
Einfluss der Struktur auf die Eigenschaften der angeregten
Zustände von Bisazomethinfarbstoffen
Bachelorarbeit
Eingereicht bei: Prof. Dr. Andreas Dreuw
Betreut von: Dipl. Chem. Jürgen Plötner
Vorgelegt von: Michael Schönnenbeck
geboren am 14.03.1984
in München
Angefertigt im Institut für Physikalische und Theoretische ChemieJohann-Wolfgang-Goethe-Universität Frankfurt a.M.
2. März 2009


Inhaltsverzeichnis
1. Motivation und Einführung ......................................................................................................................1
2. Photochemische Grundlagen.....................................................................................................................2
2.1 Elektronische Anregung von Molekülen............................................................................................2
2.2 Verhalten von Molekülen im elektronisch angeregten Zustand.......................................................2
2.2.1 Fluoreszenz...................................................................................................................................2
2.2.2 Phosphoreszenz............................................................................................................................3
2.2.3 Internal Conversion.....................................................................................................................4
2.2.4 Intramolekulare Reaktionen im angeregten Zustand...............................................................5
3. Theoretische Methodologie........................................................................................................................6
3.1 Quantenchemische Methoden.............................................................................................................6
3.2 Hartree-Fock-Theorie.........................................................................................................................7
3.3 Configuration Interaction (CI)...........................................................................................................8
3.4 Møller-Plesset-Störungstheorie...........................................................................................................8
3.5 Dichtefunktionaltheorie (DFT)...........................................................................................................9
3.5.1 Das erste Hohenberg-Kohn-Theorem........................................................................................9
3.5.2 Das zweite Hohenberg-Kohn-Theorem....................................................................................10
3.5.3 Der Kohn-Sham-Ansatz.............................................................................................................11
3.6 Zeitabhängige Dichtefunktionaltheorie (TDDFT)...........................................................................12
3.7 Übersicht über verwendete Methoden und Basissätze....................................................................13
4. Berechnungen an 2,2'-Dihydroxy-1,1'-diphenylaldazin.........................................................................14
4.1 Vergleich von Methoden und Funktionalen.....................................................................................14
4.2 Rotation um die CNNC-Bindung im Grundzustand.......................................................................20
4.3 Rotation um die CNNC-Bindung im ersten angeregten Zustand...................................................20
4.4 Intramolekularer Protonentransfer im angeregten Zustand..........................................................24
5. Berechnungen an 1-(Ethen-2-ol)-1'-(1'-methylprop-1'-en-2'-ol)-aldazin..............................................26
5.1 Vergleich von Basissätzen..................................................................................................................26
5.2 Suche nach stabilen Gleichgewichtsstrukturen...............................................................................28
5.3 Rotation um die CNNC-Bindung im Grundzustand.......................................................................30
5.4 Rotation um die CNNC-Bindung im ersten angeregten Zustand...................................................31
5.5 Intramolekularer Protonentransfer im angeregten Zustand .........................................................35
6. Zusammenfassung und Ausblick.............................................................................................................41
7. Literaturverzeichnis.................................................................................................................................43


Michael Schönnenbeck 02.03.09 Bachelorarbeit
1. Motivation und Einführung
In der vorliegenden Arbeit sollen die angeregten Zustände verschiedener Bisazomethinfarbstoffe untersucht
werden. Den Anreiz dafür lieferten bisherige Untersuchungen an Pigment Yellow 101 (P.Y.101, Abbildung
1), dass eine überraschend komplexe Photochemie aufweist. Diese beinhaltet die Konkurrenz zwischen cis-
trans-Isomerisierung und intramolekularem Protonentransfer im angeregten Zustand (ESIPT). Ein weiteres
interessantes Phänomen ist die Fluoreszenz des Moleküls, die bei organischen Pigmenten im Festkörper in
der Regel ausbleibt.1,2,3,4
Ziel der Untersuchungen ist es, herauszufinden inwiefern sich die Struktur auf das optische Verhalten der
Bisazomethinfarbstoffe auswirkt. Dazu wurden Berechnungen an folgenden P.Y.101-Derivaten mit
verkleinerten Systemen durchgeführt:
1
Abbildung 1: Lewisstruktur von Pigment Yellow 101.
Abbildung 2: 1: 2,2'-Dihydroxy-1,1'-diphenylaldazin und 2: 1-(Ethen-2-ol)-1'-(1'-methylprop-1'-en-2'-ol)-aldazin.

Michael Schönnenbeck 02.03.09 Bachelorarbeit
2. Photochemische Grundlagen
In diesem Abschnitt sollen zum Einen die elektronische Anregung von Molekülen, und zum Anderen die
intramolekularen Reaktionen im angeregten Zustand erläutert werden. Diese beinhalten strahlende und
strahlungslose Übergänge in niedrigere elektronische Zustände, sowie Geometrieänderungen der Moleküle.
2.1 Elektronische Anregung von Molekülen
Organische Moleküle mit ausgedehnten π-Systemen können einfallendes Licht im Bereich von 1250 nm bis
125 nm unter Anregung des Elektronensystems absorbieren. Der Spektralbereich für elektronische Anregung
beinhaltet also hauptsächlich ultraviolettes und sichtbares Licht, sowie einen kleinen Anteil des infraroten
Bereiches. Bei der Absorption wird die Energie eines einfallenden Photons vom Elektronensystem
aufgenommen. Das führt dazu, dass Elektronen aus einem besetzten in ein unbesetztes Orbital angehoben
werden. Wird das absorbierende Molekül mit weißem Licht bestrahlt, erscheint es für das menschliche Auge
in der Komplementärfarbe des absorbierten Lichtes.5
Damit ein elektronischer Übergang zwischen zwei Zuständen Si und Sj erlaubt ist, muss für das elektronische
Übergangsdipolmoment gelten:
=⟨Si∣ ∣S j ⟩≠0 (1)
Dies hat zur Folge, dass nur Übergänge zwischen Zuständen unterschiedlicher Symmetrie stattfinden können
(gerade (g) nach ungerade (u) bzw. u nach g). Bei dem Übergang bleibt die Multiplizität erhalten.6,7
Elektronische Übergänge gehorchen dem Franck-Condon-Prinzip.8,9 Aufgrund der ca. 2000 Mal größeren
Masse der Kerne reagieren diese träge auf die Änderung der Elektronenstruktur, sodass der Übergang
„senkrecht“ erfolgt. Die Anregung endet im sogenannten Franck-Condon-Punkt der Energiehyperfläche des
angeregten Zustandes. Dieser entspricht in der Regel auch einem vibronisch angeregten Zustand. Wenn die
kinetische Energie an die Umgebung abgegeben werden kann, relaxiert das Molekül in den
Schwingungsgrundzustand.5
2.2 Verhalten von Molekülen im elektronisch angeregten Zustand
2.2.1 Fluoreszenz
Die einfachste Möglichkeit für ein Molekül in den Grundzustand zurückzukehren ist die Fluoreszenz.
Nachdem das Molekül angeregt wurde, relaxiert es zuerst in den Schwingungsgrundzustand und
anschließend innerhalb von 10-10 bis 10-7 s in den elektronischen Grundzustand. Dabei wird die überschüssige
Energie in Form von Strahlung abgegeben. In der Regel ist die emittierte Strahlung gegenüber der
2
Michael Schönnenbeck 02.03.09 Bachelorarbeit
absorbierten Strahlung rotverschoben (Stokes-Shift).6
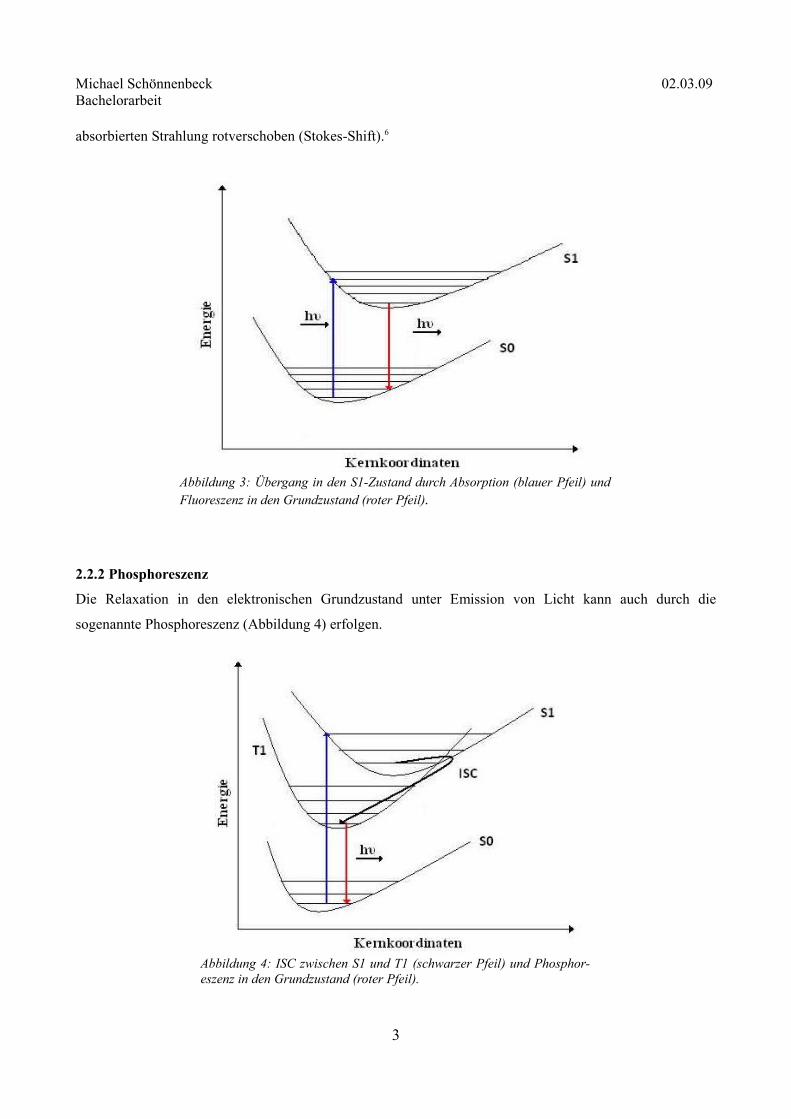
2.2.2 Phosphoreszenz
Die Relaxation in den elektronischen Grundzustand unter Emission von Licht kann auch durch die
sogenannte Phosphoreszenz (Abbildung 4) erfolgen.
3
Abbildung 3: Übergang in den S1-Zustand durch Absorption (blauer Pfeil) und Fluoreszenz in den Grundzustand (roter Pfeil).
Abbildung 4: ISC zwischen S1 und T1 (schwarzer Pfeil) und Phosphor-eszenz in den Grundzustand (roter Pfeil).

Michael Schönnenbeck 02.03.09 Bachelorarbeit
Phosphoreszenz tritt auf, wenn sich die Potentialflächen eines angeregten Singulettzustandes und eines
Triplettzustandes überschneiden. Dies wird als ISC (engl. Intersystem Crossing) bezeichnet. Hierbei wird das
Spinumkehrverbot (vgl. Abschnitt 2.1) gebrochen und das Molekül geht strahlungslos in den Triplettzustand
über. Damit es wieder in den Grundzustand gelangen kann, muss sich die Multiplizität erneut ändern, wobei
die Energie analog zur Fluoreszenz in Form von rotverschobener Strahlung abgegeben wird. Allerdings
dauert der Vorgang aufgrund der geringen Übergangswahrscheinlichkeit zwischen Singulett- und
Triplettzustand wesentlich länger als die Fluoreszenz (10-2 bis 104 s).6
2.2.3 Internal Conversion
Kommt es zu einer Kreuzung zweier Potentialflächen, z.B. der des Grundzustandes (S0) und des ersten
angeregten Zustandes (S1), kann ein strahlungsloser Übergang stattfinden. Man spricht dann von einer
internal conversion (IC). Ein Molekül kann beispielsweise von S0 nach S1 angeregt werden, und anschließend
entlang einer Normalmode in den Grundzustand zurückkehren, wenn sich die Potentialflächen entlang der
Reaktionskoordinate kreuzen. Die überschüssige Energie wird dann in Form von Wärme an die Umgebung
abgegeben (Abbildung 5a)).5
Bei einer Kreuzung zweier angeregter Potentialflächen kann es passieren, dass das Molekül in einen Zustand
relaxiert, aus dem ein Übergang in den Grundzustand optisch verboten ist. Da ein solcher Zustand
spektroskopisch nicht nachweisbar ist, bezeichnet man ihn auch als „dunklen Zustand“ (engl. Dark State,
Abbildung 5b)). Ein Übergang in den Grundzustand ist nur durch Stoßdeaktivierung unter Wärmeabgabe
möglich. Daher kann es - besonders in der Gasphase - sehr lange dauern, bis der angeregte Zustand zerfällt.7
4
Abbildung 5: a) Strahlungslose Relaxation in den Grundzustand und b) Übergang in einen dark State.

Michael Schönnenbeck 02.03.09 Bachelorarbeit
2.2.4 Intramolekulare Reaktionen im angeregten Zustand
Die gegenüber dem Grundzustand veränderte Elektronenstruktur eines angeregten Moleküls kann dazu
führen, dass intramolekulare Reaktionen stattfinden, die im Grundzustand nicht möglich sind. Dies kann z.B.
ein intramolekularer Protonentransfer (ESIPT, Excited State Intramolecular Proton Transfer) von einer OH-
Gruppe auf ein Stickstoffatom sein. Solche Mechanismen sind in der Natur häufig wiederzufinden.
Beispielsweise wurde festgestellt, dass die DNA in der Lage ist, durch einen ESIPT aus dem angeregten
Zustand unmittelbar in den Grundzustand zu relaxieren. Dies geschieht durch eine IC entlang der O-H-
Reaktionskoordinate. Die DNA ist so in der Lage, sich vor Mutation durch Lichteinstrahlung zu schützen.10
In der Natur gibt es noch zahlreiche weitere Reaktionen, die auf der optischen Anregung von Biomolekülen
beruhen, von denen z.B. das Sehvermögen der Augen oder die Photosynthese der Pflanzen abhängen.
Eine weitere intramolekulare Reaktion im angeregten Zustand ist die cis-trans-Isomerisierung.
Beispielsweise lässt sich das Azobenzolmolekül durch Lichteinstrahlung gezielt um die N=N-Bindung
isomerisieren. Das Molekül wird durch die Lichteinstrahlung in einen elektronisch angeregten Zustand
versetzt und geht anschließend, je nach eingestrahlter Wellenlänge, in die cis- oder trans-Konformation über.
Diese herausragende Eigenschaft wird in der Technik bereits für wiederbeschreibbare CDs bzw. DVDs
verwendet. In der Forschung erhofft man sich, Azobenzolderivate als Nanoschalter oder als Antrieb für
Nanomotoren einzusetzen. Tatsächlich ist es bereits gelungen, die gezielte Bewegung von Mikrokügelchen
aus Polystyrol auf Flüssigkristalloberflächen zu induzieren, die mit Azobenzol dotiert sind.11 Weitere
technische Anwendungsgebiete, die auf der optischen Anregung basieren, sind z.B. die Fotografie,
Leuchtdioden (LEDs), Laser, Lichtsensoren uvm.
5

Michael Schönnenbeck 02.03.09 Bachelorarbeit
3. Theoretische Methodologie
In diesem Abschnitt sollen die in der vorliegenden Arbeit verwendeten mathematischen Methoden zur
Untersuchung der Grund- und der angeregten Zustände verschiedener Bisazomethinfarbstoffderivate
erläutert werden. Auf eine genaue Herleitung wird verzichtet, nähere Informationen zu den einzelnen
Methoden sind in den jeweiligen Literaturverweisen enthalten.
3.1 Quantenchemische Methoden
Um den Grundzustand eines N-Elektronensystems mit K Kernkoordinaten quantenmechanisch zu
beschreiben, müssen die Wechselwirkungen aller Elektronen untereinander, aller Kerne untereinander, sowie
die Wechselwirkungen zwischen allen Elektronen und Kernen berechnet werden. In der Praxis geschieht dies
durch Lösen der zeitunabhängigen Schrödingergleichung12
H R,rR,r =ER,rR,r . (2)
Hier ist R,r die Wellenfunktion des Systems in Abhängigkeit der Kernkoordinaten R und der
Elektronenkoordinaten r. Der Hamiltonoperator H R,r beschreibt die kinetische Energie aller Elektronen,
alle Kern-Kern- und Elektron-Elektron-Wechselwirkungen, sowie alle Wechselwirkungen zwischen den
Kernen und Elektronen. Die Schrödingergleichung ist jedoch nur für ein Zweikörperproblem, z.B. ein
Wasserstoffatom exakt lösbar. Um sie auch für ein System mit mehreren Kernen und Elektronen lösen zu
können, muss von geeigneten Näherungen Gebrauch gemacht werden.
Die grundlegende Näherung, die in jeder quantenmechanischen Rechnung verwendet wird, ist die Born-
Oppenheimer-Näherung13. Diese macht von der Tatsache Gebrauch, dass sich ein Elektron aufgrund seiner
etwa 2000 mal kleineren Masse wesentlich schneller bewegt als ein Kern, und sich somit unmittelbar an die
Änderung der Kernkoordinaten anpasst. Die Born-Oppenheimer-Näherung erlaubt also eine Separation der
elektronischen und der nuklearen Wellenfunktion. Somit kann die Schrödingergleichung auf die Form
H Rf ,rRf ,r=E Rf ,rRf ,r (3)
gebracht werden, die nun von den Elektronenkoordinaten r und den fixierten Kernkoordinaten Rf abhängt.
Damit kann die Schrödingergleichung numerisch gelöst werden.
Die Born-Oppenheimer-Näherung verliert jedoch ihre Gültigkeit, wenn die Bewegungen der Kerne und
Elektronen nicht mehr getrennt betrachtet werden können. Das ist beispielsweise der Fall, wenn eine
konische Durchschneidung zweier Potentialflächen vorliegt. In einem solchen Fall muss genau untersucht
werden, inwiefern die Lösung der Schrödingergleichung unter Verwendung der Born-Oppenheimer-
Näherung noch zuverlässige Ergebnisse liefert.6,14
6

Michael Schönnenbeck 02.03.09 Bachelorarbeit
3.2 Hartree-Fock-Theorie
Ein wichtiges Verfahren zur Lösung der Schrödingergleichung ist die Hartree-Fock-Theorie14. Die
Gesamtwellenfunktion wird hier mittels Linearkombination aus den Einelektronenwellenfunktionen des
Wasserstoffatoms konstruiert, die durch die Spinorbitale i gegeben sind. Diese können z.B. Slater- oder
Gaussianorbitale sein.
Das Pauli-Prinzip6 besagt, dass die Gesamtwellenfunktion antisymmetrisch bezüglich der Vertauschung
zweier Elektronen ist. Eine wichtige Konsequenz daraus ist, dass zwei Elektronen nicht den gleichen
Zustand besetzen können. Um die Antisymmetriebedingung zu erfüllen, werden die Spinorbitale in eine
Slaterdeterminante geschrieben, um so die Gesamtwellenfunktion zu erhalten:
1,2,3,... ,N=∣a,b,... ,l ⟩ (4)
Um nun die minimale Energie des N-Elektronensystems zu erhalten, wird die Schrödingergleichung in die
Hartree-Fock-Gleichung überführt, aus der sich die Einelektronenorbitale i berechnen lassen:
F aia=ii a (5)
Die i sind hier die Orbitalenergien im Rahmen des Koopmans-Theorems15, das besagt, dass die
Orbitalenergie genau der ersten Ionisierungsenergie des Elektronensystems entspricht. Diese Aussage ist
allerdings nur näherungsweise gültig, da sie Korrelationseffekte und die bei der Ionisierung auftretenden
Relaxationseffekte vernachlässigt. Die Hartree-Fock-Gleichung beschreibt also die Energien der Orbitale i,
besetzt mit dem Elektron a. F ist der sogenannte Fock-Operator, der sich in den Einelektronen-
Hamiltonoperator ha und die Zweielektronenoperatoren J und K zerlegen lässt. J ist der
Coulomboperator, der die elektrostatische Wechselwirkung zwischen dem Elektron a im Orbital i und
Elektron b im Orbital j beschreibt. Der Austauschoperator K beschreibt die nichtklassischen
Spinwechselwirkungen zwischen den beiden Elektronen. Zusammengefasst lautet der Fock-Operator:
F a=ha∑j
[2 J j a− K ja] (6)
Die Operatoren J und K beschreiben ein sogenanntes Meanfield-Potential HF , in dem sich das Elektron
a bewegt. Gleichung 6 gilt allerdings nur für den „Closed-Shell“-Fall, bei dem nur Systeme mit
geschlossenen Schalen betrachtet werden, deren Molekülorbitale also mit jeweils zwei Elektronen besetzt
sind.
Die Hartree-Fock-Gleichungen werden durch die SCF-Methode (Self-Consistant-Field) gelöst. Dazu werden
sie zunächst in die sogenannten Roothan-Gleichung16 überführt:
FC=SC (7)
7

Michael Schönnenbeck 02.03.09 Bachelorarbeit
Dabei ist F die Fock-Matrix, C die Koeffizientenmatrix, die es im SCF-Verfahren zu bestimmen gilt, und
S die Überlappungsmatrix, die die Überlappungsintegrale der Wellenfunktionen enthält. enthält die
einzelnen Orbitalenergien i . Das SCF-Verfahren wird iterativ gelöst: man beginnt mit einem geratenen
Wert für C , setzt ihn in Gleichung 7 ein und erhält einen neuen und besseren Wert für C . Dieser Vorgang
wird solange wiederholt, bis die Rechnung konvergiert.6,14
3.3 Configuration Interaction (CI)
Der Unterschied zwischen der exakten und der mittels Hartree-Fock berechneten Grundzustandsenergie ist
die Korrelationsenergie. Sie findet ihre Ursache in der nicht berücksichtigten Elektronenkorrelation und ist
somit ein Maß für den Fehler der Hartree-Fock-Rechnungen. Die exakte Wellenfunktion des elektronischen
Grundzustandes oder eines angeregten Zustandes kann durch Linearkombination aller Grundzustands- und
angeregten Slaterdeterminanten konstruiert werden:
0E=C00∑
a,p
Capa
p∑abpq
Ca,bp,qa,b
p,q ∑abcpqr
Ca,b,cp,q,ra,b,c
p,q,r...(8)
Die Indizes a, b und c bezeichnen hier die Elektronen in den Grundzustandsorbitalen; p, q und r bezeichnen
die Elektronen in den virtuellen Orbitalen. Der Hamiltonoperator wird in der Basis der angeregten
Determinanten dargestellt. Die anschließende Diagonalisierung der Matrix und Einsetzen in das
Variationsprinzip liefert die minimale Energie des Systems. Diese Methode bezeichnet man als
Configuration Interaction (CI, engl. Konfigurationswechselwirkung). In der Praxis ist die Gleichung 8 nur
für sehr kleine Systeme numerisch lösbar, da der Rechenaufwand exponentiell mit der Systemgröße ansteigt.
Daher werden statt einer Full-CI-Rechnung in der Regel nur Einfachanregungen (CIS), Doppelanregungen
(CID) oder Einfach- und Doppelanregungen (CISD) betrachtet.6,14
3.4 Møller-Plesset-Störungstheorie
Der Nachteil von ab-initio-Methoden, die auf der Konfigurationswechselwirkung (ausgenommen Full-CI
und CIS) beruhen, liegt darin, dass sie nicht größenkonsistent sind (size-consistensy-Problem, siehe [6],
[14]). Eine größenkonsistente Alternative zur Berücksichtigung der Elektronenkorrelation liefert die
Störungstheorie. Dabei werden die exakten Energieeigenwerte des N-Elektronensystems in einer Potenzreihe
entwickelt:
∣i ⟩=∣i ⟩∑n=0
∞
cn∣in ⟩ (9)
Ei=∑n=0
∞
Ein (10)
8

Michael Schönnenbeck 02.03.09 Bachelorarbeit
Die auf der Hartree-Fock-Theorie basierenden Störungsrechnungen sind als Møller-Plesset-Störungstheorie17
bekannt. Der Hamiltonoperator
H0= F V (11)
wird in den Fock-Operator F (Gl. 6) und den Störoperator
V=∑i j
1rij−∑
i
iHF
(12)
aufgeteilt, der alle Elektron-Elektron-Wechselwirkungen beinhaltet, die von der Hartree-Fock-Methode nicht
berücksichtigt werden. Damit ergibt sich die Hartree-Fock-Energiekorrektur zweiter Ordnung (MP2):
E02=∑
abi j
∣⟨ia∣∣ jb ⟩∣2
i j−a−b(13)
Der Vorteil von MP2-Rechnungen ist, dass der Rechenaufwand in der Regel geringer ist als bei CI-
Verfahren. Der Nachteil ist, dass die Störungstheorie nicht variationell ist und somit keine obere Grenze zur
Energie liefert.6,14
3.5 Dichtefunktionaltheorie (DFT)
Einen wichtigen Durchbruch zur Berechnung des Grundzustandes eines N-Elektronensystems lieferte die
Dichtefunktionaltheorie. Hierbei soll auf die Verwendung von Atomorbitalen verzichtet werden, und statt
dessen die Elektronendichte als zentrale Größe zur Berechnung des Systems verwendet werden. Der Vorteil
liegt darin, dass dabei nicht mehr auf eine Wellenfunktion x1,x2,. ..xN zurückgegriffen werden muss, die
von N Raumvektoren abhängt. Statt dessen soll die Energie durch ein Funktional der Elektronendichte [R]
ausgedrückt werden, dass lediglich von einem Raumvektor R abhängt. Somit kann die Dimension der zu
berechnenden Integrale um das N-fache reduziert werden. Die DFT ist im Prinzip eine exakte Methode,
allerdings lassen sich die Austausch- und Korrelationseffekte in der Praxis nicht genau bestimmen und
werden daher in einem Austausch-Korrelations-Funktional Exc [ ] zusammengefasst, dass ausschließlich
von der Elektronendichte abhängt. Die entscheidenden Grundlagen zur Dichtefunktionaltheorie wurden 1964
durch die Hohenberg-Kohn-Theoreme18 aufgestellt.
3.5.1 Das erste Hohenberg-Kohn-Theorem
Das erste Hohenberg-Kohn-Theorem besagt, dass sich alle beobachtbaren Größen eines
Vielelektronensystems im Grundzustand eindeutig durch die Elektronendichte berechnen lassen. Der dafür
geführte Beweis basiert auf folgendem Widerspruch: Gäbe es zwei verschiedene Potentiale V und V ' , die
9

Michael Schönnenbeck 02.03.09 Bachelorarbeit
zur gleichen Elektronendichte führen, würde das Variationsprinzip für die zugehörigen Energien E0 und E0'
zu folgendem Ergebnis führen:
E0E0'E0E0
' (14)
Da die Elektronendichte alle Informationen über die Elektronenzahl N, die Kernladungszahl und die
Kernabstände enthält, lässt sich daraus der Hamiltonoperator und somit die Wellenfunktion und die
Grundzustandsenergie, sowie alle weiteren Observablen eines Vielelektronensystems berechnen. Die
Grundzustandsenergie ist also ein Funktional der Elektronendichte 0 und lässt sich in ihre Komponenten
zerlegen:
E0[0]=T [0]V ee [0]V Ke [0]=∫V rr drT [0]V ee [0] (15)
Das Funktional E0[0] liefert die totale Energie des Grundzustandes, T [0] die kinetische
Elektronenenergie, Vee [0] die Elektron-Elektron-Wechselwirkung und V Ke[0] die Kern-Elektron-
Wechselwirkung des N-Elektronensystems. Die Funktionale T [0] und Vee [0] sind für jedes N-
Elektronensystem gleich und werden zum Hohenberg-Kohn-Funktional zusammengefasst:
FHK=T[0]Vee [0] (16)
Das Funktional Vee [0] lässt sich wiederum in den klassischen Coulombanteil J [0] und den Austausch-
Korrelations-Anteil V XC[0] aufteilen, womit sich für das Hohenberg-Kohn-Funktional folgende
Schreibweise ergibt:
FHK=T[0]J [0]V XC [0] (17)
Die Herausforderung der Dichtefunktionaltheorie besteht darin, Näherungen für die Funktionale T [0] und
VXC [0] zu finden, die die kinetische Energie und die Austausch-Korrelations-Energie geeignet beschreiben.
Der klassische Coulombanteil hingegen ist exakt berechenbar.14
3.5.2 Das zweite Hohenberg-Kohn-Theorem
Das zweite Hohenberg-Kohn-Theorem besagt, dass das Hohenberg-Kohn-Funktional genau dann die
niedrigste Energie für den Grundzustand eines N-Elektronensystems liefert, wenn die eingesetzte
Elektronendichte der exakten Grundzustandsdichte des Systems entspricht. Dies bedeutet, dass man durch
Einsetzen einer geratenen Elektronendichte 0,G eine Energie erhält, die eine obere Grenze zur exakten
Grundzustandsenergie liefert. Für das Variationsprinzip folgt damit:
E0[0]≤E0[0,G]=T[0,G]J [0,G]V XC [0,G] (18)
10

Michael Schönnenbeck 02.03.09 Bachelorarbeit
3.5.3 Der Kohn-Sham-Ansatz
Die anfänglichen Probleme der Dichtefunktionaltheorie bestanden darin, ein Funktional zu finden, dass die
kinetische Energie der Elektronen ausreichend gut wiedergibt, da diese nicht allein durch die
Elektronendichte beschrieben werden kann. Die Konsequenz daraus ist, dass keine chemischen Bindungen
und somit keine Moleküle beschrieben werden können. Den Ausweg dafür lieferte der 1965 veröffentlichte
Kohn-Sham-Ansatz.19 Die Idee besteht darin, das wechselwirkende N-Teilchensystem als nicht-
wechselwirkendes N-Teilchensystem darzustellen, dass die gleiche Elektronendichte besitzt wie das
wechselwirkende. Dazu wird ein Satz aus N Molekülorbitalen in eine Slaterdeterminante geschrieben, die die
Elektronendichte des Systems wiedergibt. Die Grundzustandsenergie des wechselwirkenden Systems ist
E0=TVKeJ V XC . (19)
Die Grundzustandsenergie setzt sich also aus der kinetischen Energie der Elektronen und einem effektiven
Potential
Veff=VKeJ V XC (20)
zusammen, aus dem sich theoretisch die exakte Grundzustandselektronendichte berechnen lässt. Dazu
müssen die nicht bekannten Korrelations- und Austauscheffekte durch geeignete Funktionale genähert
werden. Diese können in drei Klassen aufgeteilt werden:
– LDA-Funktionale (Lokal Density Approximation), die ausschließlich von der Elektronendichte
abhängen. Sie basieren auf dem Modell eines Elektronengases, das sich in Gegenwart einer positiven
Ladung befindet, und eine endliche Elektronendichte besitzt, die an jedem Ort einen konstanten Wert
annimmt. Dies ist in der Realität allerdings nur bei metallischen Leitern der Fall, trotzdem liefern die
LDA-Funktionale für kleine Systeme, wie z.B. Na2 gute Ergebnisse.
– GGA-Funktionale (Generalized Gradient Approximation), bei denen das Austauschfunktional als
Funktional der Elektronendichte und eines Gradienten der Elektronendichte geschrieben wird. Durch
den Zusatz des Gradienten wird das asymptotische Verhalten der Elektronendichte besser
beschrieben.
– Hybridfunktionale, bei denen der Dichtefunktionaltheorie Hartree-Fock-Anteile beigemischt werden.
Der Rechenaufwand der Dichtefunktionaltheorie ist ähnlich dem der Hartree-Fock-Methode, jedoch wird die
Elektronenkorrelation durch die DFT wesentlich besser beschrieben. Ein weiterer Vorteil besteht darin, dass
die Funktionale frei wählbare Parameter beinhalten, durch die sie an empirische Daten angepasst werden
können. Z.B. sind die Parameter des LYP-Funktionals (Lee; Yang; Parr, 1989) so gewählt, dass sie die
Korrelationsenergie des Heliumatoms möglichst gut beschreiben.20 Das hat wiederum den Nachteil, dass
DFT-Rechnungen genau dann versagen, wenn Effekte auftreten, die von den Funktionalen nicht
11

Michael Schönnenbeck 02.03.09 Bachelorarbeit
berücksichtigt werden.20
3.6 Zeitabhängige Dichtefunktionaltheorie (TDDFT)
Die Eigenschaften angeregter Zustände wurden bei dieser Arbeit hauptsächlich mit Hilfe der zeitabhängigen
Dichtefunktionaltheorie (TDDFT, Time-Dependent Density Functional Theory) berechnet. Die Grundlagen
der TDDFT sollen hier nur kurz angesprochen werden. Ausführliche Beschreibungen der Methode sind in
[21] enthalten.
Um eine Beschreibung angeregter Zustände durch die Elektronendichte zu realisieren, muss die
Dichtefunktionaltheorie in eine zeitabhängige Form überführt werden. Dies leisten die
Runge-Gross-Theoreme22, die besagen, dass ein direkter Zusammenhang zwischen dem zeitabhängigen
externen Potential und der zeitabhängigen Elektronendichte besteht. Analog zu den Hohenberg-Kohn-
Theoremen hat dies zur Folge, dass sich alle zeitabhängigen Eigenschaften eines N-Elektronensystems
eindeutig durch die zeitabhängige Elektronendichte beschreiben lassen. Dies führt direkt zu den sogenannten
„Time-Dependent-Kohn-Sham-Gleichungen“.
Unter TDDFT versteht man heute allerdings eine Methode zur Berechnung elektronisch angeregter
Zustände. Diese erhält man als „linear response“ der Grundzustandselektronendichte auf ein zeitlich
oszillierendes elektrisches Feld nach Fouriertransformation aus der Zeit in die Energiedomäne.
Die TDDFT liefert im allgemeinen qualitativ gute Ergebnisse für Anregungsenergien, die mit Ergebnissen
aufwendigerer Methoden wie z.B. Coupled Cluster (CC) vergleichbar sind. Dennoch hat die TDDFT
Schwierigkeiten, bestimmte Klassen von angeregten Zuständen richtig zu beschreiben. Dies sind z.B. Charge
Transfer- (CT) und Rydbergzustände, Doppelanregungen und große π-Systeme. Diese sind zum Teil auf
mangelhafte Korrelations-Austausch-Funktionale, aber auch auf fundamentale theoretische Ursachen
zurückzuführen.
Da die in der vorliegenden Arbeit untersuchten Moleküle π-Systeme besitzen, die sich über das gesamte
Molekül erstrecken, muss zunächst genau analysiert werden, wie genau die Ergebnisse der Berechnungen
zutreffen. Da diese Problematik stark von der Wahl der Funktionale abhängt, bietet es sich an, die
Berechnungen zum Vergleich an verschiedenen Funktionalen durchzuführen. So zeigte es sich beispielsweise
bei den Berechnungen an P.Y.101, dass sich Hybridfunktionale mit hohem HF-Anteil, wie z.B. BHLYP,
besser zur Charakterisierung von CT-Zuständen eignen, als reine GGA-Funktionale wie BP86.4
12

Michael Schönnenbeck 02.03.09 Bachelorarbeit
3.7 Übersicht über verwendete Methoden und Basissätze
Zur Berechnung der Wellenfunktionen mit Turbomole wurden in der vorliegenden Arbeit folgende
Basissätze verwendet:
SVP - Split Valence Polarisation
DZP - Double Zeta Polarisation
TZVP - Triple Zeta Valence Plus Polarisation
QZVP - Quadruple Zeta Valence Plus Polarisation
Für die TDDFT-Rechnungen wurden folgende Funktionale verwendet, um die Änderungen der Spektren mit
zunehmendem Hartree-Fock-Anteil zu untersuchen und anschließend die Reaktionen im angeregten
zuständen ausreichend genau zu beschreiben.
SVWN - Slater, Vosko, Wilk, Nuisar (LDA)
BP - Becke-88-Austausch und Perdew 86 Korrelation (GGA)
BLYP - Becke-88-Austausch und Lee-Yang-Parr-Korrelation (GGA)
B3LYP - Becke-3-Parameter: Becke-88-Austausch mit 20% Hartree-Fock-Anteil und Lee-Yang-Parr-
Korrelation (Hybridfunktional)
BHLYP - Becke-Half-and-Half: Becke-88-Austausch mit 50% Hartree-Fock-Anteil und Lee-Yang- Parr-
Korrelation (Hybridfunktional)
Zum Vergleich mit den TDDFT-Funktionalen wurden ab-initio-Rechnungen mit folgenden Methoden
durchgeführt:
CIS - Einfach angeregte Konfigurationswechselwirkungen
MP2 - Møller-Plesset-Störungstheorie, Hartree-Fock-Energiekorrektur zweiter Ordnung
Zusätzlich wurden für die Untersuchungen der Rotationskoordinate von 2 Rechnungen mit Algebraic
Diagrammatic Construction (ADC) 23 durchgeführt, das in Qchem implementiert ist. Hierfür wurde der 6-
31G*-Basissatz verwendet.
Zur grafischen Darstellung von Molekülorbitalen wurde Spartan 2004 verwendet.
13

Michael Schönnenbeck 02.03.09 Bachelorarbeit
4. Berechnungen an 2,2'-Dihydroxy-1,1'-diphenylaldazin
Im ersten Teil der Arbeit wurden verschiedene Berechnungen am P.Y.101-Derivat 1 durchgeführt, um
Ähnlichkeiten und Unterschiede in den Eigenschaften der elektronisch angeregten Zustände von P.Y.101 zu
untersuchen. Beginnend mit der Optimierung des Grundzustandes wurden zunächst mehrere DFT-
Funktionale miteinander verglichen. Außerdem wurde der Grundzustand mit Hartree-Fock und MP2
optimiert. Zusätzlich wurden die die UV- und IR-Spektren des Moleküls berechnet (Abschnitt 4.1)
Da bei den Untersuchungen von P.Y.101 eine cis-trans-Isomerisierung im angeregten Zustand beobachtet
wurde, sollte die Rotation um den CNNC-Torsionswinkel auch hier untersucht werden (Abschnitt 4.3). Die
angeregte Potentialfläche von P.Y.101, die mit dem GGA-Funktional BP86 berechnet wurde, zeigt ein
artifizielles Minimum bei der gewinkelten Struktur. Daher wurden zu den Untersuchungen an 2,2'-
Dihydroxy-1,1'-diphenylaldazin neben dem GGA-Funktional BLYP auch das Hybridfunktional BHLYP
verwendet, um eventuell auftretende Artefakte zu erkennen. Um einen vollständigen Vergleich zu erzielen,
wurde auch die Rotation um den CNNC-Torsionswinkel im Grundzustand genau untersucht (Abschnitt 4.2).
Außerdem wurde der Protonentransfer im ersten angeregten Zustand von einer OH-Gruppe auf die
Bisazomethinbindung untersucht (Abschnitt 4.4), da dieses Phänomen ebenfalls bei P.Y.101 beobachtet
wurde.
4.1 Vergleich von Methoden und Funktionalen
Zur Berechnung des Grundzustandes von 2,2'-Dihydroxy-1,1'-diphenylaldazin wurde zunächst die trans-
Konformation des Moleküls optimiert. Für die Funktionalstudie wurde der SVP-Basissatz gewählt. Die
Grundzustandsoptimierung und die Berechnung der Anregungsenergien mit TDDFT erfolgten mit den
Funktionalen SVWN, BP, B3LYP und BHLYP. Der Grundzustand wurde außerdem mit Hartree-Fock und
MP2 optimiert. Die Anregungsenergien der Hartree-Fock-Geometrien wurden mit CIS berechnet, die der
MP2-Geometrien mit TDDFT/B3LYP/SVP.
Alle Geometrieoptimierungen führten zu einer planaren Gleichgewichtsstruktur in trans-Stellung und diol-
Form. Aus den Ergebnissen wurde ein Vergleichsspektrum erstellt, das die UV-Absorptionsbanden des
Moleküls zeigt (Abbildung 6). Die aus den TDDFT-Spektren resultierenden UV-Spektren sind mit
zunehmendem Hartree-Fock-Anteil blauverschoben. Die mit B3LYP-Funktional berechneten
Anregungsenergien der MP2-Optimierungen weisen ein ähnliches Spektrum auf, wie das der B3LYP-
Optimierung.
14

Michael Schönnenbeck 02.03.09 Bachelorarbeit
15
Tabelle 1: Übergangsenergien und Oszillatorstärken zu den Geometrieoptimierungen im Grundzustand.
TS Energie [nm] Orbitale Typ
SVWN S1 475 0.32297 HOMO - LUMO π−π∗S2 455 0.00000 HOMO-1 - LUMO π−π∗S3 421 0.00096 HOMO-3 - LUMOS4 359 0.72133 HOMO-2 - LUMO π−π∗S5 331 0.00000 HOMO - LUMO+1 π−π∗S6 304 0.04110 HOMO-1 - LUMO+1 π−π∗
BP S1 467 0.32404 HOMO - LUMO π−π∗S2 448 0.00000 HOMO-1 - LUMO π−π∗S3 407 0.00126 HOMO-3 - LUMOS4 360 0.70558 HOMO-2 - LUMO π−π∗S5 334 0.00000 HOMO - LUMO+1 π−π∗
S6 306 0.03170 HOMO-1 - LUMO+1 π−π∗
B3LYP S1 383 0.56783 HOMO - LUMO π−π∗S2 353 0.00000 HOMO-1 - LUMO π−π∗S3 335 0.00252 HOMO-3 - LUMOS4 307 0.58093 HOMO-2 - LUMO π−π∗S5 278 0.00000 HOMO - LUMO+1 π−π∗
S6 256 0.01868 HOMO-1 - LUMO+1 π−π∗
BHLYP S1 315 0.83165 HOMO - LUMO π−π∗S2 280 0.00000 HOMO-1 - LUMO π−π∗S3 271 0.00371 HOMO-4 - LUMO π−π∗S4 256 0.41280 HOMO-2 - LUMO π−π∗S5 223 0.00000 HOMO-3 - LUMO
S6 220 0.00000 HOMO - LUMO+2 π−π∗
HF/CIS S1 265 0.94930 HOMO - LUMO π−π∗S2 234 0.00000 HOMO-1 - LUMO π−π∗S3 224 0.16624 HOMO-2 - LUMO π−π∗S4 219 0.00480 HOMO-4 - LUMOS5 214 0.00000 HOMO-3 - LUMO π−π∗S6 186 1.13708 HOMO-1 - LUMO+3 π−π∗
MP2/B3LYP S1 379 0.56430 HOMO - LUMO π−π∗S2 350 0.00000 HOMO-1 - LUMO π−π∗S3 327 0.00265 HOMO-4 - LUMOS4 307 0.58179 HOMO-2 - LUMO π−π∗S5 277 0.00000 HOMO-3 - LUMO π−π∗S6 256 0.01754 HOMO - LUMO+2 π−π∗
Osz. [au]
n-π∗
n-π∗
n-π∗
n-π∗
n-π∗
n-π∗

Michael Schönnenbeck 02.03.09 Bachelorarbeit
Alle Methoden finden einen optisch erlaubten S1-Zustand mit π-π*-Charakter (vgl. Tabelle 1). Die
verbotenen n-π*-Übergänge werden von den TDDFT-Funktionalen als S3-Zustand gefunden. Während
TDDFT mit lokalen Funktionalen und B3LYP den n-π*-Zustand als S3 liefert, ist dieser S5 bzw. S4, wenn
das BHLYP-Funktional bzw. CIS verwendet wird. Das CIS-Spektrum der HF-Grundzustandsoptimierung
weicht von den anderen Spektren ab und ist stark blauverschoben. Diese Überschätzung der
Anregungsenergien durch CIS lässt sich darauf zurückführen, dass die zur Berechnung der
Anregungsenergien verwendeten virtuellen Orbitale einem zusätzlichen Elektron entsprechen und nicht
einem angeregten. Dies führt zu einer für diesen Zweck zu hohen Orbitalenergie.
Neben der Abhängigkeit der Anregungsenergien von dem gewählten Funktional wurde außerdem der
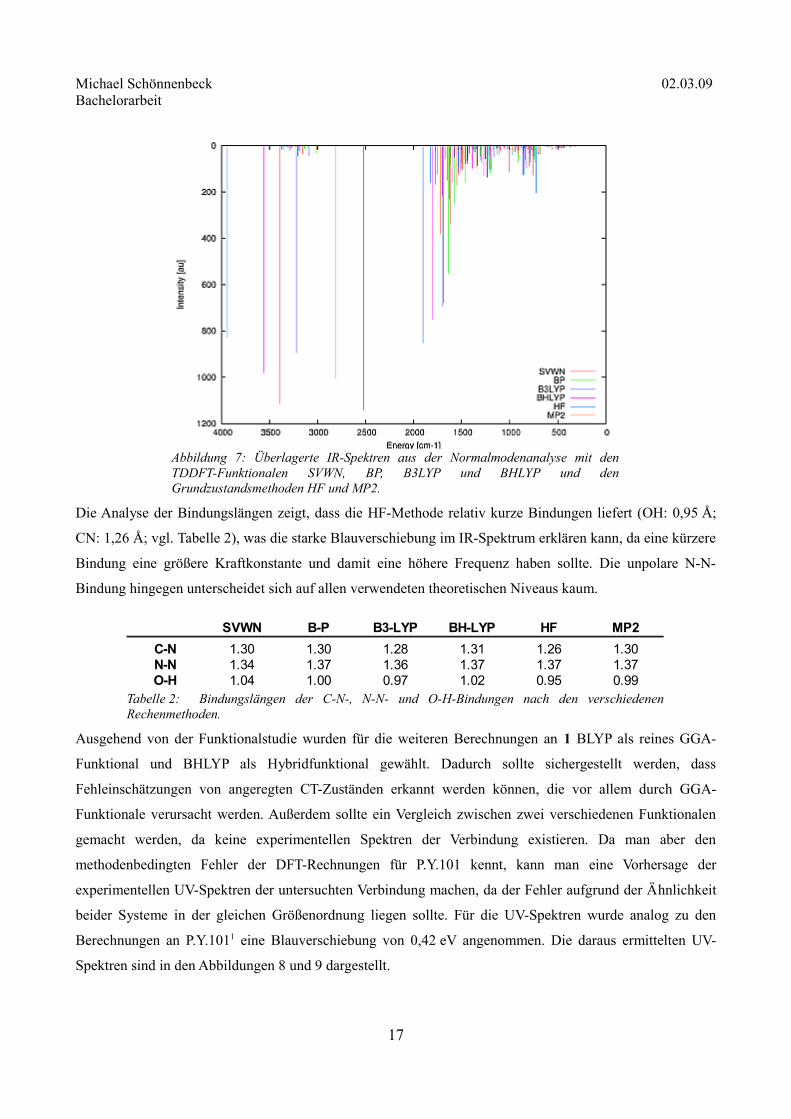
Einfluss auf die Schwingungsspektren studiert. Dazu wurde eine Normalmodenanalyse durchgeführt. Auch
hier wurde ein Vergleichsspektrum aus den Anregungsenergien im IR-Bereich erstellt (Abbildung 7).
16
Abbildung 6: Vergleichsspektrum aus den mit den TDDFT-Funktionalen SVWN, BP, B3LYP und BHLYP und den Grundzustandsmethoden HF und MP2 berechneten Grundzustandsgeometrien. Die Anregungsenergien der HF-Geometrien wurden mit CIS berechnet, die der MP2-Geometrien mit TDDFT/B3LYP/DZP.
Michael Schönnenbeck 02.03.09 Bachelorarbeit
Die Analyse der Bindungslängen zeigt, dass die HF-Methode relativ kurze Bindungen liefert (OH: 0,95 Å;
CN: 1,26 Å; vgl. Tabelle 2), was die starke Blauverschiebung im IR-Spektrum erklären kann, da eine kürzere
Bindung eine größere Kraftkonstante und damit eine höhere Frequenz haben sollte. Die unpolare N-N-
Bindung hingegen unterscheidet sich auf allen verwendeten theoretischen Niveaus kaum.
Ausgehend von der Funktionalstudie wurden für die weiteren Berechnungen an 1 BLYP als reines GGA-
Funktional und BHLYP als Hybridfunktional gewählt. Dadurch sollte sichergestellt werden, dass
Fehleinschätzungen von angeregten CT-Zuständen erkannt werden können, die vor allem durch GGA-
Funktionale verursacht werden. Außerdem sollte ein Vergleich zwischen zwei verschiedenen Funktionalen
gemacht werden, da keine experimentellen Spektren der Verbindung existieren. Da man aber den
methodenbedingten Fehler der DFT-Rechnungen für P.Y.101 kennt, kann man eine Vorhersage der
experimentellen UV-Spektren der untersuchten Verbindung machen, da der Fehler aufgrund der Ähnlichkeit
beider Systeme in der gleichen Größenordnung liegen sollte. Für die UV-Spektren wurde analog zu den
Berechnungen an P.Y.1011 eine Blauverschiebung von 0,42 eV angenommen. Die daraus ermittelten UV-
Spektren sind in den Abbildungen 8 und 9 dargestellt.
17
Abbildung 7: Überlagerte IR-Spektren aus der Normalmodenanalyse mit den TDDFT-Funktionalen SVWN, BP, B3LYP und BHLYP und den Grundzustandsmethoden HF und MP2.
Tabelle 2: Bindungslängen der C-N-, N-N- und O-H-Bindungen nach den verschiedenen Rechenmethoden.
SVWN B-P B3-LYP BH-LYP HF MP2
C-N 1.30 1.30 1.28 1.31 1.26 1.30N-N 1.34 1.37 1.36 1.37 1.37 1.37O-H 1.04 1.00 0.97 1.02 0.95 0.99

Michael Schönnenbeck 02.03.09 Bachelorarbeit
Für berechnete IR-Spektren mit BP 86 ist in der Literatur20 ein Korrekturfaktor von 0,9945 zu finden. Da für
das BHLYP-Funktional kein Literaturwert zu finden ist, wurde hierfür der gleiche Korrekturfaktor wie für
B3LYP angenommen (0,9614). Für den vorgenommen Lorentzfit wurde eine Halbwertsbreite von 60 cm-1
angenommen. Daraus ergeben sich die Schwingungsspektren in den Abbildungen 10 und 11
18
Abbildung 8: Vorhergesagtes UV-Spektrum aus den mit DFT/BHLYP/DZP berechneten Geometrien. Für den Lorentzfit wurde eine Halbwertsbreite von 50 nm angenommen. Die berechneten Banden wurden um 0,42 eV (blau) korrigiert.
Abbildung 9: Vorhergesagtes UV-Spektrum aus den mit DFT/BP/DZP berechneten Geometrien. Für den Lorentzfit wurde eine Halbwertsbreite von 50 nm angenommen. Die berechneten Banden wurden um 0,42 eV (blau) korrigiert.

Michael Schönnenbeck 02.03.09 Bachelorarbeit
19
Abbildung 10: Vorhergesagtes IR-Spektrum aus den mit DFT/BLYP/DZP berechneten Geometrien. Für den Lorentzfit wurde eine Halbwertsbreite von 60 cm- 1 angenommen. Die berechneten Banden mit dem Faktor 0,9614 korrigiert.
Abbildung 11: Vorhergesagtes IR-Spektrum aus den mit DFT/BLYP/DZP berechneten Geometrien. Für den Lorentzfit wurde eine Halbwertsbreite von 60 cm- 1 angenommen. Die berechneten Banden mit dem Faktor 0,9945 korrigiert.

Michael Schönnenbeck 02.03.09 Bachelorarbeit
4.2 Rotation um die CNNC-Bindung im Grundzustand
Um das Verhalten im Grundzustand zu untersuchen, wurden die Energien entlang der Rotationskoordinate
um die CNNC-Bindung berechnet. Dazu wurde der Diederwinkel in 10°-Schritten von 0 - 180° festgehalten,
während die restlichen Freiheitsgrade frei optimiert wurden. Da die Fehleinschätzung von CT-Zuständen im
Grundzustand nicht auftreten, wurde hier lediglich mit dem BHLYP-Funktional gerechnet. Für die
Grundzustandsgeometrien wurden die ersten fünf Anregungsenergien berechnet. Für diese und für alle
weiteren Berechnungen an 1 wurde der DZP-Basissatz verwendet.
Die Potentialkurve entlang der Rotationskoordinate (Abbildung 12) hat im Grundzustand ein Minimum bei
180° (trans) und ein Maximum bei 0° (cis). Eine Rotationsbarriere liegt nicht vor. Die Anregung in den S1-
Zustand ist erlaubt und findet bei 312 nm statt. Die Potentialkurve des ersten angeregten Zustandes hat bei
90° ein Maximum. Die Gleichgewichtsstrukturen befinden sich bei 0° und bei 180°, wobei das globale
Minimum bei 180° um 0,25 eV günstiger ist als das lokale bei 0°. Die Rotationsbarriere für eine trans-cis-
Isomerisierung beträgt 0,31 eV. Bei alleiniger Betrachtung der Grundzustandsoptimierung ist also nach der
elektronischen Anregung in den S1-Zustand keine Isomerisierung zur trans-Konformation zu erwarten.
4.3 Rotation um die CNNC-Bindung im ersten angeregten Zustand
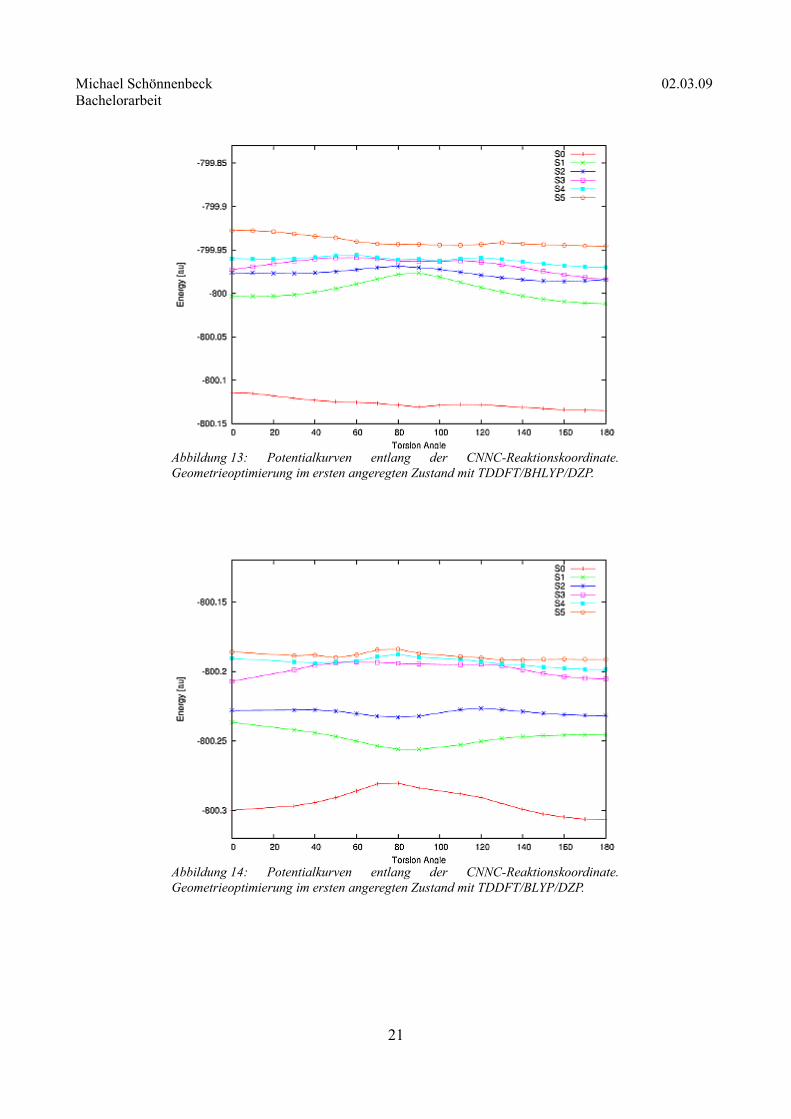
Um das Verhalten des Moleküls im ersten angeregten Zustand zu untersuchen, wurden zunächst die
Geometrien entlang der CNNC-Rotationskoordinate optimiert. Dazu wurde ebenfalls das BHLYP- und
zusätzlich das BLYP-Funktional verwendet, um eventuelle CT-Zustände zu identifizieren.
20
Abbildung 12: Potentialkurve entlang der CNNC-Reaktionskoordinate im Grundzustand und den ersten fünf angeregten Zuständen. Geometrieoptimierung im Grundzustand mit TDDFT/BHLYP/DZP.
Michael Schönnenbeck 02.03.09 Bachelorarbeit
21
Abbildung 14: Potentialkurven entlang der CNNC-Reaktionskoordinate. Geometrieoptimierung im ersten angeregten Zustand mit TDDFT/BLYP/DZP.
Abbildung 13: Potentialkurven entlang der CNNC-Reaktionskoordinate. Geometrieoptimierung im ersten angeregten Zustand mit TDDFT/BHLYP/DZP.

Michael Schönnenbeck 02.03.09 Bachelorarbeit
Die mit BHLYP berechnete Potentialkurve des ersten angeregten Zustandes (Abbildung 13) zeigt qualitativ
das gleiche Verhalten wie die der Grundzustandsoptimierung: Das Maximum ist bei 90° zu finden, die trans-
Konformation ist um 0,24 eV energetisch günstiger als die cis-Konformation. Die Rotationsbarriere beträgt
0,96 eV. Die Fluoreszenz aus der cis-Konformation findet bei 409 nm statt, die aus der trans-Konformation
bei 370 nm.
Ein völlig anderes Verhalten des Moleküls im S1-Zustand zeigt die mit BLYP optimierte Potentialkurve
(Abbildung 14). Sie hat ein Minimum bei 90°, das um 0,29 eV günstiger ist als das trans-Isomer. Da bei
allen bisherigen Untersuchungen an P.Y.1011,2,3,4 und 2,2'-Dihydroxy-1,1'-diphenylaldazin eine große
Rotationsbarriere gefunden wurde, liegt die Vermutung nahe, dass es sich hier um einen falsch
eingeschätzten CT-Zustand handelt. Tatsächlich zeigen die Molekülorbitale bei 90° einen starken CT-
Charakter (Abbildung 15).
Um dies genauer zu untersuchen, wurden auf die optimierten BLYP-Geometrien Singlepointrechnungen mit
dem BHLYP-Funktional, und umgekehrt Singlepointrechnungen mit dem BLYP-Funktional auf die BHLYP-
Geometrien, durchgeführt (Abbildung 16 und 17). Die Singlepointrechnungen mit BHLYP ergeben eine
Korrektur der BLYP-Optimierungen. Das Molekül hat eine Rotationsbarriere von 0.42 eV und die stabilste
Geometrie in trans-Stellung. Die BLYP-Singlepointrechnungen auf die BHLYP-Optimierungen weisen zwar
bei 90° eine um 0,57 eV geringere Rotationsbarriere von 0,39 eV auf, als die BHLYP-Optimierung selbst,
jedoch wird qualitativ das gleiche Verhalten wiedergegeben.
22
Abbildung 15: Molekülorbitale zum S1-Übergang bei 90° und 180° zur Potentialkurve entlang der CNNC-Rotationskoordinate mit TDDFT/BLYP/DZP.

Michael Schönnenbeck 02.03.09 Bachelorarbeit
Zusammenfassend lässt sich sagen, dass das Minimum der BLYP-Optimierungen ein CT-Artefakt ist und das
Molekül nach der optischen Anregung weiterhin als trans-Isomer vorliegt. Die Relaxation in den
Grundzustand unter Fluoreszenz wäre anhand der BHLYP-Optimierungen bei 370 nm zu erwarten.
23
Abbildung 16: Singlepointrechnungen mit TDDFT/BHLYP/DZP auf die optimierten BLYP-Geometrien.
Abbildung 17: Singlepointrechnungen mit TDDFT/BLYP/DZP auf die optimierten BHLYP-Geometrien.

Michael Schönnenbeck 02.03.09 Bachelorarbeit
4.4 Intramolekularer Protonentransfer im angeregten Zustand
Um auch den Protonentransfer im ersten angeregten Zustand zu untersuchen, wurde die OH-Bindungslänge
als weitere Reaktionskoordinate gewählt. Die Geometrieoptimierungen wurden von 0,95 bis 1,5 Å mit einer
Schrittweite von 0,05 Å durchgeführt.
24
Abbildung 18: Potentialkurven entlang der O-H-Koordinate. Geometrieoptimierung im ersten angeregten Zustand mit TDDFT/BHLYP/DZP.
Abbildung 19: Potentialkurven entlang der O-H-Koordinate. Geometrieoptimierung im ersten angeregten Zustand mit TDDFT/BLYP/DZP. Die Optimierung bei 0,95 Å ist nicht konvergiert; der Punkt wurde grafisch interpoliert.

Michael Schönnenbeck 02.03.09 Bachelorarbeit
Die Potentialkurve der BHLYP-Geometrieoptimierungen (Abbildung 18) hat ein lokales Minimum bei 1,1 Å.
Die bei 1,5 Å gefundenen Geometrie zeigt jedoch eine um 0,07 eV günstigere Energie. Da die Barriere für
den Protonentransfer nur 0,05 eV beträgt, ist eine Gleichgewichtsverteilung zwischen der diol- und der keto-
Form zu erwarten, bei der das Gleichgewicht stark auf die Seite des keto-Isomers verschoben ist.
Ein anderes Verhalten zeigt die Potentialkurve der BLYP-Geometrieoptimierungen (Abbildung 19). Die S1-
Kurve zeigt einen deutlichen Energiegewinn durch den Protonentransfer und weist keine Barriere auf. Nach
der optischen Anregung des Moleküls ist also ein Übergang des Moleküls in das keto-Isomer zu erwarten.
25

Michael Schönnenbeck 02.03.09 Bachelorarbeit
5. Berechnungen an 1-(Ethen-2-ol)-1'-(1'-methylprop-1'-en-2'-ol)-aldazin.
In diesem Teil der Arbeit sollen die elektronischen Zustände des P.Y.101-Derivats 2 untersucht werden. Das
Molekül stellt ein stark verkleinertes System der Farbstoffe dar, bei dem die planaren Phenyl- bzw.
Naphthyleinheiten vollständig entfernt wurden. Analog zum Vorgehen bei Derivat 1 wurde die Rotation um
die zentrale CNNC-Bindung im Grund- (Abschnitt 5.3) und im angeregten Zustand (Abschnitt 5.4), sowie
der intramolekulare Protonentransfer im ersten angeregten Zustand (Abschnitt 5.5) untersucht, um
Ähnlichkeiten und Unterschiede in den Eigenschaften der angeregten Zustände zu finden. Dabei wurden
ebenfalls die Funktionale BLYP und BHLYP verwendet.
5.1 Vergleich von Basissätzen
Um den geeigneten Basissatz für die Geometrieoptimierungen des Moleküls zu finden, wurde der
Grundzustand zunächst in cis- und trans-Stellung mit dem BLYP-Funktional frei optimiert. Für die
Optimierung wurden die SVP-, DZP-, TZVP- und QZVP-Basissätze verwendet und die Anregungsenergien
in einem UV-Spektrum miteinander verglichen.
Die optimierten BLYP-Geometrien im Grundzustand in cis- (Abbildung 20) und trans-Stellung (Abbildung
21) zeigen ähnliche Spektren. Die Berechnungen finden jeweils einen verbotenen S1- und einen erlaubten
S2-Übergang mit vergleichbaren Intensitäten.
26
Abbildung 20: Vergleich der UV-Spektren zu den Geometrieoptimierungen im Grundzustand in cis-Stellung mit TDDFT/BLYP und den Basissätzen SVP, DZP, TZVP und QZVP.

Michael Schönnenbeck 02.03.09 Bachelorarbeit
Auf die optimierten BLYP-Grundzustandsstrukturen wurden Singlepointrechnungen mit dem BHLYP-
Funktional und DZP-Basissatz durchgeführt, und ebenfalls im UV-Spektrum verglichen.
Auch die Spektren der Singlepointrechnungen mit BHLYP (Abbildung 22 und 23) zeigen sehr ähnliche
Verschiebungen. Im Gegensatz zu den Ergebnissen die das BLYP-Funktional liefert, ist der S1-Übergang
hier erlaubt und der S2-Übergang verboten.
27
Abbildung 21: Vergleich der UV-Spektren zu den Geometrieoptimierungen im Grundzustand in trans-Stellung mit TDDFT/BLYP und den Basissätzen SVP, DZP, TZVP und QZVP.
Abbildung 22: Vergleich der UV-Spektren aus den Singlepointrechnungen mit BHLYP auf die mit BLYP optimierten Geometrien in cis-Stellung.
Michael Schönnenbeck 02.03.09 Bachelorarbeit
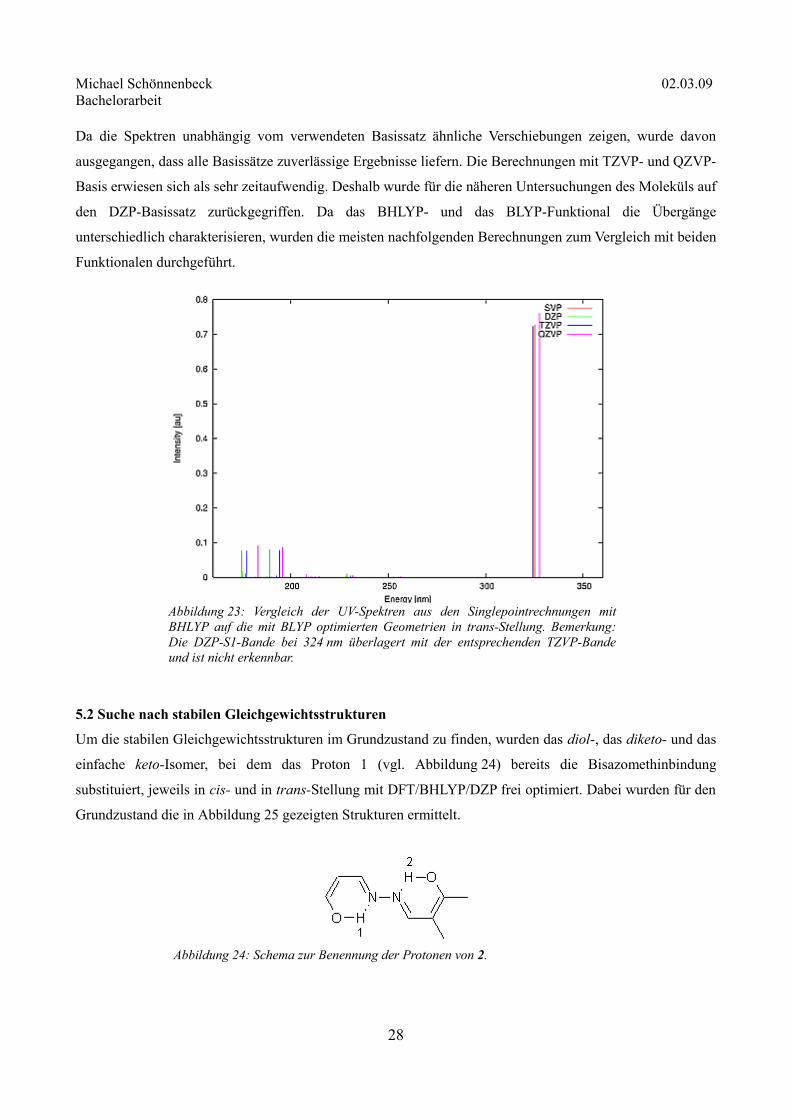
Da die Spektren unabhängig vom verwendeten Basissatz ähnliche Verschiebungen zeigen, wurde davon
ausgegangen, dass alle Basissätze zuverlässige Ergebnisse liefern. Die Berechnungen mit TZVP- und QZVP-
Basis erwiesen sich als sehr zeitaufwendig. Deshalb wurde für die näheren Untersuchungen des Moleküls auf
den DZP-Basissatz zurückgegriffen. Da das BHLYP- und das BLYP-Funktional die Übergänge
unterschiedlich charakterisieren, wurden die meisten nachfolgenden Berechnungen zum Vergleich mit beiden
Funktionalen durchgeführt.
5.2 Suche nach stabilen Gleichgewichtsstrukturen
Um die stabilen Gleichgewichtsstrukturen im Grundzustand zu finden, wurden das diol-, das diketo- und das
einfache keto-Isomer, bei dem das Proton 1 (vgl. Abbildung 24) bereits die Bisazomethinbindung
substituiert, jeweils in cis- und in trans-Stellung mit DFT/BHLYP/DZP frei optimiert. Dabei wurden für den
Grundzustand die in Abbildung 25 gezeigten Strukturen ermittelt.
28
Abbildung 23: Vergleich der UV-Spektren aus den Singlepointrechnungen mit BHLYP auf die mit BLYP optimierten Geometrien in trans-Stellung. Bemerkung: Die DZP-S1-Bande bei 324 nm überlagert mit der entsprechenden TZVP-Bande und ist nicht erkennbar.
Abbildung 24: Schema zur Benennung der Protonen von 2.

Michael Schönnenbeck 02.03.09 Bachelorarbeit
Das cis-keto-, das trans-keto und das trans-diol-Isomer behielten die planare Ausgangsstruktur bei. Die
Optimierungen der cis-diketo- und der trans-diketo-Ausgangsstruktur lieferten gewinkelte
Gleichgewichtsstrukturen mit einem CNNC-Torsionswinkel von 117° bzw. -117°. Dies ist auf die Abstoßung
der freien Elektronenpaare der Stickstoffatome zurückzuführen, deren Optimierung eine leichte
Pyramidalisierung ergab. Die Optimierung der cis-diol-Struktur lieferte ebenfalls eine gewinkelte
Gleichgewichtsstruktur mit einem CNNC-Torsionswinkel von 66°, die sich durch die sterische
Wechselwirkung der OH-Gruppen erklären lässt.
Auch im ersten angeregten Zustand wurden die Isomere der Verbindung mit TDDFT/BHLYP/DZP frei
optimiert. Alle Isomere behielten im Gegensatz zum Grundzustand die planare Ausgangsstruktur bei. Das
liegt daran, dass der Doppelbindungscharakter der N-N-Bindung im S1-Zustand stärker ist als in S0, was die
planare Struktur begünstigt.
29
Abbildung 25: Stabile Strukturen des Derivats 1 aus den Geometrieoptimierung im Grundzustand mit DFT/BHLYP/DZP. Die Beschriftung bezeichnet die Ausgangsstrukturen der Geometrieoptimierungen.

Michael Schönnenbeck 02.03.09 Bachelorarbeit
Ebenfalls im S1-Zustand wurden die diol- und die keto-Form des Moleküls in cis- und trans-Stellung mit
TDDFT/BLYP/DZP frei optimiert. Beide diol-Isomere behielten hier die planare Ausgangsstruktur bei. Die
Optimierungen der keto-Isomere lieferten gewinkelte Strukturen mit Torsionswinkeln von jeweils 90°.
Hierbei handelt es sich um energetisch unterschätzte CT-Zustände. Aufgrund der artifiziellen Energieminima
wurden bei den weiteren Berechnungen an 2 weiterhin beide Funktionale verwendet.
5.3 Rotation um die CNNC-Bindung im Grundzustand
Zur Berechnung der Potentialkurve entlang der CNNC-Rotationskoordinate des keto-Isomers im
Grundzustand wurde das BHLYP-Funktional gewählt (vgl. Abbildung 26). Dabei wurde die stabilste
Struktur in trans-Stellung gefunden. Beim Torsionswinkel von 30° befindet sich ein lokales Minimum, dass
sich nur durch die sterischen Wechselwirkungen der cis-ständigen Wasserstoffatome der
Bisazomethinbindung erklären lässt. Das globale Maximum der Kurve liegt bei 100°. Die Rotationsbarriere
für eine Isomerisierung zur planaren trans-Struktur beträgt lediglich 0,07 eV. Bei einer freien Optimierung
der planaren cis-Struktur mit DFT/BHLYP/DZP (vgl. Abschnitt 5.2) wurde diese ebenfalls als stabile
Struktur charakterisiert, die aber eine höhere Energie aufweist als das 30°-Isomer. Es ist somit eine
Gleichgewichtsverteilung zwischen dem 30°- und dem trans-Isomer zu erwarten. Der erlaubte Übergang in
den ersten angeregten Zustand in trans-Stellung findet bei 304 nm statt, während das Molekül bei 30° bei
316 nm absorbiert.
30
Abbildung 26: Potentialkurve entlang der CNNC-Reaktionskoordinate im Grundzustand und den ersten fünf angeregten Zuständen. Geometrieoptimierung im Grundzustand mit DFT/BHLYP/DZP.

Michael Schönnenbeck 02.03.09 Bachelorarbeit
Das Maximum des angeregten Zustandes befindet sich bei 90°. Das cis-Isomer ist hier etwas stabiler als das
trans-Isomer. Die Rotationsbarriere beträgt 0,55 eV. Dies entspricht in etwa den Ergebnissen der
Berechnungen an 2 (vgl. Abschnitt 4.2).
5.4 Rotation um die CNNC-Bindung im ersten angeregten Zustand
Um die Aussagen über die Isomerisierung im S1-Zustand zu vertiefen, wurden die Geometrien entlang der
CNNC-Koordinate im ersten angeregten Zustand zunächst mit TDDFT/BLYP/DZP optimiert (Abbildung
29). Die Berechnung einer vollständigen Potentialkurve erwies sich jedoch als äußerst kompliziert, da sie
entlang der Rotationskoordinate in mindestens eine konische Durchschneidung läuft. Entlang der
Rotationskoordinate findet im Bereich von 20° bis 40° ein zweiter Protonentransfer auf die
Bisazomethinbindung statt. In diesem Bereich liegt das Molekül als diketo-Isomer vor. Dies führt beim
Übergang von 10° nach 20° zu einem sprunghaften Energiegewinn von 0,21 eV. Im Grundzustand steigt die
Energie in diesem Bereich um 1,42 eV. Ein weiterer Einbruch der Energien im Grundzustand ist im Bereich
von 80° bis 100° zu erkennen. Die Energie fällt hier sprunghaft um 1.22 eV ab.
Um das Verhalten entlang der reinen Rotationskoordinate zu zeigen, wurden die kritischen Punkte grafisch
interpoliert (Abbildung 28). Es handelt sich dabei nicht mehr um einen minimalen Energiepfad. Im S1-
Zustand ist wieder ein artifizielles Minimum bei 70° zu erkennen, das auf einen CT-Zustand zurückzuführen
ist. Im Grundzustand ist ein ähnliches Verhalten zu erkennen, wie bei der Grundzustandsoptimierung mit
31
Abbildung 27: Potentialkurve entlang der CNNC-Reaktionskoordinate im Grundzustand und den ersten fünf angeregten Zuständen. Geometrieoptimierung im ersten angeregten Zustand mit TDDFT/BLYP/DZP.

Michael Schönnenbeck 02.03.09 Bachelorarbeit
TDDFT/BHLYP/DZP. Allerdings befindet sich hier kein lokales Minimum bei 30°. Das trans-Isomer ist
wieder günstiger als das cis-Isomer; zwischen beiden liegt eine Rotationsbarriere von 0,95 eV vor. Das
Maximum liegt hier bei 50°.
32
Abbildung 28: Potentialkurve entlang der CNNC-Reaktionskoordinate im Grundzustand und den ersten fünf angeregten Zuständen. Geometrieoptimierung im ersten angeregten Zustand mit TDDFT/BLYP/DZP. Die Punkte zwischen 10° und 50° und zwischen 70° und 110° wurden grafisch interpoliert.
Abbildung 29: Potentialkurve entlang der CNNC-Reaktionskoordinate im Grundzustand und den ersten fünf angeregten Zuständen. Geometrieoptimierung im ersten angeregten Zustand mit TDDFT/BHLYP/DZP.

Michael Schönnenbeck 02.03.09 Bachelorarbeit
Die Potentialkurve wurde ebenfalls mit TDDFT/BHLYP/DZP berechnet (Abbildung 29). Sie läuft hier
offensichtlich in mehrere konische Durchschneidungen. Die methylierten Kohlenstoffe sind pyramidalisiert,
so dass die Methylgruppen leicht aus der Ebene hervorstehen. Auch der OH-substituierte Kohlenstoff wird
pyramidalisiert. Aufgrund der konischen Durchschneidungen konvergierten die Rechnungen in den
Bereichen von 30° bis 60° und von 120° bis 160° nicht. Im S0-Potential besteht ein Energieanstieg von
2,41 eV zwischen der optimierten Struktur bei 20° und der bei 70°, im S1 Potential sinkt die entsprechende
Energie um 0,44 eV, was ein deutliches Zeichen dafür ist, dass es sich um konische Durchschneidungen
mehrerer Normalmoden handelt, da die Optimierung kein Energieminimum finden konnte.
Um ein besseres Bild der Rotation zu erhalten, wurden die Energien der Strukturen von 30° bis 160° mit
Singlepointrechnungen bestimmt (Abbildung 30). Die S1-Potentialkurve entspricht hier eher den erwarteten
Ergebnissen. Die cis-Struktur ist mit einem Energieunterschied von 0,04 eV etwas günstiger als die trans-
Struktur. Zwischen den beiden Isomeren befindet sich eine Rotationsbarriere von 0,49 eV. Die Fluoreszenz in
den Grundzustand aus der cis-Konformation ist bei 382 nm zu erwarten, die aus der trans-Konformation bei
349 nm
Zusätzlich wurden die angeregten Zustände der S1-optimierten BHLYP-Geometrien mit
Singlepointrechnungen mit ADC 2S (Abbildung 31) und ADC 2X (Abbildung 32) berechnet. Die
zugehörigen Grundzustandsenergien wurden mit MP2 berechnet. Der Grundzustand der MP2-
Singlepointrechnungen verhält sich ähnlich zu dem der Grundzustandsoptimierungen mit BHLYP
(Abbildung 26). Bei 30° befindet sich ein lokales Minimum, das aber eine wesentlich höhere
33
Abbildung 30: Singlepointrechnungen mit TDDFT/BHLYP/DZP entlang der CNNC-Rotationskoordinate ausgehend von den optimierten S1-Strukturen bei 20° und 170°.

Michael Schönnenbeck 02.03.09 Bachelorarbeit
Rotationsbarriere (0.34 eV) zur trans-Struktur aufweist, die um 0.22 eV stabiler ist. Im ersten angeregten
Zustand befindet sich bei 30° ebenfalls ein lokales Minimum mit einer cis-trans-Rotationsbarriere von
0,42 eV. Die trans-Struktur ist hier um 0,03 eV stabiler als die Gleichgewichtsstruktur bei 30°.
34
Abbildung 32: Singlepointrechnungen auf die mit TDDFT/BHLYP/DZP optimierten S1-Geometrien entlang der CNNC-Rotationskoordinate mit ADC 2X. Die Grundzustandsenergien wurden mit MP2 berechnet.
Abbildung 31: Singlepointrechnungen auf die mit TDDFT/BHLYP/DZP optimierten S1-Geometrien entlang der CNNC-Rotationskoordinate mit ADC 2S. Die Grundzustandsenergien wurden mit MP2 berechnet.

Michael Schönnenbeck 02.03.09 Bachelorarbeit
Der erste angeregte Zustand, dessen Energie mit ADC 2X berechnet wurde, hat ebenfalls ein lokales
Minimum bei 30°. Das globale Minimum liegt hier bei 180° und ist um 0,03 eV günstiger als das lokale bei
30°. Das Maximum liegt bei 100°. Die cis-trans-Rotationsbarriere beträgt 0,42 eV. Die Absorption in den
ersten angeregten Zustand ist in 30°-Stellung bei 519 nm zu erwarten und in trans-Stellung bei 480 nm. Dies
ist ein deutlicher Unterschied zu allen bisherigen Berechnungen an 1 und 2: Das Molekül absorbiert hier im
sichtbaren Bereich. Aufgrund der Rotationsbarriere ist nach der optischen Anregung in den S1-Zustand aus
den beiden Gleichgewichtsstrukturen keine Isomerisierung entlang der CNNC-Koordinate zu erwarten. Der
S3-Zustand wird bei den planaren Strukturen bei 0° und bei 180° als Doppelanregung identifiziert. Der
Doppelanregungscharakter nimmt jedoch mit zunehmender Drehung um die CNNC-Bindung ab. Bei der
90°- Struktur überwiegt der Einfachanregungscharakter. An diesem Punkt sind der S2 und der S3-Zustand
nahezu entartet. Die Form der Molekülorbitale ändert sich in diesem Bereich leicht, jedoch findet keine
Kreuzung der Zustände statt.
Bei den Berechnungen der Potentialkurve entlang der Rotationskoordinate mit ADC 2S (Abbildung 31)
wurden qualitativ die gleichen Minima und das gleiche Maximum im S1-Zustand gefunden. Die trans-
Struktur ist ebenfalls um 0.03 eV günstiger als die Struktur bei 30°. Die Barriere für eine cis-trans-
Isomerisierung beträgt hier jedoch nur 0,19 eV. Die Anregung des trans-Isomers in den S1-Zustand ist bei
379 nm zu erwarten, die des Isomers bei 30° bei 402 nm.
Qualitativ geben ADC 2S und ADC 2X das gleiche Verhalten der angeregten Zustände wieder. Nur die S3-
und S4-Zustände zeigen leichte Unterschiede, da diese einen starken Doppelanregungscharakter besitzen, der
durch ADC 2X besser beschrieben wird. Auch die Potentialkurven der mit TDDFT/BHLYP/DZP
berechneten angeregten Zustände zeigen kaum Unterschiede zu den mit ADC berechneten Potentialkurven.
Diese Studie zeigt, dass die mittels TDDFT getroffenen Schlussfolgerungen im Großen und Ganzen richtig
sind, da der S1-Zustand keinen Doppelanregungscharakter besitzt und somit durch TDDFT gut beschrieben
werden kann.
5.5 Intramolekularer Protonentransfer im angeregten Zustand
Weiterhin sollte die Übertragung der Protonen auf die Bisazomethinbindung im S1-Zustand untersucht
werden. Als Ausgangspunkt für die Berechnungen wurde das diol-Isomer gewählt, und zunächst die
Übertragung beider OH-Protonen getrennt betrachtet. Anschließend wurde der Übergang vom keto- zum
diketo-Isomer untersucht. Dabei wurde davon ausgegangen, dass das Proton 1 (vgl. Abbildung 24) bereits
auf die Bisazomethinbindung übertragen wurde. Den Grund dafür lieferte die zweifach protonierte Struktur,
die bei den Untersuchungen der Potentialkurve entlang der CNNC-Rotationskoordinate gefunden wurde
(vgl. Abschnitt 5.4). Vorweg ist anzumerken, dass viele der Rechnungen aufgrund der konischen
Durchschneidungen nicht konvergierten. Daher wurden die fehlenden Energien mit Singlepointrechnungen
35
Michael Schönnenbeck 02.03.09 Bachelorarbeit
bestimmt.
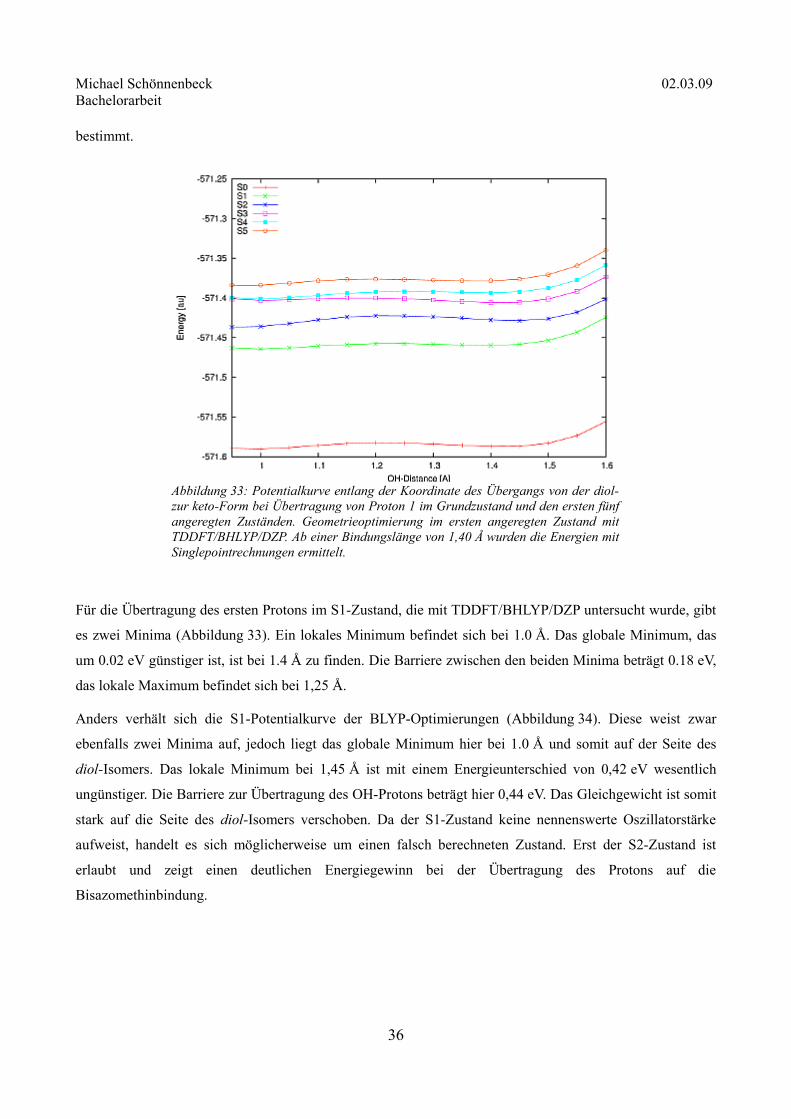
Für die Übertragung des ersten Protons im S1-Zustand, die mit TDDFT/BHLYP/DZP untersucht wurde, gibt
es zwei Minima (Abbildung 33). Ein lokales Minimum befindet sich bei 1.0 Å. Das globale Minimum, das
um 0.02 eV günstiger ist, ist bei 1.4 Å zu finden. Die Barriere zwischen den beiden Minima beträgt 0.18 eV,
das lokale Maximum befindet sich bei 1,25 Å.
Anders verhält sich die S1-Potentialkurve der BLYP-Optimierungen (Abbildung 34). Diese weist zwar
ebenfalls zwei Minima auf, jedoch liegt das globale Minimum hier bei 1.0 Å und somit auf der Seite des
diol-Isomers. Das lokale Minimum bei 1,45 Å ist mit einem Energieunterschied von 0,42 eV wesentlich
ungünstiger. Die Barriere zur Übertragung des OH-Protons beträgt hier 0,44 eV. Das Gleichgewicht ist somit
stark auf die Seite des diol-Isomers verschoben. Da der S1-Zustand keine nennenswerte Oszillatorstärke
aufweist, handelt es sich möglicherweise um einen falsch berechneten Zustand. Erst der S2-Zustand ist
erlaubt und zeigt einen deutlichen Energiegewinn bei der Übertragung des Protons auf die
Bisazomethinbindung.
36
Abbildung 33: Potentialkurve entlang der Koordinate des Übergangs von der diol- zur keto-Form bei Übertragung von Proton 1 im Grundzustand und den ersten fünf angeregten Zuständen. Geometrieoptimierung im ersten angeregten Zustand mit TDDFT/BHLYP/DZP. Ab einer Bindungslänge von 1,40 Å wurden die Energien mit Singlepointrechnungen ermittelt.

Michael Schönnenbeck 02.03.09 Bachelorarbeit
37
Abbildung 34: Potentialkurve entlang der Koordinate des Übergangs von der diol- zur keto-Form bei Übertragung von Proton 1 im Grundzustand und den ersten fünf angeregten Zuständen. Geometrieoptimierung im ersten angeregten Zustand mit TDDFT/BLYP/DZP. Ab einer Bindungslänge von 1,40 Å wurden die Energien mit Singlepointrechnungen ermittelt.
Abbildung 35: Potentialkurve entlang der Koordinate des Übergangs von der diol- zur keto-Form bei Übertragung von Proton 2 im Grundzustand und den ersten fünf angeregten Zuständen. Geometrieoptimierung im ersten angeregten Zustand mit TDDFT/BHLYP/DZP. Die Energien bei 0,95 Å, 1,0 Å und 1,6 Å wurden mit Singlepointrechnungen bestimmt.

Michael Schönnenbeck 02.03.09 Bachelorarbeit
Die BHLYP-Potentialkurve zur Übertragung des Protons 2 im S1-Zustand (Abbildung 35) besitzt ein
globales Minimum bei 1,55 Å, dass um 0,18 eV günstiger ist als das lokale Minimum bei 1,0 Å. Das
Maximum befindet sich bei 1,15 Å. Die Barriere für den Protonentransfer ist mit 0,05 eV relativ klein, so
dass eine Gleichgewichtsverteilung zu erwarten ist, die stark in die Richtung des keto-Isomers verschoben
ist.
Die mit BLYP-optimierte Potentialkurve (Abbildung 36) weist im ersten angeregten Zustand bei 1,25 Å
einen plötzlichen Energieabfall von 0.78 eV auf. An dieser Stelle wurde eine Gleichgewichtsstruktur mit
einem CNNC-Torsionswinkel von 87° gefunden, während bei 1,20 Å eine planare Struktur vorliegt. Die
gewinkelte Struktur bei 1,25 Å besitzt einen starken CT-Charakter. Die erste Anregungsenergie von 1,19 eV
(bei 1,0 Å) ist im Vergleich zur Berechnung mit dem BHLYP-Funktional (3,05 eV) viel zu niedrig, was
ebenfalls für einen falsch charakterisieren Zustand spricht. Außerdem weist der S1-Zustand keine
nennenswerte Oszillatorstärke auf. Bei 1,0 Å ist ein Minimum, dass in etwa dem der BHLYP-Optimierungen
entspricht. Die Kurve fällt ab 1,25 Å kontinuierlich ab, da es sich aber um einen artifiziellen Zustand handelt,
kann man keine Aussage darüber treffen, ob der Protonentransfer tatsächlich stattfindet. Der S2-Zustand
hingegen ist erlaubt und zeigt einen deutlichen Energiegewinn mit zunehmendem OH-Abstand. Vermutlich
handelt es sich hierbei um den tatsächlichen S1-Zustand.
38
Abbildung 36: Potentialkurve entlang der Koordinate des Übergangs von der diol- zur keto-Form bei Übertragung von Proton 2 im Grundzustand und den ersten fünf angeregten Zuständen. Geometrieoptimierung im ersten angeregten Zustand mit TDDFT/BLYP/DZP. Die Rechnung bei 1,55 Å ist nicht konvergiert, daher wurde der Punkt interpoliert.
Michael Schönnenbeck 02.03.09 Bachelorarbeit
Beim Vergleich der mit TDDFT/BHLYP/DZP berechneten Potentiale zur Übertragung der Protonen 1 und 2
zeigt sich, dass die Übertragung des zweiten Protons günstiger ist als die des ersten. Zum Einen weist die
Potentialkurve für den Transfer von Proton 2 eine geringere Barriere und einen größeren Energiegewinn auf,
zum Anderen ist die keto-Form um 0,19 eV stabiler. Ein sicherer Vergleich der BLYP-Potentialkurven lässt
sich aufgrund der gefundenen CT-Artefakte nicht machen. Wenn die erste Anregung tatsächlich in den hier
gefundenen S2-Zustand erfolgt, ist von einem Transfer der Protonen 1 und 2 auszugehen. Dies müsste jedoch
auf höheren theoretischen Niveaus untersucht werden. Im Rahmen dieser Arbeit war dies jedoch aus
Zeitgründen nicht möglich.
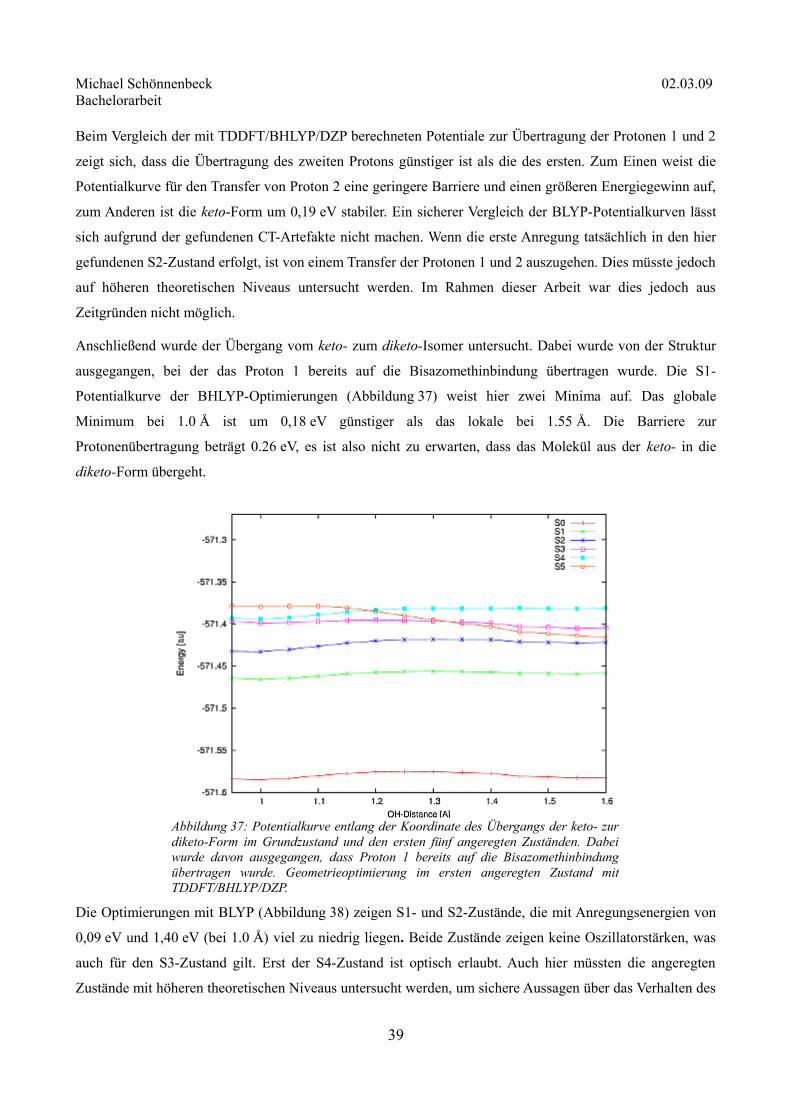
Anschließend wurde der Übergang vom keto- zum diketo-Isomer untersucht. Dabei wurde von der Struktur
ausgegangen, bei der das Proton 1 bereits auf die Bisazomethinbindung übertragen wurde. Die S1-
Potentialkurve der BHLYP-Optimierungen (Abbildung 37) weist hier zwei Minima auf. Das globale
Minimum bei 1.0 Å ist um 0,18 eV günstiger als das lokale bei 1.55 Å. Die Barriere zur
Protonenübertragung beträgt 0.26 eV, es ist also nicht zu erwarten, dass das Molekül aus der keto- in die
diketo-Form übergeht.
Die Optimierungen mit BLYP (Abbildung 38) zeigen S1- und S2-Zustände, die mit Anregungsenergien von
0,09 eV und 1,40 eV (bei 1.0 Å) viel zu niedrig liegen. Beide Zustände zeigen keine Oszillatorstärken, was
auch für den S3-Zustand gilt. Erst der S4-Zustand ist optisch erlaubt. Auch hier müssten die angeregten
Zustände mit höheren theoretischen Niveaus untersucht werden, um sichere Aussagen über das Verhalten des
39
Abbildung 37: Potentialkurve entlang der Koordinate des Übergangs der keto- zur diketo-Form im Grundzustand und den ersten fünf angeregten Zuständen. Dabei wurde davon ausgegangen, dass Proton 1 bereits auf die Bisazomethinbindung übertragen wurde. Geometrieoptimierung im ersten angeregten Zustand mit TDDFT/BHLYP/DZP.

Michael Schönnenbeck 02.03.09 Bachelorarbeit
Moleküls treffen zu können.
Da der diol-keto-Übergang des Protons 2 energetisch günstiger ist als der des Protons 1, wäre es sinnvoll, bei
zukünftigen Untersuchungen auch die Potentialkurven für den keto-diketo-Transfer des Protons 1 zu
untersuchen.
40
Abbildung 38: Potentialkurve entlang der Koordinate des Übergangs der keto- zur diketo-Form im Grundzustand und den ersten fünf angeregten Zuständen. Dabei wurde davon ausgegangen, dass Proton 1 bereits auf die Bisazomethinbindung übertragen wurde. Geometrieoptimierung im ersten angeregten Zustand mit TDDFT/BLYP/DZP.

Michael Schönnenbeck 02.03.09 Bachelorarbeit
6. Zusammenfassung und Ausblick
Die Berechnungen entlang der Rotationskoordinate am Molekül 1 (Abschnitt 4.2) zeigen im Grundzustand
eine stabile Geometrie in trans-Stellung. Im ersten angeregten Zustand wurden sowohl für das cis- als auch
das trans-Isomer stabile Gleichgewichtsstrukturen gefunden, wobei die trans-Konfiguration die energetisch
günstigere ist. Bei den Berechnungen der angeregten Zustände mit TDDFT/BLYP/DZP wurden artifizielle
Energieminima für die gewinkelten Strukturen gefunden, die auf die kategorische Unterschätzung von CT-
Zuständen durch reine GGA-Funktionale zurückzuführen ist. Die artifiziellen Minima konnten jedoch durch
den Vergleich mit TDDFT/BHLYP/DZP eindeutig identifiziert werden. Absorption und Fluoreszenz des
Moleküls finden im UV-Bereich statt.
Weiterhin ist davon auszugehen, dass im angeregten Zustand ein intramolekularer Protonentransfer
(Abschnitt 4.3) stattfindet, wobei anhand der Berechnungen mit TDDFT/BHLYP/DZP von einer
Gleichgewichtsverteilung zwischen der diol- und der keto-Form auszugehen ist.
Die Untersuchung der Rotation um den CNNC-Torsionswinkel des Moleküls 2 lieferte im Grundzustand
(Abschnitt 5.3) Energieminima in cis- und trans-Stellung mit einer hohen Rotationsbarriere. Auch hier ist
das trans-Isomer das energetisch günstigere. Für die Rotation im ersten angeregten Zustand (Abschnitt 5.4)
wurden ebenfalls Minima in cis- und trans-Stellung gefunden. Auch hier treten CT-Artefakte bei den
Untersuchungen mit dem BLYP-Funktional auf, die aber durch den Vergleich mit TDDFT/BHLYP/DZP
bereinigt werden konnten.
Über den Protonentransfer im S1-Zustand (Abschnitt 5.5) lassen sich nur anhand der Untersuchungen mit
TDDFT/BHLYP/DZP klare Aussagen treffen, da die mit BLYP-Funktionalen berechneten Potentialkurven
für die ersten angeregten Zustände nicht korrekt beschrieben wurden. Die mit BHLYP berechneten
Potentialkurven zeigen, dass die Übertragung des Protons 2 auf die Bisazomethinbindung etwas günstiger ist
als die des Protons 1. Der Übergang in die diketo-Form ist nicht zu erwarten, da das keto-Isomer günstigere
Energien aufweist.
Die Untersuchungen an den Molekülen 1 und 2 zeigen, dass sich reine GGA Funktionale nicht zur
Beschreibung von Bisazomethinfarbstoffen eignen, da hier entlang der CNNC-Reaktionskoordinate CT-
Zustände auftreten, die nicht korrekt beschrieben werden können. Die Verwendung von Hybridfunktionalen
hingegen liefert zuverlässige Ergebnisse, die die angeregten Zustände ausreichend gut beschreiben. Das zeigt
sich beim Vergleich zu den Studien an P.Y.101, bei dem die gleichen Probleme durch die Verwendung von
GGA-Funktionalen auftreten. Die Berechnungen mit ADC bestätigen die korrekte Beschreibung durch das
Hybridfunktional BHLYP.
41

Michael Schönnenbeck 02.03.09 Bachelorarbeit
Für weiterführende Studien an Bisazomethinfarbstoffen auf dem TDDFT-Niveau sollten ausschließlich
Hybridfunktionale verwendet werden. Dies können zum Einen weitere Untersuchungen des
Protonentransfers im angeregten Zustand sein, bei dem zuerst das Proton 2 und anschließend Proton 1
übertragen wird. Außerdem sollte untersucht werden, inwiefern sich die Substitution an den Strukturen 1 und
2 durch verschiedene funktionelle Gruppen auf die Eigenschaften der angeregten Zustände auswirken. Da es
eine große Anzahl an Bisazomethinfarbstoffen gibt, sind auch für diese genauere Studien von großem
Interesse.
42

Michael Schönnenbeck 02.03.09 Bachelorarbeit
7. Literaturverzeichnis
1: A. Dreuw; J. Plötner; L. Lorenz; J. Wachtveitl; J. E. Djanhan; J. Brüning; T. Metz; M. Bolte; M. U.
Schmidt, Angew. Chem., 2005,44, 7783-7786
2: J. Plötner; A. Dreuw, Phys. Chem. Chem. Phys., 2006,8, 1197-1204
3: L. Lorenz; J. Plötner; V. V. Matylitsky; A. Dreuw; J. Wachtveitl, J. Phys. Chem. A, 2007,111, 10891-10898
4: J. Plötner; A. Dreuw, Chem. Phys., 2008,347, 472-482
5: G. Wedler, Lehrbuch der Physikalischen Chemie, 5. Auflage,Wiley-VCH, 2004
6: P. Atkins; R. Friedman, Molecular Quantum Mechanics, 1. Auflage,Oxford, 2005
7: P. Atkins; J. Paula, Physikalische Chemie, 4. Auflage,Wiley-VCH, 2005
8: J. Franck, Trans. Faraday Soc., 1926,21, 536-542
9: E. Condon, Phys. Rev., 1928,32, 858-872
10: A. L. Sobolewski; W. Domcke; C. Dedonder-Lardeux; C. Jouvet, Phys. Chem. Chem. Phys., 2002,4,
1093-1100
11: A. Kusar; H. Nagano; T. Ogata; T. Nonaka; S. Kurihara, Angew. Chem., 2009,48, in Print
12: E. Shrödinger, Phys. Rev., 1926,28, 1049-1070
13: M. Born; J. R. Oppenheimer, Ann. Phys, 1927,38, 457-484
14: A. Szabo; N. S. Ostlund, Modern Quantum Chemistry, 1. Auflage,Dover, 1996
15: T. Koopmans, Physica, 1934,1, 104-113
16: C. C. J. Roothaan, Rev. Mod. Phys, 1951,23, 69
17: C. Møller; M. S. Plesset, Phys. Rev., 1934,46, 618-622
18: P. Hohenberg; W. Kohn, Phys. Rev. B, 1964,136, 864-871
19: W. Kohn; L. J. Sham, Phys. Rev. A, 1965,140, 1133
20: W. Koch; M.C. Holthausen, A Chemist's Guide to Density Functional Theory, 2. Auflage,Wiley-VCH,
2002
21: A. Dreuw; M. Head-Gordon, Chem. Rev., 2005,105, 4009-4037
22: E. Runge; E. K. U. Gross, Phys. Rev. Lett., 1984,52, 997-1000
43







