cell Cycle Control and Carcinogenesis
Im Neuenheimer Feld 242
D-69120 Heidelberg
Education
1987 PhD at the Universities of Saarland, Saarbrücken and Lund, Sweden
1987-1990 Post-Doctoral Work at the Institute of Molecular Pathology (IMP), Vienna
(Austria) with Prof. Max L. Birnstiel
1991-1995 Post-Doctoral Work at the European Molecular Biology Laboratory (EMBL),
Heidelberg, Germany with Drs. Giulio Draetta and Eric Karsenti
Professional postions held
1996- Head of the Research Group “Cell Cycle Control and Carcinogenesis”
2000 Habilitation Faculty of Biosciences, University of Heidelberg
2012 Professor of Cell Biology, Faculty of Biosciences, University of Heidelberg
Honors, Awards
1984-1985 Fellowship from the DAAD (University of Lund, Sweden)
1987 EMBO fellowship (University of Nijmegen, The Netherlands)
1991-1993 Research fellowship from the German Research Foundation (DFG)
(EMBL, Heidelberg)
Selected Publications
Warnke S., Kemmler S., Hames RS, Tsai HL, Hoffmann-Rohrer U., Fry AM, and Hoffmann
I.
(2004) The polo-like kinase-2 is required for centriole duplication in mammalian cells.
Curr Biol. 14:1200-1207.
Cizmecioglu, O., Arnold, M., Bahtz, R., Settele, F., Ehret, L., Haselmann-Weiß, U., Antony,
C.
and Hoffmann, I. (2010) Cep152 acts as a scaffold for recruitment of Plk4 and CPAP to
the centrosome. J Cell Biol. 191: 731-739.
Timofeev, O., Cizmecioglu, O., Settele F., Kempf, T. and Hoffmann I. (2010) Cdc25
phosphatases are required for timely assembly of Cdk1/CyclinB complex at the G2/M
transition, J. Biol. Chem. 285:16978-16990.
Bahtz, R., Seidler, J., Arnold, M., Haselmann-Weiss, U., Antony, C., Lehmann, W and
Hoffmann, I., (2012) GCP6 is a substrate of Plk4 and required for centriole duplication,
J Cell Sci, 125:486-96.
Cizmecioglu, O, Krause, A., Bahtz, R., Malek, N. and Hoffmann, I. Phosphorylation of
Fbxw7/hCdc4

Identifizierung und Charakterisierung von Phosphatase-Inhibitoren mit
antiproliferativer Wirkung zur Therapie von Krebserkrankungen
Prof. Dr. Ingrid Hoffmann, DKFZ, Heidelberg
Proteinphosphatasen und -kinasen steuern durch reversible Proteinphosphorylierung eine
Vielzahl zellulärer Mechanismen. Die Blockierung der Aktivität spezifischer Kinasen und vor
allem die von Phosphatasen stellen daher einen wichtigen, neuen Ansatz mit breiten
Anwendungsmöglichkeiten in der Krebsforschung dar. Bislang gibt es jedoch nur wenige
selektive und effektive Inhibitoren von Proteinphosphatasen. Solche Substanzen besäßen
allerdings entscheidende therapeutische Vorteile, da das Substratspektrum von
Proteinphosphatasen unterschiedlich von dem der Kinasen ist.
Cdc25-Phosphatasen dephosphorylieren und aktivieren dadurch Cyclin-abhängige Kinasen
(CDK) an wichtigen Übergängen des Zellzyklus, den Übergängen in die S-Phase und die
Mitose. Die Proteinmengen von Cdc25A und B aber nicht die von Cdc25C liegen dereguliert
bei verschiedenen Krebsarten vor, u.a. bei Brustkrebs, Pankreastumoren und dem
nichtkleinzelligen
Bronchialkarzinom. Die Charakterisierung kürzlich identifizierter
niedermolekularer Inhibitoren der humanen Cdc25 Phosphatasen steht im Mittelpunkt des
Forschungsvorhabens.
Nach der Durchführung von drei Medium-Throughput Screens verschiedener Bibliotheken
niedermolekularer Inhibitoren liegen uns mittlerweile 42 Inhibitoren von Cdc25A vor, die 8
verschiedenen Strukturklassen angehören. Diese Inhibitoren hemmen die Phosphatase-
Aktivität von Cdc25A in vitro und in vivo und blockieren die Zellproliferation in HeLa- und
MCF7-Zellen (IC50 ≤ 50 μM). Es wurden weitere humane Zelllinien getestet, die ähnliche
Ergebnisse lieferten. Eine Reihe der gefundenen Inhibitoren sind spezifisch für Cdc25A,
andere hemmen alle drei Cdc25-Phosphatasen. In Zusammenarbeit mit Prof. Markus
Kalesse (Hannover) wurde eine Reihe von strukturoptimierten Wirkstoffen einer
Strukturklasse im Hinblick auf die Hemmung von Cdc25A in vivo und in FACS-Scan
Analysen untersucht. Das weitere Vorgehen besteht darin, eine Reihe von
strukturoptimierten Substanzen in humanen Krebszelllinien zu testen. Darüber hinaus ist
geplant, diese Inhibitoren in humanen Tumoren in vitro Xenograftmodellen zu untersuchen
(Zusammenarbeit mit Prof. Nisar Malek, Tübingen).
Unter Einsatz der Inhibitoren soll die genaue Funktion der Cdc25-Phosphatasen am Eintritt
der Zelle in die Mitose entschlüsselt werden. Unser langfristiges Ziel ist es, die Inhibitoren
von Cdc25-Phosphatasen zukünftig als Therapeutika bei der Krebsbehandlung einzusetzen

Prof. Dr. Markus Kalesse
Institut für organische Chemie
Schneiderberg 1b
30167 Hannover
Wir haben es uns zur Aufgabe gemacht, Wirk- und Naturstoffe weiterentwickeln, um diese
für die medizinische Anwendung nutzbar zu machen. Aufgrund der überdurchschnittlich
hohen Erfolgswahrscheinlichkeit von Naturstoffen in den Indikationsgebieten Infektion und
Krebs wird der Fokus auf diese medizinischen Ausrichtungen gelegt. Projekte der
Wirkstoffforschung sind hoch interdisziplinär und können daher nur in Zusammenarbeit von
Forschergruppen unterschiedlicher Fächer bearbeitet werden. Dabei müssen medizinische
Herausforderungen in biochemische Fragestellungen übersetzt und durch chemische
Modifizierung an den Wirk- bzw. Naturstoffen gelöst werden. Zu diesem Zweck arbeiten wir
mit medizinischen, biochemischen und biologischen Arbeitskreise der Arbeitsgruppe
„Wirkstoffentwicklung“ zusammen.
Diese Probleme der Wirkstoffforschung liegen zum großen Teil in der tradierten Trennung
von chemischer Synthese und biosynthetischer Produktion von Naturstoffen. Im biologisch-
medizinischen Bereich liegt der Bruch der Forschungsgebiete zwischen den Gruppen, die
biologische Expertisen besitzen, und denen, die gezielt chemische Modifizierungen an
Substanzen vornehmen. Erschwerend kommt hinzu, dass bei der chemischen Entwicklung
von Wirkstoffen oft die biomedizinische Expertise fehlt, die eine gezielte Optimierung von
Wirkstoffen erst möglich macht.
In der Zusammenarbeit mit der Arbeitsgruppe „Wirkstoffentwicklung“ sollen die oben
genannten Probleme angegangen, die Trennung zwischen den Fachdisziplinen überwunden
und neue Verfahren zur Generierung optimierter Wirkstoffe und molekularer Therapieansätze
entwickelt werden. Durch die frühe Einbindung biologischer und medizinischer Gruppen
kann das Potenzial von Naturstoffen als mögliche Wirkstoffe schnell erkannt und
zielgerichteter weiterentwickelt werden.
In Zusammenarbeit mit der Gruppe von PD Dr. Ingrid Hoffmann am DKFZ werden derzeit
Cdc25-Inhibitoren als mögliche anti-Tumor-Verbindungen entwickelt.
Neben der Weiterentwicklung von Proteasom-Inhibitoren (Argyrin) haben wir eine Synthese
von Pellasoren erfolgreich abschließen können.
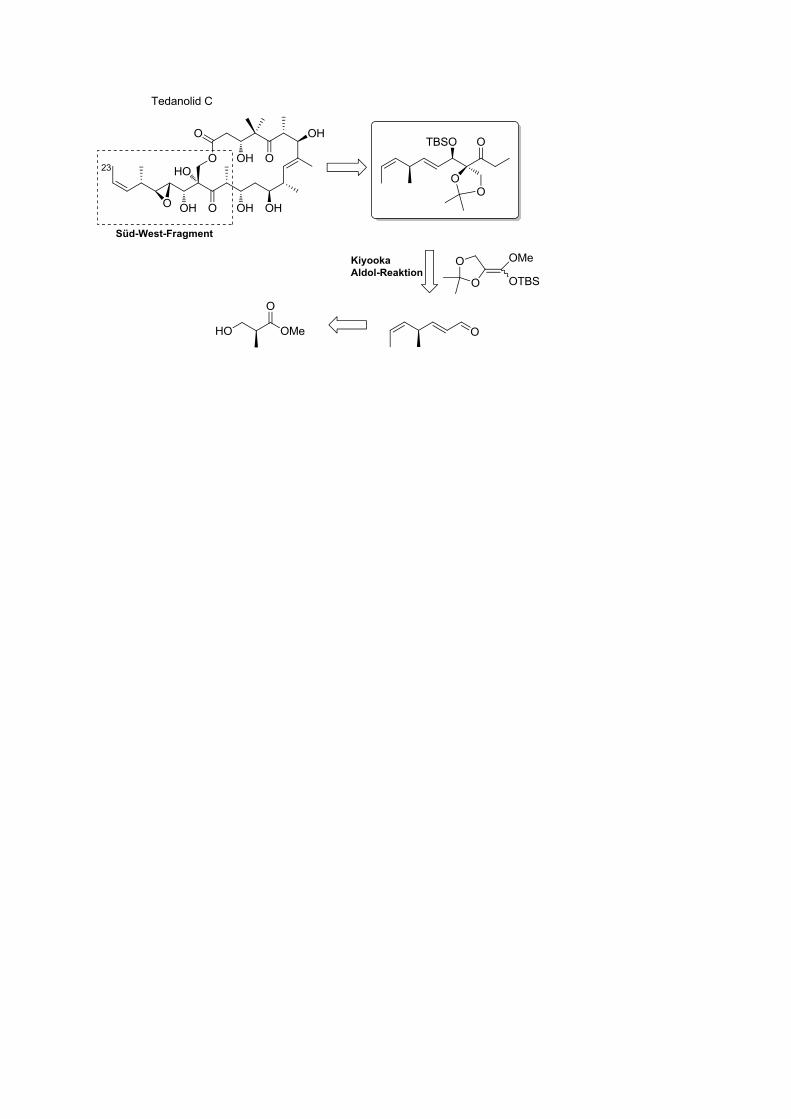
Des weiteren konnte das Süd-West-Fragment von Tedanolid C stereoselektiv aufgebaut
werden.

Prof. Dr. rer. nat. Dietmar Manstein
Institute for Biophyical Chemistry
Hannover Medical School (MHH)
Education
INSTITUTION AND LOCATION DEGREE YEAR FIELD OF STUDY
Universität Hannover and MHH, Hannover
Ruprecht-Karls-Universität Heidelberg
Ruprecht-Karls-Universität Heidelberg
Diplom
Dr. rer. nat.
Dr. rer. nat.
habil.
1983
1986
1999
Biochemistry
Biochemistry
Biochemistry and
Biophysics
Professional Appointments
1983
1984 – 1987
1987 – 1990
1990 – 1996
1996 – 2002
2002 –
2003 –
Predoctoral Trainee, EMBL, Heidelberg (Jürg Rosenbusch)
Research Associate, Max-Planck-Institute for Medical Research, Heidelberg
(Roger S. Goody) and Department of Biological Chemistry, University of Michigan,
Ann Arbor, USA (Vincent Massay)
Postdoctoral Fellow, Departments of Cell Biology and Developmental Biology,
Stanford University School of Medicine, Stanford, USA (James A. Spudich)
Group Leader, Division of Physical Biochemistry, National Institute for Medical
Research, London, U.K.
Group Leader (C3), Dept. of Biophysics, Max-Planck-Institute for Medical Research,
Heidelberg
Director, Institute for Biophysical Chemistry, MHH
Director, Research Division for Structure Analysis, MHH
Head, Research Core Facility for Laser Microscopy, MHH
Chair of Preclinical Research & Education, MHH
Member, MHH Senate

Honors / Positions
1985
1987 – 1989
1992 –
2002
2002
2006 –
2008 –
2008 –
2008 –
2010 –
2011 –
DFG Fellowships, University of Michigan Medical School (6 months)
DFG Fellowship, Stanford University School of Medicine
Executive Editor, Journal of Muscle Research and Cell Motility
Wellcome Trust University Award (not accepted)
Primo et unico loco, Chair of Physiological Chemistry, Ruhr-University-Bochum
(not accepted)
Speaker, DFG-Research Unit “Molecular Mechanisms of Cell Motility”
(FOR629)
Editor, FEBS Letters
Member, ERA-Instruments Scientific Advisory Board
Member, German Committee for Synchrotron Research (KFS)
Member, Steering Committee for the Establishment of the Centre for Structural
Systems Biology (CSSB) at the DESY campus, Hamburg
Editor FEBS OpenBio

Key Publications
Behrmann, E., Müller, M., Penczek, P.A., Mannherz, H.G., Manstein, D.J.*, and Raunser, S.*
(2012). Subnanometer-Resolution Structure of the Rigor Actin-Tropomyosin-Myosin Complex Cell
150, 327-338. IF 32.403
Preller, M., Chinthalapudi, K., Martin, R., Knölker, H.-J., and Manstein, D.J. (2011). Inhibition of
Myosin ATPase Activity by Halogenated Pseudilins: A Structure-Activity Study. J. Med. Chem. 54,
3675-3685. IF 5.248
Montessuit S., Somasekharam S.P., Terrones O., Lucken-Ardjomande S., Herzig S,
Schwarzenbacher R., Manstein D.J., Bossy-Wetzel E., Basanez G., Meda P. and Martinou J.-C.
(2010). Membrane Remodeling Induced By The Dynamin Related GTPase Drp1 Stimulates Bax
Oligomerization. Cell 142, 889-901. IF 32.403
Fedorov, R., Böhl, M., Tsiavaliaris, G., Hartmann, F. K., Taft, M. H., Baruch, P., Brenner, B.,
Martin, R., Knölker, H.-J., Gutzeit, H. O., Manstein, D. J.. (2009). The mechanism of
pentabromopseudilin inhibition of myosin motor activity. Nat. Struct. Mol. Biol. 16, 80-88. IF
12.273
Knoth, T., Warburg, K., Katzka, C., Rai, A., Wolf, A., Brockmeyer, A., Janning, P., Reubold, T. F.,
Eschenburg, S., Manstein, D. J., Hubel, K., Kaiser, M., H. Waldmann (2009). The Ras Pathway
Modulator Melophlin A Targets Dynamins. Angew. Chem. Int. Ed. Engl. 48, 7240-7245. IF 11.829
Tsiavaliaris, G., Fujita-Becker, S. and Manstein, D. J. (2004). Molecular engineering of a
backwards-moving myosin motor. Nature 427, 558-561. IF 34.480
Patent Applications
D.J. Manstein, M. Preller, M. Furch, M. Kalesse, Markus, N. Diaz Gomez “Novel means and
methods for treating malaria and other parasitic disorders” PCT/EP2011/007305.3.; Filing Date: 8. 9.
2011
D.J. Manstein, R. Fedorov, G. Tsiavaliaris, H.-J. Knölker, R. Martin, J. Kirst, H. O. Gutzeit, M. Böhl,
M. Furch „Means for treating myosin-related diseases“ (WO/2009/065600), International Application
No.: PCT/EP2008/009891; US 2011/0105554 A1; International Filing Date: 21.11.2008; Publication
Date: 28.05.2009

Prof. Dr. Thomas Mayer
Dept.of Molecular Genetics
University of Konstanz
78457 Konstanz, Germany
Education
INSTITUTION AND
LOCATION
DEGREE YEAR(s) FIELD OF STUDY
University of Heidelberg
ZMBH Heidelberg
Study
Dissertation
1989-
1993
1994-
1997
Molecular Biology
Molecular Biology
Professional Appointments and Scientific Career
1998 – 2002
202-2007
Since 2007
Postdoctoral Fellow (DFG Emmy Noether Fellow), Harvard Medical School,
Institute of Chemical Biology, Boston, USA, in the lab of Prof. T. Mitchison
Emmy-Noether Group Leader at the Max-Planck Institute of Biochemistry,
Martinsried, Germany
Full-Professor at the University of Konstanz
Honors / Positions
2007
2000
Walther-Flemming-Medaille der Deutschen Gesellschaft für
Zellbiologie
Heinz Maier-Leibnitz-Award

Key Publications
Tischer, T. Hörmanseder, E., and Mayer, T.U. (2012). The APC/C Inhibitor XErp1/Emi2 Is
Essential for Xenopus Early Embryonic Divisions. Science. 2012 Sep 27. [Epub ahead of
print]
Catarinella, M., Grüner, T., Strittmatter, T,, Marx, A., and Mayer, T.U. (2009). BTB-1: The first
small molecule inhibitor of the mitotic motor protein Kif18A, Angew. Chem. Int. Ed., 48, 9072-
9076
Hummer, S. and Mayer, T.U. (2009) Cdk1 negatively regulates midzone localization of the
mitotic kinesin Mklp2 and the chromosomal passenger complex. Curr Biol, 19, 607-12
Mayr, M., Hümmer, S., Bormann, J., Grüner, T., Adio, S., Woehlke, G., and Mayer, T.U.
(2007). The human kinesin Kif18A is a motile microtubule depolymerase essential for
chromosome congression.
Curr. Biol., 17, 488-498.
Leuenberger, M.G., Sanz, M.A, Leßmann,T., Voigt,T., Lopez-Canet,M., Menninger, S.,
Müller,O., Hümmer, S., Bormann, J., Mayer , T.U., and Waldmann, H. (2006). Natural product-
derived modulators of cell cycle progression and viral entry by enantioselective oxa Diels-
Alder reactions on the solid phase.
Chem Biol., 14, 443-451.
Rauh, N.R., Schmidt, A., Bormann, J., Nigg, E.A. and Mayer, T.U. (2005) Calcium triggers exit
from meiosis II by targeting the APC/C inhibitor XErp1 for degradation. Nature, 437, 1048-52
Perlman, Z.E., Mitchison, T.J., and Mayer, T.U. (2005). High-content screening and
mechanistic profiling of drug activity in an automated centrosome duplication assay.
Chembiochem., 6, 145-151.
Mayer, T.U., Kapoor, T.M., Haggarty, S.J, King, R.W., Schreiber, S.L., and Mitchtison, T.J.
(1999). Small molecule inhibitor of spindle bipolarity identified in a phenotype-based screen.
Science, 286, 971-974.

Die genetische Integrität eines jeden Organismus‘ hängt von der fehlerfreien Segregation der
Chromosomen während der Mitose ab. Fehler in diesem Prozess führen zu Aneuploidien, welche
zur Tumorentstehung beitragen können. Die funktionelle Untersuchung mitotischer Proteine
wird durch deren komplexe Regulation und die hohe Dynamik der Chromosomentrennung
erschwert. Niedermolekulare Moleküle eignen sich, aufgrund ihrer schnellen und oftmals
reversiblen Wirkungsweise, hervorragend zur Untersuchung dynamischer Prozesse. Für die
Identifizierung geeigneter Substanzen werden in unserem Labor protein- und zellbasierte
Screens durchgeführt.
Die Inhibierung des mitotischen Motorproteins Eg5 durch Monastrol, führt zur Ausbildung
monopolarer Spindeln. Um einen besseren Einblick in den Mechanismus des Spindelaufbaus zu
gewinnen, wurde ein zellbasierter Screen nach Verbindungen durchsucht, welche den
monopolaren Phänotyp in Eg5-inhibierten Zellen supprimieren können. Mit Hilfe einer
automatisierten Bilderkennungssoftware, die in der Lage ist, bipolare von monopolaren Spindeln
zu unterscheiden, wurden 20.000 Verbindungen einer sogenannten Pharmakophor-Bibliothek im
Duplikat durchsucht. Dabei wurde eine Verbindung, genannt 28H16, identifiziert, welche in
Konzentrations-abhängiger Weise, die Bildung bipolarer Spindeln in Monastrol-behandelten
HeLa-Zellen induziert. In vitro Untersuchungen ergaben, dass Monastrol, in Anwesenheit von
28H16, in der Lage ist, die ATPase-Aktivität von Eg5 zu inhibieren. Dieses Ergebnis belegt,
dass 28H16 nicht direkt mit der Eg5-inhibierenden Wirkung von Monastrol interferiert. Um
auszuschließen, dass der Phänotyp von 28H16 darauf beruht, die Aufnahme von Monastrol in die
Zelle zu verhindern bzw. dessen Export aus der Zelle zu fördern, wurde untersucht, ob 28H16
auch den durch die Depletion von Eg5 induzierten monopolaren Phänotyp unterdrücken kann.
Diese Untersuchungen ergaben, dass 28H16 tatsächlich in der Lage ist, die Bildung bipolarer
Spindeln in Eg5-depletierten Zellen zu induzieren.
Im Fokus gegenwärtiger Forschung steht die Identifizierung des Zielproteins von 28H16. Hierzu
wurden zum einen in vitro untersucht, ob 28H16 Antagonisten von Eg5 inhibiert. Diese
Untersuchungen ergaben jedoch keine Erkenntnisse über das Zielprotein von 28H16. Daher
besteht der nächste Schritt in der Affinitätsaufreinigung der Bindungspartner von 28H16. Erste
Struktur-Aktivitäts-Untersuchungen brachten Erkenntnisse darüber, über welche funktionelle
Gruppe, 28H16 immobilisiert werden kann, ohne jedoch die biologische Aktivität der
Verbindung zu verlieren. Eine strukturell ähnliche, jedoch biologisch inaktive Verbindung, dient
als negative Kontrolle der massenspektrometrischen Identifizierung von 28H16
Bindungspartnern. Aufgrund des sehr spezifischen Phänotyps, gehen wir davon aus, dass 28H16
ein wertvolles Hilfsmittel für die Untersuchung des bipolaren Spindelaufbaus in Säugetierzellen
sein wird. Diese Erkenntnisse werden auch zu einem besseren Verständnis darüber beitragen,
wie Defekte im Spindelaufbau zu der Entstehung aneuploider Zellen führen.

PD Dr. Patrick Michl
Universitätsklinikum Gießen und Marburg
Baldingerstr 1
35033 Marburg
Identification of targets acting synergistically with erlotinib in pancreatic cancer using a
kinome-wide loss-of-function screen
Background: Pancreatic cancer is characterized by a high degree of resistance to
chemotherapy. The EGFR inhibitor erlotinib is the only small-molecule inhibitor which was
shown to provide a small survival benefit in combination with cytotoxic chemotherapy.
Objectives: Identification of targets whose inhibition acts synergistically with erlotinib,
thereby overcoming drug resistance in pancreatic cancer and identification of drugs
interfering with these targets.
Methods: We employed a kinome-wide siRNA-based loss-of-function screen in pancreatic
cancer cells in the presence or absence of erlotinib to identify kinases that are synergistically
lethal with erlotinib. Targets were characterized individually by various in vitro assays using
knock-down and overexpression strategies.
Results: 9 out of 779 tested kinases led to a synthetic lethality in combination with erlotinib.
The effect on cell viability could be verified individually after knock-down of all 9 kinases.
Two of them, the kinases SNF1L and RPS6KA2, were characterized in greater detail. Knock-
down of SNF1L predominantly induced apoptosis, whereas overexpression conferred
significant rescue from drug-induced apoptosis. RPS6K2 was shown to act downstream ERK,
and knock-down of RPS6K2 by siRNA or by using a specific inhibitor led to cell cycle arrest,
whereas RPS6K2 activation significantly enhanced cell cycle progression. Work is in
progress to set up a compound screen to identify drugs that can interfere with RPS6K2
activity in vivo.
Conclusion: By applying a synergistic lethality screen using a kinome-wide RNAi-library
approach, we identified SNF1L and RPS6KA2 as potential drug targets whose inhibition
synergistically enhanced the effect of erlotinib on tumor cell growth and survival. These
kinases therefore represent promising drug candidates suitable for the development of specific
inhibitors for pancreatic cancer therapy.
Outlook: A compound screen will be applied to identify inhibitors which are feasible for in
vivo application in appropriate mouse models to explore synergistic targeting of EGFR and
RPS6KA2.

PD Dr. Markus Möhler
Medical Center of Johannes
Gutenberg-University Mainz
First Department of Internal Medicine
Langenbeckstraße 1
55101 Mainz
University Education
1986-1992 Heidelberg University and organisation of lectures entitled ‘Ethics in
Medicine‘
1991 Junior assistant at Basel University Hospital, Switzerland
1992 Albert Einstein College of Yeshiva University, New York
1993 USMLE
1994 Doctoral dissertation supervised by Nobel Laureate Prof. Dr. H. zur
Hausen, German Cancer Research Center
1993 – 1996 Medical Residency at Heidelberg University
1997 – 1998 2-year Post-Doc at the German Cancer Research Center, Division of Tumor
Biochemistry and Applied Tumor Virology, Germany
Since 1999 Mainz University Hospital, Gastroenterology, Nephrology; Prof. P.R. Galle
Degree Internal Medicine and ESMO Certificate
Colorectal cancer group member (Arbeitsgemeinschaft Internistische
Onkologie)
February 2005 Junior Professorship (Habilitation; PhD), Degree of Gastroenterology,
Consultant for Gastroenterology and GI Oncology (Oberarzt)
2007 Steering board for gastric cancer in DGVS
Nov. 2007 EORTC GI Board Member, Organisation of EORTC GI Group Meeting in
Mainz
2008-2010 Team leader of the German Upper GI Cancer group of (AIO)

2008-2011 Coordinator of Nation-wide guideline in diagnosis and therapy of gastric
cancer
since 2009 Head of Gastrointestinal Oncology Unit at Mainz University
2012 Professorship at Mainz University
Grants/Awards
1998–1993 Grant from the Cusanuswerk Study Foundation
1995 Doctorate Students Award by DGHO, Hamburg
1996 Travel Award of the American Association of Cancer Research
2003-2005 3 Merit Awards of the American Society of Clinical Oncology or ASCO GI
CG Schmidt- Award West-German Tumor-Center, Essen
2007 Best Poster Award of Gastrointestinal Oncology, Kreta, Greece
Memberships
American Society of Clinical Oncology (ASCO),European Society of Medical Oncology
(ESMO),
GI Group of EORTC steering board,German Society of Intern Oncology (AIO),
Editorial Support for Journals and Institutions
International Journal of Cancer, Lancet Oncology,BMC Cancer, Clinical Cancer Research
Cancer Research etc.
Appraisals for Societies
Arbeitsgemeinschaft Internistische Onkologie AIO, Deutsche Forschungsgemeinschaft,
German Cancer Society, Cancer UK, Sander Stiftung, Belgian Cancer Society, Spanish
RETIC,EORTC etc.

Novel 3-Azaindolyl-4-Aryl Maleimides, so called Moguntinones
– new selective inhibitors for treating solide tumours
Moehler M. 1
, Maderer A.1, Plutizki S.
2, Khillimberger K.
1, Kindler, T.
1,Dannhardt G.
2,
1University of Mainz, Departments of Internal Medicine, Mainz, Germany
2University of Mainz, Institute of Pharmacy, Mainz, Germany
Tumor growth and metastasis is highly associated with the overexpression of protein kinases
(PK) regulating tumor cell growth, apoptosis resistance and prolonged cell survival. Novel
azaindolyl-maleimides have been characterized and developed by the Mainz University
research team. These so called Moguntinones are new innovative, synthetic designed small
molecules, which are a combination of three natural products. These patent-protected kinase
inhibitors, invented by the Institute of Pharmacy and our Department of Internal Medicine,
Mainz. display a new generation of targeted inhibitors for tumour progression, angiogenesis
suppression and tumor resistance.
So far, the substances were analysed by kinase assays and HET-CAM as well as in vitro in
human colon cancer and gastric cancer cell lines for RNA and protein expression levels.
Different viability and apoptosis assays were performed in the tumour cells, which were
incubated with Moguntinones and different cytostatic drugs. Additionally, intracellular
signalling pathways were analysed. In vitro data were further verified in a human xenograft
NOD/SCID mouse model.
Moguntinones showed clear anti-angiogenic effects in HET-CAM assays and inhibitory
activities in IC50 kinase assays. After generating additional substances with little structural
changes and better biological effects, these Moguntinones alone induced apoptosis only in
higher concentrations (>10µM). Furthermore, stronger synergistic effects for induction of
apoptosis were observed in lower concentrations (<10µM) in combination with classical
cytostatic drugs like irinotecan. The signalling pathways AKT or FAK were totally inhibited
in incubated tumour cells. The in vivo mouse model again showed significant reductions in
tumour growth and tumour weight. Even more, comparable suppressive effects of
Moguntinones in KRAS, BRAF and PI3KCA-mutated colon and gastric cancer cell models
were identified. Taken together, these novel azaindolyl-aryl-maleimides, members show
remarkable inhibition of angiogenesis in tandem with potent inhibitory efficacy against
different tyrosine kinases, related to cell growth, proliferation, survival, cell migration and
angiogenesis in malignant tissues.
Additionally, inhibition of VEGFR-2 and FMS-like tyrosine kinase-3 (FLT-3) was detected in
nanomolar ranges. Further investigations disclosed a likewise inhibitory potency against
glycogen synthase kinase-3 (GSK-3). In addition, the compounds exhibited potent anti-
proliferative and pro-apoptotic properties in different carcinoma cell lines, such MOLM-14.
Based on the so far presented high potency of moguntinones on in vitro assays and in vivo
chick embryo assay, prospective studies are on the way to improve the pharmacological
profile leading to novel drug candidates. The experiments argue also for a high potency of
these substances to complement standard combinations and to overcome possible tumour
resistance mechanisms. Thus, the consortium aims to develop these substances in clinical
phase I studies.

Prof. Dr. rer. nat. Roland Hans Stauber
Molecular and Cellular Oncology/Mainz Screening Center (MSC),
Medical University Mainz
Langenbeckstr. 1, D-55016 Mainz
Phone: +49 6131-17-7002
Fax: +49 6131-17-6671
e-Mail: [email protected]
EDUCATION
INSTITUTION AND LOCATION DEGREE YEAR(s) FIELD OF
STUDY
University of Würzburg Dipl. Biology 1989 Molecular
Virology
University of Würzburg Dr. rer. nat. 1994 Molecular
Virology
University of Erlangen Priv. Doz. 1999 Virology
PROFESSIONAL APPOINTMENTS
1994 - 1997 Postdoctoral Fellow, National Cancer Institute (USA)
1997 - 2001 Group Leader, Institut für Klinische und Molekulare Virologie, University
Erlangen-
2001 - 2006 Group Leader, Coordinator of the NGFN1+2 CancerNet Georg-Speyer-Haus,
Frankfurt
since 2006 Univ.-Prof. for Molecular and Cellular Oncology (W2), Medical University Mainz
since 2012 Coordinator of the Chemical BioMedicine consortium in Mainz
AWARDS
1994 - 2000 BMBF-Fellowship Infectiology
1999 Habilitation Fellowship Walter und Sibylle Kalkhof-Rose-Stiftung
2010 Alexander Karl Award for Cancer Research
5 KEY PUBLICATIONS
1. Bier C, Knauer SK, Wünsch D, et al. Allosteric inhibition of Taspase1's
pathobiological activity by enforced dimerization in vivo. The FASEB J 2012;fj.11-
202432.
2. Bier C, Knauer SK, Klapthor A, et al. Cell-based analysis of structure-function
activity of threonine aspartase 1. J Biol Chem 2011;286(4):3007-17.
3. Tenzer S, Docter D, Rosfa S, et al. Nanoparticle Size Is a Critical Physicochemical
Determinant of the Human Blood Plasma Corona: A Comprehensive Quantitative
Proteomic Analysis. ACS nano 2011;5(9):7155-67.

4. Engels K, Knauer SK, Loibl S, et al. NO signaling confers cytoprotectivity through the
survivin network in ovarian carcinomas. Cancer Res 2008;68(13):5159-66.
5. Stauber RH, Mann W, Knauer SK. Nuclear and cytoplasmic survivin: molecular
mechanism, prognostic, and therapeutic potential. Cancer Res 2007;67(13):5999-6002.
Chemistry-enforced strategies to target the cancer relevant protease Taspase1
Proteases play critical roles in numerous (patho)biological processes, including neoplastic
diseases that in principle could be medicated by chemico-genetic protease modulators.
Hence, proteases are clinically relevant and accepted drug targets for the pharmaceutical
industry. In this respect, human Taspase1 encodes a protease of 420 amino acids (aa)
cleaving substrates in trans by recognizing a conserved peptide motif. Taspase1 is
overexpressed in numerous liquid and solid human cancers suggesting that Taspase1 is
coopted to promote and sustain tumorigenesis. However, the detailed molecular mechanisms
how Taspase1 drives oncogenesis are still not understood. Unfortunately, Taspase1’s
catalytic activity is not affected by common protease inhibitors and neither small molecule
inhibitors nor effective in vitro and in vivo assays for this enzyme were available to dissect
Taspase1’s pathobiological impact in vivo 1.
To overcome these limitations, we seeked to target Taspase1’s oncogenic potential by: (i)
developing a cell-based Taspase1 assays (CBA) to study Taspase1 function in living cells; (ii)
adapting the CBA on a HTS platform to identify potential small molecule Taspase1 inhibitors;
(iii) testing strategies aiming to inhibit Taspase1 by chemical decoys modulating Taspase1’s
dimerization.
(i) The spatial and functional division into the nucleus and the cytoplasm was exploited to
design a translocation-based Taspase1-biosensor (BioTasp) assay. The BioTasp indicator
protein is composed of GST, GFP, combinations of a nuclear import (NLS) and an export
signal (NES), combined with a
Taspase1 cleavage site. The fusion
proteins localize predominantly to the
cytoplasm, whereas Taspase1-
mediated cleavage liberates the NES,
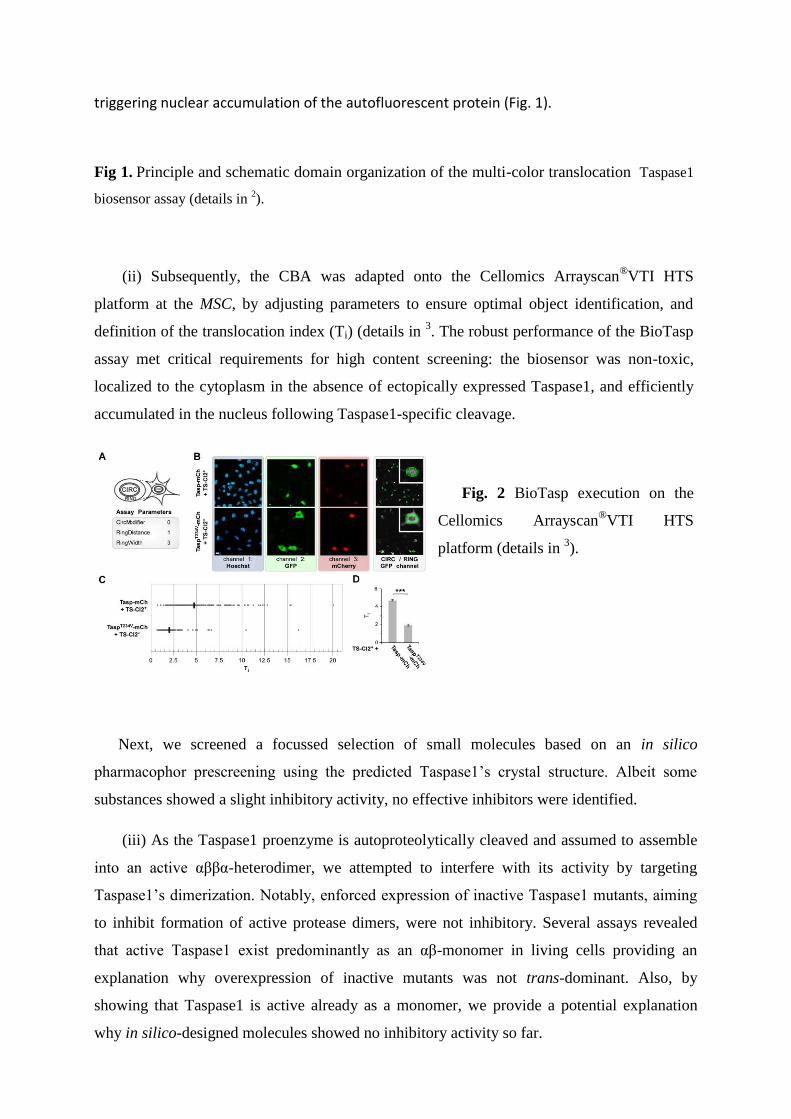
triggering nuclear accumulation of the autofluorescent protein (Fig. 1).
Fig 1. Principle and schematic domain organization of the multi-color translocation Taspase1
biosensor assay (details in 2).
(ii) Subsequently, the CBA was adapted onto the Cellomics Arrayscan®VTI HTS
platform at the MSC, by adjusting parameters to ensure optimal object identification, and
definition of the translocation index (Ti) (details in 3. The robust performance of the BioTasp
assay met critical requirements for high content screening: the biosensor was non-toxic,
localized to the cytoplasm in the absence of ectopically expressed Taspase1, and efficiently
accumulated in the nucleus following Taspase1-specific cleavage.
Fig. 2 BioTasp execution on the
Cellomics Arrayscan®
VTI HTS
platform (details in 3).
Next, we screened a focussed selection of small molecules based on an in silico
pharmacophor prescreening using the predicted Taspase1’s crystal structure. Albeit some
substances showed a slight inhibitory activity, no effective inhibitors were identified.
(iii) As the Taspase1 proenzyme is autoproteolytically cleaved and assumed to assemble
into an active αββα-heterodimer, we attempted to interfere with its activity by targeting
Taspase1’s dimerization. Notably, enforced expression of inactive Taspase1 mutants, aiming
to inhibit formation of active protease dimers, were not inhibitory. Several assays revealed
that active Taspase1 exist predominantly as an αβ-monomer in living cells providing an
explanation why overexpression of inactive mutants was not trans-dominant. Also, by
showing that Taspase1 is active already as a monomer, we provide a potential explanation
why in silico-designed molecules showed no inhibitory activity so far.

partners the To alternatively test the biological consequences of enforced dimerization,
we engineered Taspase1 variants containing the Jun/Fos dimerization motif. In absence of
the respective interaction protease fusions were fully active,
while enforcing dimerization by co-expression significantly
inhibited processing of several target proteins in living
cells. Our results support strategies aiming to inhibit the
cancer promoting activity of Taspase1 by the identification
of chemical decoys enforcing its dimerization 4,5
(Fig. 3).
Fig. 3. Model illustrating allosteric inhibition of Taspase1
by chemical-enforced dimerization (details in 5).
Collectively, we provided novel insights into the pathobiological functions of Taspase1
and developed assay systems tailored to systematically identify the first Taspase1 inhibitors.
Therefore, we are planning to employ cell based HTS using synthetic as well as natural
compound libraries in collaboration with AIO partners.
Cooperating partners:
HIPS
FMP Berlin
Prof. Dr. Thines/Möhler (Mainz)/Bode (Frankfurt)
Prof. Dr. Rademann/Leipzig
References
(1)Stauber, R. H.; Bier, C.; Knauer, S. K. Cancer Research 2012, doi: 10.1158/0008-
5472.CAN-12-0150.
(2)Bier, C.; Knauer, S. K.; Klapthor, A.; Schweitzer, A.; Rekik, A.; Kramer, O. H.;
Marschalek, R.; Stauber, R. H. J Biol Chem 2011, 286, 3007-17.
(3)Knauer, S. K.; Fetz, V.; Rabenstein, J.; Friedl, S.; Hofmann, B.; Sabiani, S.; Schröder,
E.; Kunst, L.; Proschak, E.; Thines, E.; Kindler, T.; Schneider, G.; Marschalek, R.; Stauber,
R. H.; Bier, C. PlosOne 2011, (DOI: 10.1371/journal.pone.0018253).
(4)Bier, C.; Hecht, R.; Kunst, L.; Scheiding, S.; Wunsch, D.; Goesswein, D.; Schneider,
G.; Kramer, O. H.; Knauer, S. K.; Stauber, R. H. PLoS One 2012, 7, e34142.
(5)Bier, C.; Knauer, S. K.; Wünsch, D.; Kunst, L.; Scheiding, S.; Kaiser, M.; Ottmann,
C.; Kramer, O. H.; Stauber, R. H. The FASEB J. 2012, fj.11-202432.








