
Endbericht zum Forschungsprojekt
„Ermittlung des kritischen Sulfatgehaltes von Beton“
(FFG Projekt 839199)
Autoren
J. Tritthart, F. Mittermayr, D. Klammer und A. Saxer
August 2014

Inhalt
1. Einleitung ..................................................................................................................... 1
2. Reaktionen von Sulfat mit Zement und Schadensformen ............................................. 3
2.1 Ettringit (3CaO.Al2O3.3CaSO4.32H2O) ......................................................................... 3
2.1.1 Übersicht hinsichtlich Ettringitschädigung .......................................................... 4
2.1.1.1 Lösungen von Natriumsulfat (Na2SO4) ..................................................... 5
2.1.1.2 Lösungen von Magnesiumsulfat (MgSO4) ................................................ 5
2.1.1.3 Lösungen von Gips (CaSO4.2H2O) ........................................................... 6
2.1.1.4 Komposit-Zemente und Sulfat-Angriff ....................................................... 6
2.1.1.5 Mechanismus der Dehnung...................................................................... 6
2.2 Thaumasit (CaSiO3.CaCO3.CaSO4. 15H2O) ................................................................. 7
3. Sulfat im österreichischen Regelwerk ........................................................................... 8
3.1 Betonrohstoffe .............................................................................................................. 8
3.2 Expositionsklassen ....................................................................................................... 8
4. Ausgangssituation und Überlegungen zum Forschungsvorhaben ...............................12
5. Versuchsprogramm .....................................................................................................16
6. Prüfkörperherstellung, -auslagerung und Probenentnahme aus
bestehenden Bauwerken .............................................................................................20
6.1 Herstellung und Auslagerung der Mörtelproben ..........................................................20
6.1.1 Laborlagerung ................................................................................................. 21
6.1.2 Auslagerung im Kanalsystem von Linz ............................................................ 21
6.1.3 Betonproben ................................................................................................... 23
6.2 Probenentnahme .........................................................................................................24
6.2.1 Kanalsystem von Linz ..................................................................................... 24
6.2.2 Tunnel mit Sulfatschäden am Beton ................................................................ 25
7. Beschreibung der Untersuchungen .............................................................................30
7.1 In Labor und Kanalsystem ausgelagerte Proben .........................................................30
7.2 Aus Kanalsystem und Tunneln entnommene Beton- und Gebirgswasserproben .........34
7.3 Für die Entwicklung einer Prüfmethode zur nasschemischen Bestimmung
der Sulfatverteilung verwendete Betonproben .............................................................35
8. Ergebnisse der im Kanalsystem von Linz durchgeführten Untersuchungen ................36
8.1 Betonproben ................................................................................................................36
8.2 Mörtelproben ...............................................................................................................41
8.2.1 Probenbeschaffenheit ........................................................................................... 41
8.2.2 Festigkeiten ..................................................................................................... 41
8.2.3 Sulfatgehalt ..................................................................................................... 44
8.2.3.1 Mittlerer Sulfatgehalt ......................................................................................44
8.2.3.2 Sulfat Profil ..............................................................................................46
8.2.4 Konzentration der Inhaltsstoffe der Porenlösung ............................................. 46

9. Ergebnisse der im Labor ausgelagerten Mörtelproben ................................................51
9.1 Experimente zur Ettringitbildung ..................................................................................51
9.1.1 Festigkeiten ..................................................................................................... 51
9.1.2 Gesamtsulfatgehalt ......................................................................................... 54
9.1.3 Sulfatverteilung innerhalb der Proben ............................................................. 59
9.1.4 Porenlösung .................................................................................................... 60
9.2 Thaumasitbildung ........................................................................................................63
10. Nasschemische Bestimmung der Sulfatverteilung in Betonproben ..............................67
10.1 Allgemeines............................................................................................................67
10.2 Betone für die Untersuchungen ..............................................................................67
10.3 Herstellung von Betonbalken und Probenentnahme ..................................................69
10.4 Probenlagerung, Untersuchung und deren Ergebnisse .............................................70
10.4.1 Bestimmung des Sulfatgehalts ............................................................................ 70
10.4.2 Ergebnisse .......................................................................................................... 70
10.5 Diskussion der Ergebnisse .....................................................................................71
11. Untersuchung von Bauwerksschäden .........................................................................73
12. Zusammenfassung .........................................................................................................80
12.1 Allgemeines............................................................................................................80
12.2 Durchgeführte Untersuchungen..............................................................................81
12.3 Ergebnisse der Untersuchungen ............................................................................84
12.3.1 Im Kanalsystem von Linz ausgelagerte Proben ............................................... 84
12.3.2 Im Labor bei 20°C ausgelagerte Proben (Ettringitbildung) ............................... 85
12.3.3 Im Labor bei 5°C ausgelagerte Proben (Thaumasitbildung) ............................ 87
12.3.4 Betonschäden in Tunneln ................................................................................ 89
13. Diskussion der Ergebnisse und Schlussfolgerungen ...................................................91
13.1 Linzer Abwasserkanäle .............................................................................................91
13.2 Untersuchungen an Laborproben und aus Tunneln entnommenen Proben ............92
13.2.1 Im Labor bei 20°C ausgelagerte Proben (Ettringitbildung) ............................... 92
13.2.2 Im Labor bei 5°C ausgelagerte Proben (Thaumasitbildung) ............................ 94
13.2.3 Aus Tunneln mit Betonschäden entnommene Proben ..................................... 95
14. Empfehlungen für die Praxis ...........................................................................................96
15. Literatur ........................................................................................................................ 100

1
1. Einleitung
Es ist seit langer Zeit bekannt, dass Sulfationen Treiberscheinungen von Beton verursachen
und ihn solcherart schädigen können [1-7]. Dennoch ist der Zusatz von Sulfat in Form von
Gips (CaSO4·2H2O) bei der Zementherstellung zur Regelung der Erstarrungszeiten
unverzichtbar, die zugemahlene Gipsmenge ist aber durch das Regelwerk limitiert. Ebenso
darf der Sulfatgehalt von Gesteinskörnungen den in der entsprechenden Norm festgelegten
Grenzwert nicht übersteigen. Wenn Beton nachträglich in Kontakt zu sulfathaltigem Wasser
oder Boden kommt, können die in der wässrigen Phase gelösten Sulfationen durch Diffusion
in den Beton eindringen und Treiberscheinungen verursachen [8-11]. Diese Schädigungsart
ist gefürchtet, weil es sehr lange dauern kann, bis der Sulfatgehalt entsprechend stark
überhöht ist und die Schädigung augenscheinlich wird (Rissbildungen, etc.). Zur Beurteilung
der Aggressivität der mit dem Beton in Kontakt stehenden Wässer bzw. Böden sind im
Regelwerk Expositionsklassen mit Sulfat-Grenzkonzentrationen festgelegt, bei deren
Überschreitung einem Angriff entweder durch betontechnische Maßnahmen vorgebeugt
werden kann oder der Kontakt zum Beton unterbunden werden muss. Für die Beurteilung der
Expositionsklasse wird von konstant bleibenden Konzentrationen ausgegangen und es sind
keine Angaben enthalten, wie die Beurteilung bei expositionsklassenüberschreitenden
Konzentrationsschwankungen erfolgen soll. Dies erscheint praxisfern, weil die
Sulfatkonzentration von natürlichen Wässern lokalen und saisonalen Schwankungen
unterliegt. Es kann daher mittels der im Regelwerk enthaltenen Grenzwerte keine gesicherte
Prognose erstellt werden, ob in einem konkreten Fall eine akute Schadensgefahr besteht. Dies
erwies sich etwa im Fall des Kanalsystems der Stadt Linz als gravierender Nachteil, wo es
zeitweise zu expositionsklassenüberschreitenden Schwankungen der Sulfatkonzentration
gekommen ist, weil in das dortige Kanalsystem nicht nur häusliche sondern auch Abwässer
aus Industriebetrieben eingeleitet werden. Da ein solcher Fall im Regelwerk nicht
berücksichtigt ist, konnte die Frage nach der Beurteilung der Expositionsklasse nicht
beantwortet werden (mangels Alternativen werden zur Abschätzung der Schädigungsgefahr
auch bei Abwässern die Grenzwerte der Norm herangezogen obwohl diese nur für natürliche
Wässer gelten).
Ferner sind im Regelwerk keine Angaben enthalten, in welchem Umfang Sulfat von dem
jeweiligen Beton aufgenommen werden kann, ohne eine Schädigung zu bewirken. Die
Schädigung des Betons ist aber nicht unmittelbar von der Sulfatkonzentration des mit ihm in
Kontakt stehenden Wassers abhängig, sondern insbesondere von der Menge des schon in
den Beton eingedrungenen Sulfates [10, 11]. Aus diesem Grund ist für Prognosen der

2
Schädigung in jedem konkreten Fall der Sulfatgehalt des Betons, bezogen auf den
Zementgehalt, von entscheidender Bedeutung.
Ein weiterer Nachteil des gültigen Regelwerkes ist darin zu sehen, dass nur der
Sulfatschädigung durch Ettringitbildung, also der Treibgefahr Rechnung getragen wird, nicht
aber der Sulfatschädigung durch Thaumasitbildung. Dies ist vermutlich deshalb so, weil die
Thaumasit-Schädigung erst seit etwa den 1990-er Jahren internationale Aufmerksamkeit
erregt hat und hauptsächlich nur bei Temperaturen unterhalb von etwa 15°C stattfindet bzw.
gefährlich wird [12-23]. Dabei kommt es zu einer Zermürbung (Aufweichung) des
Zementsteins [7, 13, 15, 16, 19, 22, 24-53]. Derlei Schäden sind in österreichischen Straßen-
bzw. Eisenbahntunneln aufgetreten und es konnte nicht gesagt werden, welche Reaktionen
bzw. Mechanismen der Betonschädigung zugrunde lagen [54-58]. Dazu kommt, dass den
Autoren keine im bestehenden Regelwerk verankerte Prüfmethode bekannt war, mit der die
Beständigkeit eines bestehenden Betons gegenüber einem Sulfatangriff getestet werden
kann. Bei hydraulischen Bindemitteln bzw. Beton-Zusatzstoffen (ÖN EN 196-1 für Zement [59],
ÖN EN 450-1 für Flugasche [60]) wird gewöhnlich davon ausgegangen, dass die
Dauerhaftigkeit eines damit hergestelltem Betons gegeben ist, wenn die am Ort der
Verwendung hinsichtlich Dauerhaftigkeit geltenden Regeln erfüllt sind. Dies ist aber nicht zu
verallgemeinern, denn in allen drei Teilen der ÖN B 3309 „Aufbereitete, hydraulisch wirkende
Zusatzstoffe für die Betonherstellung“ ist die Forderung enthalten, dass die Sulfatbeständigkeit
im Zuge der Erstprüfung mittels des Verfahrens von Koch-Steinegger nachgewiesen werden
muss (beschrieben im Anhang B der ÖN B 3309-1) [61].
In Zusammenhang mit dem Sulfatangriff auf Beton bestanden also eine Reihe offener Fragen
und die Hauptzielsetzung des gegenständlichen Forschungsvorhabens war, herauszufinden
a) wie die Expositionsklasse bei schwankenden Sulfatkonzentrationen im angreifenden
Wasser beurteilt werden soll, b) wie schnell der Sulfatgehalt in Abhängigkeit von der
Zementsorte und Rezeptur (W/Z-Wert, etc.) ansteigt, c) ab welchem Sulfatgehalt der Beton
schadhaft wird und Schutzmaßnahmen erforderlich werden sowie d) welche Mechanismen der
Thaumasitbildung zugrunde liegen und wie eine Thaumasitschädigung vermieden werden
kann. Dazu bedurfte es einer zielorientierten Vorgangsweise unter Einbeziehung von
detaillierten mineralogischen, chemischen und mikrostrukturellen Untersuchungen in Hinblick
auf die Bildung von Sulfatverbindungen wie Ettringit, Gips und Thaumasit. Wichtig erschien
ferner die Änderung des Volumens von Proben in Abhängigkeit von deren Sulfatgehalt. Ein
verbessertes Verständnis der Angriffsmechanismen a) bei nicht konstanter Sulfatanlieferung
durch das angreifende Wasser und b) bei Temperaturen unter etwa 15°C und der Anwesenheit

3
von Karbonat wurde als die Basis angesehen, um in weiterer Folge verbesserte Testmethoden
entwickeln bzw. Angaben hinsichtlich der im Einzelfall erforderlichen Schutzmaßnahmen
ableiten zu können.
Nachfolgend wird zunächst ein kurzer, schematischer Überblick über die zur Ettringit- bzw.
Thaumasitbildung führenden Reaktionen gegeben und danach auf das bestehende Regelwerk
eingegangen. Nach Diskussion der wichtigsten offenen Fragen wird der Plan für die
Untersuchungen im Labor und von Schäden an Bauwerken besprochen. Danach werden die
durchgeführten Versuche und die erhaltenen Ergebnisse sowie die daraus gezogenen
Schlüsse beschrieben. Letztlich wird noch auf Empfehlungen für die Praxis eingegangen, die
aus den Ergebnissen abgeleitet wurden.
2. Reaktionen von Sulfat mit Zement und Schadensformen
2.1 Ettringit (3CaO.Al2O3.3CaSO4.32H2O)
Während der Anfangsphase der Hydratation von Zement reagieren das im Zementklinker
enthaltene Tricalciumaluminat (3CaO.Al2O3; kurz C3A genannt) bzw. die aluminatisch-
ferritischen Klinkerbestandteile mit dem als Erstarrungsverzögerer zugemahlenen Gips, wobei
sich Mischkristalle der Zusammensetzung 3CaO(Al2O3,Fe2O3)3CaSO4.xH2O bilden, die auch
als AFt-Phase bezeichnet werden (A für Al2O3; F für Fe2O3 und t für tri, also 3CaSO4), deren
eines Endglied der Ettringit ist:
3CaO.Al2O3 + 3CaSO4 + 32H2O 3CaO.Al2O3.3CaSO4.32H2O (Ettringit; auch als
Trisulfat bezeichnet)
Da der Sulfatgehat von Zement nicht so hoch ist, dass alles in normalem Klinker enthaltene
C3A in Ettringit umgewandelt werden kann, bildet sich aus dem primär gebildeten Ettringit
mit dem restlichen C3A des sufatärmere "Monosulfat":
3CaO.Al2O3.3CaSO4.32H2O + 2(3CaO.Al2O3) + 4H2O 3(3CaO.Al2O3.CaSO4.12H2O)
„Monosulfat“
Ist nach der Umwandlung des Ettringit in Monosulfat noch immer überschüssiges C3A
vorhanden, so reagiert es unter Aufnahme von Ca(OH) 2:

4
3CaO.Al2O3+Ca(OH)2+(x-1)H2O 4CaO.Al2O3.xH2O (x = 13 oder 19; „Aluminathydrat“)
Wenn Beton mehr Sulfat enthält, als im zugemahlenen Gips enthalten ist, etwa wenn Zement
mit Gips vermischt wird oder wenn nachträglich auf den erhärteten Beton sulfathaltige Wässer
einwirken, reagieren die aluminatischen Phasen des hydratisierten Zementes zu Ettringit. Da
so krasse Fehler wie das Mischen von Zement mit Gips nur sehr selten vorkommen (irrtümlich
oder aus Unkenntnis der Unverträglichkeit der beiden Bindemittel), hat nahezu nur das
nachträgliche Eindringen von Sulfat praktische Bedeutung. Das Sulfat dringt dabei im Wege
der Diffusion in den Beton ein. Ettringit hat ein niedriges spezifisches Gewicht und benötigt
daher viel Platz. Zunächst wird der vorhandene Porenraum (Kapillarporen) gefüllt, weshalb es
in der Anfangsphase des Angriffs zu einer Abnahme der Porosität des Betons und zu einer
Zunahme seiner Festigkeit kommt. Erst danach werden die Porenwände durch die weiterhin
stattfindende Ettringitbildung weggedrückt und es kommt zu einem Dehnprozess, der zunächst
zu Rissbildungen führt und bis zum Zerfall des Betons führen kann. Da der Diffusionstransport
des Sulfats von außen in den Beton hinein ein sehr langsamer Prozess ist, dauert es lange bis
Schäden erkennbar werden. Wenn etwa die an der Baustelle vorhandenen Wässer bzw.
Böden nicht vor Baubeginn untersucht wurden und keine vorbeugenden Schutzmaßnahmen
getroffen werden, ist das Bauwerk längst fertig, bis es nach und nach zu Schäden kommt.
Diese Schadensform ist daher gefürchtet, kann heute aber durch Beachtung und Einhaltung
des Regelwerkes vermieden werden [62].
2.1.1 Übersicht hinsichtlich Ettringitschädigung
Die nachfolgende kurze Übersicht zur Ettringitschädigung ist im Wesentlichen aus dem Buch
„Cement Chemistry“ von H. F. W. Taylor entnommen [63]. Danach besteht Übereinstimmung,
dass die, durch fortwährend eindringendes Sulfat verursachte Ettringitbildung zur Dehnung
des Betons, zu Rissbildungen, Festigkeitsverlust und letztlich zum völligen Zerfall des Betons
führen kann. Es besteht auch Übereinstimmung, dass ein sehr wesentlicher Faktor, der die
Stärke des Angriffs beeinflusst, die Geschwindigkeit ist, mit der Sulfationen in den Beton
eindiffundieren können. Je weniger porös der Beton ist (niedriger W/B-Wert, gute Verdichtung,
etc.) umso langsamer kann Sulfat eindiffundieren und umso höher ist der Widerstand gegen
einen Angriff. Während in Forschungsarbeiten das Hauptaugenmerk zumeist auf das
Dehnungsverhalten und die Rissbildung gelegt wurde, zeigt die Praxis, dass die
Festigkeitseinbußen bedeutender sind. Die Schädigung kann durch einen dichten Beton mit
geringer Permeabilität und durch Verwendung eines Zementes mit erhöhtem Sulfatwiderstand

5
(HS-Zement, C3A-arm bzw. frei) oder - mit Vorbehalt - durch Komposit-Zemente minimiert
werden. Auch ist allgemein anerkannt, dass die Art des eindringenden Sulfatsalzes von nicht
unerheblichem Einfluss ist. Nachfolgend werden die wichtigsten Zusammenhänge kurz
geschildert.
2.1.1.1 Lösungen von Natriumsulfat (Na2SO4)
Zuerst wird das eindringende Sulfat von vorhandenem Monosulfat gebunden, das dadurch in
Ettringit umgewandelt wird. Der Ettringit bildet sich in Form von Mikrokristallen aus, die mit den
Calciumsilikathydraten (C-S-H-Phase) eng verwachsenen und mittels Röntgendiffraktometrie
(XRD) gut erfassbar sind, aber beim Scannen im Elektronenmikroskop nicht als eigene Phase
ausgemacht werden können, obwohl Mikrosondenanalysen auf ihre Anwesenheit hinweisen.
In Oberflächennähe fällt das Ca/Si-Verhältnis der C-S-H-Phase ab, der Ca(OH)2 Gehalt wird
reduziert und Gips gebildet, der teilweise mit der C-S-H-Phase vermischt ist und teilweise in
Form von kleinen Adern vorliegt, die oft parallel zur Oberfläche verlaufen. In weiterer Folge
bilden sich innerhalb der Bindemittelmatrix Risse, die oft mit den Gipsadern
zusammenhängen.
Für die Ettringitbildung aus dem Monosulfat sind Ca+2-, SO4-2-Ionen und Wasser notwendig.
Das Sulfat kommt vom angreifenden Wasser, das Calcium vom Ca(OH)2 und - nach dessen
Verbrauch - von der C-S-H-Phase. In einem späteren Stadium wird Gips gebildet, wofür wieder
Ca+2-ionen benötigt werden. Daher wird angenommen, dass Na2SO4-Lösungen mit der Zeit
auch die C-S-H-Phase angreifen (das Ca/Si-Verhältnis nimmt ab). Versuche mit Pasten von
erhärtetem C3S haben gezeigt, dass die Pasten von einer 0,15 molarer Na2SO4-Lösung (~14
000 mg SO4-2/l) nur langsam angegriffen wurden, dass aber konzentrierte Lösungen auch C2S-
Pasten angreifen.
2.1.1.2 Lösungen von Magnesiumsulfat (MgSO4)
Die Lagerung von Zementpasten bzw. –mörteln führt an der Oberfläche zur Bildung einer
beinahe durchgehenden Schicht von einem Gemisch aus Mg(OH)2 (Brucit) und CaSO4 (Gips),
mit dem Brucit auf der Außenseite. An den Kanten von würfeligen Prüfkörpern war der Angriff
stärker und es hat sich auch ein Magnesiumsilikathydrat gebildet. Aus röntgenographischen
und elektronenmikroskopischen Untersuchungen wurde abgeleitet, dass es sich dabei um
einen schlecht kristallisierten Serpentin handelt (3MgO.2SiO2.2H2O), dessen Mg/Si-Verhältnis
zwischen 4:1 und 1:1 liegt. Die Entkalkung der C-S-H-Phase ist stärker ausgeprägt als beim
Angriff durch Na2SO4 und führt letztlich zur völligen Zerstörung der C-S-H-Phase mit der

6
Bildung eines Silica-Gels. Die stärkere Entkalkung der C-S-H-Phase beruht auf der geringen
Löslichkeit des Brucits, dessen Bildung auch zu einer Abnahme des pH-Wertes der
Porenlösung führen kann. Bei Verwendung eines erhöht Sulfat-beständigen Zementes werden
die Entkalkung der C-S-H-Phase und die Rissbildung stark verringert.
2.1.1.3 Lösungen von Gips (CaSO4.2H2O)
Obwohl die Löslichkeit von Gips im Vergleich mit Na2SO4 nicht besonders gut ist, kann auch
eine Gipslösung einen Angriff bewirken. Alle Ionen (Ca+2, SO4-2), die zur Ettringitbildung aus
Monosulfat benötigt werden, sind ja in der Lösung vorhanden. Eine Entkalkung der C-S-H-
Phase muss hier nicht stattfinden. Natürliche Zuschläge, die Gips enthalten, führen zu einem
sogenannten internen Angriff. Dabei wird Gips durch Ca(OH)2 ersetzt und das Sulfat wird im
entstehenden Ettringit gebunden, dessen Bildung Treiben verursachen kann.
2.1.1.4 Komposit-Zemente und Sulfat-Angriff
Der teilweise Ersatz von Klinker durch mineralische Zumahlstoffe kann die Sulfatbeständigkeit
verbessern bzw. die Stärke des Angriffs verringern. Sie wirken sich im Allgemeinen gegen
einen Angriff durch Na2SO4-Lösungen günstiger aus als gegen einen Angriff durch MgSO4-
Lösungen. Wegen der langsameren Erhärtung von Komposit-Zementen hängt der positive
Effekt aber stark von der Nachbehandlung vor dem Kontakt zu Sulfatlösungen ab. Wie
ausgeführt, beruht ein großer Teil der positiven Wirkung von puzzolanischen bzw. latent
hydraulischen Zumahlstoffen auf der gegenüber normalem Portlandzement dichteren
Bindemittelmatrix und dem dadurch bewirkten langsameren Eindringen von Sulfat.
2.1.1.5 Mechanismus der Dehnung
Die Expansion wird zumeist mit der Zunahme des Feststoffvolumens erklärt, das mit der
Ettringitbildung verbunden ist. Diese ist aber ungefähr gleich groß wie die, die bei der
Hydratation von C3S und der Bildung der C-S-H-Phase und Ca(OH)2 eintritt. Von den
existierenden Hypothesen zum Ettringit-Treiben sind die nachfolgenden drei Hypothesen am
wichtigsten:
1.) Die Rissbildung wird durch ein richtungsorientiertes Kristallwachstum verursacht.
2.) Ettringit Kristalle kolloidaler Dimensionen nehmen Wasser auf, wodurch ein Druck
entsteht, ähnlich dem, der durch Osmose verursacht wird.
3.) Die Expansion wird durch Änderungen in den Quellungseigenschaften des Zementgels

7
verursacht, wofür die Ettringitbildung nebensächlich ist und möglicherweise nur einen
indirekten Einfluss hat.
Welche der Hypothesen den größten Wert hat, kann nicht gesagt werden, weil es dazu keine
übereinstimmende Meinung gibt. Der Umstand, dass die Dehnung, wie festgestellt wurde,
nicht gleichzeitig mit der Ettringitbildung stattfindet sondern erst später, spricht gegen die erste
Hypothese. Ein seriöser Einwand, warum auch die zweite Hypothese anzuzweifeln ist, liegt
darin, dass es schwer verständlich ist, warum die im C-S-H-Gel verteilten Ettringit-Kristalle
Wasser stärker anziehen sollten als das Gel selbst. Insgesamt spricht daher viel für die 3.
Hypothese, weil das Aufsaugen von Wasser durch ein Gel starke Expansionsdrücke erzeugen
kann. Eine Möglichkeit liegt darin, dass das für die Ettringitbildung unmittelbar benötigte
Wasser aus der umgebenden C-S-H-Phase stammt, welche dadurch dehydriert wird und dass
es erst dann zur Expansion kommt, wenn Wasser von außen eingesaugt wird.
2.2 Thaumasit (CaSiO3.CaCO3.CaSO4. 15H2O)
Wie die chemische Formel von Thaumasit zeigt, ist kein Aluminat als Bindungspartner des
Calciums enthalten, sondern neben dem Sulfat noch Karbonat und Silikat. Außerdem enthält
die Verbindung relativ viel gebundenes Wasser. Das Karbonat kann von der Auflösung
Karbonatischer Gesteinskörnungen oder von Wässern stammen, die mit dem Beton in Kontakt
stehen und die CO2 aus der Luft aufgenommen haben oder aus anderen Gründen Karbonat
enthalten. Das Silikat stammt vom hydratisierten Tri- bzw. Di-Calciumsilikat des
Zementklinkers (C3S, C2S). Da Calciumsilikathydrate die Festigkeitsträger des Zementsteines
sind, führt die Thaumasitbildung zu einem Verlust der Festigkeit des Betons bis hin zu dessen
völligem „Aufweichen“ aber zu keiner Volumsinstabilität. Diese Art der Betonschädigung ist
erst seit einigen Jahrzehnten bekannt und wurde in Österreich zuerst von W. Lukas 1975
beschrieben, der bei der Untersuchung von Schäden an der Spritzbetonauskleidung von
Stollen die Bildung von Thaumasit und Woodfordit als Ursache der Zerstörung festgestellt hat
[64]. Darin ist angegeben, dass sich zuerst aus dem C3A bis zu dessen vollständigen
Verbrauch Ettringit gebildet hat und dass es danach zu einer Umwandlung in Thaumasit kam.
Woodfordit ist darin als ein „SiO2 haltiger Ettringit“ bezeichnet. Heute gilt als gesichert, dass
Thaumasit entweder aus Ettringit oder direkt durch Reaktion von Sulfat mit Karbonat (aus CO3-
2-Ionen oder aus atmosphärischem CO2) und Silikat entstehen kann [12, 13, 16, 21, 34, 65,
66]. Am direkten Weg kommt es zu nachstehenden Reaktionen:
3CaO.2SiO2.3H2O (Tobermorit) + 2(CaSO4.2H2O) + 2CaCO3 + 24H2O

8
2[CaSiO3.CaCO3.CaSO4.15H2O] (Thaumasit) + Ca(OH)2
bzw:
3CaO.2SiO2.3H2O + 2(CaSO4.2H2O) + CaCO3 + CO2 + 23H2O
2[CaSiO3.CaCO3.CaSO4.15H2O]
Bei Bildung aus Ettringit wird das Aluminat über komplizierte Reaktionen durch Silikat ersetzt
und Karbonat eingebaut [16]. Der Vorgang führt aber zu keiner kontinuierlichen Zunahme des
Silikatgehaltes im Ettringit denn es gibt eine Reihe von Mischkristallenen mit unterschiedlichen
Aluminat-, Silikat- und Karbonatgehalt. Die zwischen den Endgliedern Ettringit bzw. Thaumasit
liegenden Verbindungen werden in einer Sammelbezeichnung als Woodfordit bezeichnet. Der
Bildungsweg über die Woodfordit Route ist als schneller beschrieben als der direkte Weg [16].
Sowohl der direkte Weg als auch die Woodfordit Route finden nach Bensted nur bei
Temperaturen unterhalb von 15°C statt [16]. Nach Schmidt et. al. ist die Bildung grundsätzlich
auch bei höheren Temperaturen möglich [22].
3. Sulfat im österreichischen Regelwerk
3.1 Betonrohstoffe
Gemäß der ÖN EN 197-1 [67] darf der SO3-Gehalt von CEM I und CEM II Zement (mit
Ausnahme des CEM II/B-T) 3,5 %, von den übrigen Zementsorten 4,5 % nicht übersteigen. In
Gesteinskörnungen für die Betonherstellung ist der Sulfatgehalt gemäß der ÖN EN 12620 [68]
limitiert und je nach Kategorie unterschiedlich [≤0,2% bei Kategorie AS0,2, ≤0,8% bei Kategorie
AS0,8 und >0,8% bei Kategorie ASangegeben (die Grenzwerte gelten für alle Gesteinskörnungen
außer Hochofenstückschlacke)]. Das Zugabewasser von Beton darf gemäß der ÖN EN 1008
[69] nicht mehr als 2000 mg SO4-2/l enthalten.
3.2 Expositionsklassen
Grundlage zur Beurteilung angreifender Wässer ist die ÖNORM EN 206-1 [70] bzw. die
ÖNORM B 4710-1 [71]. Die EN 206-1 wurde als Rahmennorm entwickelt, weil „der Betonbau

9
in Europa unter verschiedenen klimatischen und geographischen Bedingungen, unter
verschiedenen Schutzniveaus und unter verschiedenen gut eingeführten regionalen
Gepflogenheiten und Erfahrungen angewandt wird“. Die ÖNORM B 4710-1 ist die nationale
Umsetzung der EN 206-1. Mit ihrem Erscheinen wurde u. a. die ÖNORM B 3305 [72] außer
Kraft gesetzt. Die für Grundwasser geltenden Werte sind in Tabelle 1 wiedergegeben. Sie
gelten für Wasser mit einer Temperatur von 5°C - 25°C und einer Fließgeschwindigkeit, die
klein genug ist, um näherungsweise hydrostatische Bedingungen anzunehmen.
Tabelle 1: Grenzwerte für Grundwasser (ÖN B 4710-1)
Angriffsart
chemisches
Merkmal
Expositionsklasse
XA1
(chemisch schwach
angreifende
Umgebung)
XA2
(chemisch mäßig
angreifende
Umgebung)
XA3
(chemisch stark
angreifende
Umgebung)
Treibend (T) SO42- mg/l ≥200 und 600 >600 und 3000 >3000 und 6000
Lösend (L) pH-Wert ≥6,5 und 5,5 <5,5 und 4,5 <4,5 und 4,0
Lösend (L) CO2 mg/l
angreifend
≥15 und 40 >40 und 100 >100 bis zur
Sättigung
Lösend (L) NH4+ mg/l ≥15 und 30 >30 und 60 >60 und 100
Lösend (L) Mg+2 mg/l ≥300 und 1000 >1000 und 3000 >3000 bis zur
Sättigung
Lösend (L) °dH 0-3 -- --
Hinsichtlich der Expositionsklasse bestimmt der schärfste Wert jedes einzelnen Merkmals die
Klasse. „Wenn zwei oder mehrere Merkmale zur selben Klasse führen, muss die Umgebung
der nächst höheren Klasse zugeordnet werden, sofern nicht in einer speziellen für diesen Fall
nachgewiesen ist, dass dies nicht erforderlich ist“. Bei Anwesenheit von Sulfat ist für die
Expositionsklasse XA1 ein maximaler Gehalt des Zementes an Tricalciumaluminat (C3A) von
3% vorgeschrieben, bei den Expositionsklassen XA2 und XA3 muss der Zement C3A-frei sein.
Da die ÖN B 3305 auch Sulfatgrenzwerte von Wässern gegenüber Beton mit normalem
Zement beinhaltet hat, erscheint es nötig, diese Norm zumindest für eine Diskussion
einzubeziehen. Die Grenzwerte sind in Tabelle 2 enthalten. Wie in der ÖN B 3305 vermerkt,
gelten die Grenzwerte im Unterschied zur ÖN B 4710-1 „für stehendes und fließendes, in
großen Mengen vorhandenes, unmittelbar angreifendes Wasser“. Dass es sich etwa bei

10
Abwässern um solches Wasser handelt, ist ein zusätzlicher Grund, der im gegenständlichen
Zusammenhang für die Einbeziehung der ÖN B 3305 spricht. Danach nimmt der Angriffsgrad
ab, „wenn das Wasser nur in geringer Menge ansteht und sich praktisch nicht bewegt, so dass
sich die angreifenden Bestandteile nur langsam erneuern können“.
Tabelle 2: Grenzwerte für die Beurteilung von Wässern gemäß ÖNORM B 3305
Angriffsgrade
nicht
angreifend
schwach
angreifend
stark
angreifend
sehr stark
angreifend
pH-Wert > 6,5 6,5-5,5 5,5-4,5 > 4,5
kalkaggressive
Kohlensäure (CO2;
mg/l)
< 15
15-30
30-60
>60
Gesamthärte (°dH) >3 3-0 tritt nicht auf tritt nicht auf
Ammonium
(NH4
+; mg/l)
<15 15-30 30-60 >60
Magnesium
(Mg+2
; mg/l)
<100 100-300 300-1500 >1500
Sulfat
(SO4
-2
mg/l)
PZ* und
EPZ**
<200 200-300 300-400 >400
Hochofen-
Zement
<400
400-500 500-600 >600
HS-
Zement***
<600 600-1500 1500-3000 >3000
*) Portlandzement; **)Eisenportlandzement; ***) erhöht sulfatbeständiger Zement (3%C3A)
Wie aus Tabelle 2 ersichtlich, werden Wässer mit einem SO4-2-Gehalt von über 200 mg/l als
aggressiv gegenüber Beton beurteilt, der mit PZ (Portlandzement; heute CEM I) bzw. EPZ
(Eisenportlandzement; heute CEM II B-S) hergestellt wurde (bei beiden Sorten keine
Begrenzung des C3A-Gehaltes). Wässer, die auf Beton einwirken, der mit einem erhöht
sulfatbeständigen Zement (HS-Zement; C3A-Gehalt: 3%) hergestellt wurde, werden bis zu
600 mg SO4-2/l als „nicht angreifend“ bewertet. Dies ist ein erheblicher Unterschied zur ÖN B
4710-1.
Bezüglich schwankender Sulfatkonzentrationen wäre noch zu erwähnen, dass in der ÖN B

11
5017 [73] der Hinweis enthalten ist, dass „die Beurteilung des chemischen Angriffs nach der
lang andauernden Belastung zu erfolgen hat“ („kurzfristige, z. B. bei Störfällen auftretende
Belastungen beanspruchen den Beton weniger und erlauben daher die Einstufung in einer um
mindestens 1 Stufe niedrigeren Klasse als dies auf Grund der Konzentration der Stoffe
vorzunehmen wäre“). Welche Studien bzw. Erfahrungswerte dieser Regelung zu Grunde
liegen, ist nicht angegeben. Alle wissenschaftlichen Untersuchungen des Sulfatangriffs, die
den Projektbeteiligten bekannt sind, wurden nicht unter schwankenden sondern bei
bestimmten SO4-2 Konzentrationen durchgeführt (im Labor bei definierten Bedingungen durch
Auslagerung in künstlich hergestellten Lösungen mit bestimmten Sulfatkonzentration bzw. in
„Naturversuchen“ durch Auslagerung der Prüfkörper in Meerwasser). Daher erschien diese
wichtige Frage trotz der in der ÖN B 5017 enthaltener Regelung als ungeklärt.
Anforderungen an die Betonqualität
Grundsätzlich wird vorausgesetzt, dass der Beton im Einklang mit den Regeln der ÖN B 4710-
1 hergestellt ist. Sowohl bei lösendem als auch treibendem Angriff ist für die Expositionsklasse
XA1 ein max. W/B-Wert von 0,55 und ein anrechenbarer Bindemittelgehalt von mindestens
300 kg/m³ vorgeschrieben. Bei XA2 darf der W/B-Wert 0,45 nicht übersteigen und der
anrechenbare Bindemittelgehalt muss mindestens 360 kg/m³ betragen. Bei XA3 muss ein HL-
SW-Beton gemäß ÖN B 5017 verwendet werden. Im Fall eines treibenden Angriffs ist für
einen, mit CEM I - Zement hergestellten Beton bei XA1 ein maximaler Gehalt des Zementes
an Tricalciumaluminat (C3A) von 3% vorgeschrieben, bei den Expositionsklassen XA2 und
XA3 muss der Zement C3A-frei sein. Im Fall, dass kein CEM I verwendet wird, ist bei den
erlaubten CEM II Zementen durch den Zementhersteller der Nachweis der Sulfatbeständigkeit
analog der ÖN B 3309 [61] zu erbringen. Außerdem darf die Zugabe von Zusatzstoffen 10 %
nicht übersteigen. Die ÖN B 3305 [72] hat selbst keine Angaben hinsichtlich der erforderlichen
Betonqualität enthalten, jedoch waren im Merkblatt 7 „Chemische Angriffe“ aus Zement und
Beton Anforderungen in Abhängigkeit des Angriffsgrades gemäß ÖN B 3305 angegeben.
Danach musste der Beton ab dem Angriffsgrad „schwach angreifend“ wasserundurchlässig
sein (W/Z-Wert gemäß der alten ÖN B 4200-10 [74] bei Nachweis am Frischbeton: 0,55) und
den übrigen Bestimmungen des Merkblattes hinsichtlich Art des Zuschlages, der Sieblinie, des
Konsistenzbereichs, der Nachbehandlung, etc. entsprechen. Beim Angriffsgrad „stark
angreifend“ war ein maximaler W/Z-Wert von 0,45 vorgeschrieben und bei „sehr stark
angreifend“ musste neben einem W/Z-Wert von 0,45 dafür Sorge getragen werden, dass kein
direkter Kontakt des Wassers mit dem Beton besteht.
Der etwa bei der Expositionsklasse XA2T vorgeschriebene niedrige W/B-Wert von ≤0,45 und

12
hohe Zementgehalt (≥ 360 kg/m³) bewirkt starke Erschwernisse bei der Betonverarbeitung,
besonders im Sommer, eine hohe Wärmeentwicklung sowie die Möglichkeit von autogenem
Schwinden und damit eine erhöhte Gefahr von Rissbildungen. In den ÖVBB-Richtlinien
„Weiße Wannen“, „Innenschalenbeton“ bzw. „Beton für Kläranlagen“ werden zur Verringerung
der Wärmeentwicklung und damit zur Verringerung der Rissneigung des Betons höhere W/B-
Werte zugelassen als in ÖNORM B 4710-1 angegeben, wenn die geforderten Eigenschaften
(Festigkeitsklasse, Beständigkeit gegen die Expositionsklasse) am Festbeton nachgewiesen
werden. Für den Nachweis der Sulfatbeständigkeit am Festbeton erschien deshalb eine
allgemeine Prüfmethode dringend erforderlich, weil damit die Herstellung eines besser
verarbeitbaren und wirtschaftlicheren Betons mit geringerem Bindemittelgehalt und
Zusatzmitteldosierung ermöglicht würde. Die Erfahrungen aus der Praxis zeigen, dass auch
Betone mit höheren W/B-Werten und gleichzeitig dichtem Betongefüge eine hohe
Widerstandsfähigkeit gegen treibenden Sulfatangriff aufweisen.
4. Ausgangssituation und Überlegungen zum Forschungsvorhaben
Schon aus der Tatsache, dass sich das Regelwerk nur auf konstante Sulfatkonzentrationen
von Wässern bzw. Böden in Kontakt zu Beton bezieht und keinerlei Hinweise enthält, wie die
Expositionsklasse bei schwankender Sulfatkonzentration erfolgen soll, ergeben sich
erhebliche Schwierigkeiten. So konnte eine Anfrage der LINZ-AG nach der Beurteilung der
Expositionklasse der im Kanalsystem der Stadt Linz enthaltenen Abwässer nicht klar
beantwortet werden, weil die darin enthaltenen Abwässer zeitweise stärkeren Schwankungen
unterliegen. Dies ist deshalb so, weil nicht nur häusliche sondern auch Abwässer von
Industriebetrieben eingeleitet werden, die zeitweise stärker sulfathaltig sind, so dass die
Sulfatkonzentration der Abwässer zumindest bereichsweise stärkeren Schwankungen
unterliegt. Im Bereich der Einleitung hat das Wasser kurzzeitig sogar mehr als 6000 mgSO4-2/l
enthalten und ein Kontakt des Betons zu dem Wasser hätte somit unterbunden werden
müssen. Auch war nicht bekannt, welche Zementsorte (C3A-frei oder nicht) verwendet wurde.
Maßnahmen zu Unterbindung des Kontakts der Abwässer zum Beton (etwa die Applikation
einer Kunststoffbeschichtung) waren aber schon deshalb nicht möglich, weil solche Arbeiten
unter Betrieb nicht ausgeführt werden können und keine Möglichkeit zur Umleitung der
Abwässer vorhanden war. Außerdem wären die damit verbundenen Kosten nicht zu
rechtfertigen, nur weil die zulässige Sulfatkonzentration überschritten war und somit keine
Sicherheit mehr bestand, dass kein Schaden entstehen kann (der Verlust der absoluten
Sicherheit bedeutet nicht, dass es deshalb notwendigerweise zu einem Schaden kommt).

13
Schäden am Beton waren keine sichtbar. Hier stellte sich die zusätzliche Frage, bis zu welchen
Sulfatgehalt des Betons kein Risiko einer Schädigung besteht. Der Schadensbildung gehen ja
das langsame Eindringen der Sulfationen und ein Anstieg des Sulfatgehaltes voraus.
Diesbezüglich ist im Regelwerk aber kein Hinweis enthalten. Daher konnte zu dem
geschilderten Sachverhalt in Linz nur die Mängel im Regelwerk und die Notwendigkeit von
Forschungsbedarf dargelegt, aber keine durchführbaren Lösungsvorschläge angeboten
werden. Der Forschungsbedarf erschien hier offensichtlich, weil die Annahme des Regelwerks
nach konstanten Schadstoffkonzentrationen im angreifenden Wasser praxisfremd ist und es
auch bei natürlichen Wässern zu saisonalen bzw. jahreszeitlichen Schwankungen der
Konzentration der Inhaltsstoffe kommt und daher die Beurteilung des Schadensrisikos auf
schwankende Konzentrationen ausgerichtet werden müsste (ist die höchste Konzentration,
der Tages-, Wochen- Monats- oder Jahresmittelwert relevant?).
Gravierende Schäden in Österreich, die durch Ettringitbildung an bestehenden Bauwerken
zufolge Kontaktes zu sulfatführenden Wässern oder Böden entstanden sind, waren den
Autoren nicht bekannt. Durch Ettringit verursachte Schäden sind meist auf Unkenntnis der
Zusammenhänge wie der Unverträglichkeit von Zement mit Gips zuzuschreiben. So ist
gelegentlich in Ausschreibungen zum Bau von Wohnhäusern der Passus enthalten, dass zur
Fixierung von Verteilerdosen der Elektroinstallation ein zementhaltiges Bindemittel verwendet
werden muss. Elektriker sind aber den Umgang mit Zement nicht gewohnt sondern benutzten
dazu meist Gips. Einerseits um der Ausschreibung nicht zuwider zu handeln und um
andererseits die Vorteile von Gips, wie dessen Geschmeidigkeit und rasches Ansteifen nicht
zu verlieren, wurde manchmal (in Ausnahmefällen) Zement mit Gips gemischt, weil sich die
handelnden Personen der Unverträglichkeit der beiden Bindemittel nicht bewusst waren.
Zumeist sind die Räume fertig verputzt oder sogar schon benutzt, wenn es zu Verwölbungen
und Rissbildungen des Putzes über Elektro-Verteilerdosen kommt. Auch kommt es
gelegentlich zu Schäden bei Berührungskontakt von Gips mit einem zementgebundenen
Mörtel, etwa wenn Fliesen auf einem Gipsputz mit einem zementgebundenen Fliesenkleber
aufgebracht werden und genügend Feuchtigkeit vorhanden ist (z. B. Toiletten, fensterlose
Badezimmer, etc.). Dabei handelt es sich allerdings um seltene und extreme Bedingungen.
Es waren aber Betonschäden in Tunneln bekannt, die durch Gebirgsstöcke mit Gipshaltigen
Bereichen führten. Diese Schäden sind nicht durch Ettringit sondern Thaumasit verursacht
worden. Dass Thaumasit im Regelwerk überhaupt nicht verankert ist, war ein weiterer Grund
für die Beantragung des Forschungsvorhabens. Eine Forschungsarbeit zur Sulfatkorrosion
durchzuführen, erschien auch deshalb interessant, weil die Überschreitung des sich aus den

14
Grenzwerten der Rohstoffe ergebenden zulässigen Sulfatgrenzwertes von Beton nicht
automatisch zu einer Schädigung führt, das Schadensbild bzw. der Schadensfortschritt je nach
Randbedingungen sehr verschieden sein kann und die Sulfatkorrosion insgesamt so vielfältig
und Variantenreich ist, dass es zu verschiedenen Details auch heute noch keine einheitliche
Auffassungen gibt [56, 75, 76].
Um den Einfluss schwankender Sulfatkonzentrationen verstehen zu lernen und gleichzeitig die
Nähe zur Praxis zu wahren, erschien es zielführend, neu hergestellte Proben mit bekannter
Rezeptur (unterschiedliche Zementsorten und Porosität) an Stellen unterschiedlicher
Sulfatkonzentrationen im Kanalsystem von Linz auszulagern (die Sulfatkonzentration des
Kanalwassers schwankt abschnittsweise zufolge der zeitweiligen Einleitung industrieller
Abwässer relativ stark) und hinsichtlich der Änderungen des Sulfat-Gehaltes, der Art der
entstehenden Sulfatverbindungen, etc. zu untersuchen. Auch der Bestimmung der
Veränderungen der Zusammensetzung der Porenlösung während der Lagerung wurde hohe
Relevanz beigemessen, weil die stattfindenden chemischen Reaktionen über die flüssige
Phase ablaufen und die Lage des sich zwischen den Feststoffen (Hydratationsprodukten) und
der flüssiger Phase (Porenlösung) einstellenden Gleichgewichtes von der Zusammensetzung
der flüssigen Phase abhängt. Hier war zu erwarten, dass sie sich durch Diffusionsausgleich
mit zunehmender Lagerungsdauer immer mehr jener der Lagerlösung annähert, weshalb für
ein tieferes Verständnis der Zusammenhänge die Bestimmung der Veränderungen der
Porenlösungszusammensetzung unbedingt nötig erschien. Solche Messungen an Beton bzw.
Mörtelproben mit baupraktisch niedrigem W/Z-Wert sind aber nach dem Kenntnisstand der
Projektbeteiligten im Zusammenhang mit der Sulfatkorrosion von Beton noch nicht
durchgeführt worden und beinhalten daher einen hohen Innovationsgehalt. Um hinsichtlich
des Einflusses schwankender Sulfatkonzentration des Wassers Aussagen zu erhalten, sollten
neue Proben auch im Labor ausgelagert werden, wobei die Sulfatkonzentration der
Lagerlösungen einerseits wechseln und andererseits konstant bleiben sollte. Die neu
hergestellten Proben sollten nicht nur unterschiedliche Rezeptur (W/Z-Wert) haben sondern
auch unterschiedliche Bindemittel beinhalten, um den Einfluss des C3A und puzzolanischer
Zumahlstoffe auf die Schädigung zu erfassen. Auch erschien es wichtig, den Beton des
Kanalsystems von Linz zu Beginn des Forschungsvorhabens und an dessen Ende zu
untersuchen, um Hinweise für die Geschwindigkeit der Sulfataufnahme zu bekommen und -
wenn möglich - Prognosen hinsichtlich der Zeit bis zum Erreichen einer kritischen Situation
erstellen zu können. Aus den Ergebnissen sollen nicht nur für den Praxisfall Linzer
Abwasserkanäle geltende Aussagen abgeleitet werden sondern auch solche von genereller
Bedeutung, etwa wie aussagekräftig im Allgemeinen die Kenntnis der Sulfatkonzentration des

15
angreifenden Wassers zusammen mit der Porosität eines Betons hinsichtlich des
Schadensrisikos ist.
Zur Klärung der Schadensmechanismen bei Thaumasitbildung sollten an Bauwerken
entstandene Schäden untersucht werden (Reaktionsprodukte, vorhandene Wässer, etc.). Aus
den Ergebnissen sollten dann Schlüsse hinsichtlich deren künftiger Vermeidung gezogen
werden können. Auch hier erschien die Einbeziehung von Laboruntersuchungen an neu
hergestellten Proben unabdingbar notwendig. Um den Einfluss der Temperatur zu
dokumentieren sollten kalksteinhaltige Proben (Karbonat in der Probe vorhanden) bei
unterschiedlichen Sulfatkonzentrationen der Lagerlösung a) im Labor bei 20°C und b) in einer
Kühltruhe bei etwa 5°C ausgelagert werden.
Abgesehen von den geschilderten Untersuchungen mangelte es auch an einer geeigneten
Methode, mit der der Sulfatgehalt von Beton nasschemisch mit der oft nötigen Feinabstufung
(<1 mm) zur Erfassung der Sulfatverteilung (bzw. anderer Schadstoffe wie Chlorid) bestimmt
kann. Dazu wurden Untersuchungen durchgeführt, die auf einer Methode aufbauten, die zur
Bestimmung des Sulfatprofils von Zementsteinen entwickelt wurde [77].
Aus all den geschilderten Gründen wurde mit der Industrie Kontakt aufgenommen und die
Einreichung eines Forschungsprojektes bei der FFG angeregt. Das Projekt wurde in weiterer
Folge seitens der Österreichischen Bautechnik Veranstaltungs GmbH (ÖBV) eingereicht. Als
Projektpartner waren beteiligt:
Industrielle Partner
-LINZ SERVICE GmbH
-Firma DSM Fine Chemicals Austria, Linz
-ÖBB
-ASFINAG Bau Management GmbH
-Vereinigung der österreichischen Zementindustrie (VÖZ)
Wissenschaftliche Stellen
-Institut für Materialprüfung und Baustofftechnologie mit angeschlossener TVFA für
Festigkeits und Materialprüfung (Technische Universität Graz)
-Institut für Angewandte Geowissenschaften (Technische Universität Graz)
-Arbeitsbereich Materialtechnologie (Univ. Innsbruck)

16
Sonstige Stellen
-Österreichischen Bautechnik Veranstaltungs GmbH (ÖBV; Beantragungen und
formelle Projektabwicklung))
Baurat DI Dr. H. Huber (Konsulent)
Die Bewilligung des mit 4 Jahren Laufzeit beantragten Projektes mit dem Titel „Ermittlung des
kritischen Sulfatgehaltes von Beton“ wurde im Juni 2009 erteilt.
5. Versuchsprogramm
Für die im Linzer Abwassersystem und im Labor in sulfathältigen Lagerlösungen
auszulagernden Proben wurden Mörtelproben a) Würfel mit 4 cm Kantenlänge und b) Prismen
der Dimension 4 x 4 x 16 cm vorgesehen. Die Würfel waren zur Untersuchung des
durchschnittlichen Sulfatgehaltes sowie des Sulfatprofils bestimmt und die Prismen zur
Bestimmung der Festigkeiten sowie zur Gewinnung von Porenlösung. Im Linzer Kanalsystem
wurden vier Auslagerungsstellen gewählt. Die Auslagerung der Proben in Sulfatlösungen im
Labor sollte bei zwei verschiedene Lagerungstemperaturen erfolgen (5°C für die Thaumasit-
und 20°C für die Ettringitbildung). Für das Studium der Geschwindigkeit des Eindringens von
Sulfat und die Schadensbildung durch Ettringit (20°C) wurden die in der EN 206-1 bzw. der
ÖN EN 4710 angegebenen Grenzkonzentrationen der Expositionsklassen XA1 (600 mg SO4-
2/l), XA2 (3000 mg SO4-2/l) und XA3 (6000 mg SO4
-2/l) gewählt. Um wechselnde
Konzentrationen zu simulieren, war vorgesehen, dass Proben in Lösungen mit 600 mg SO4-2/l
und 6000 mg SO4-2/l getaucht werden und dass die Proben zunächst in wöchentlichem
Abstand von der einen in die andere Lagerlösung eingetaucht werden (entspricht einer
mittleren Sulfatkonzentration von 3300 mg/l; d. i. die Expostitionsklasse XA2). Die
Lagerlösungen mussten von Zeit zu Zeit erneuert werden (zu Beginn jeden Monat, nach 1 Jahr
Laufzeit alle 2 Monate und ab 2,5 Jahren alle 3 Monate), um eine annähernd konstante
Sulfatkonzentration der Lagerlösungen über den gesamten Versuchszeitraum sicher zu
stellen. Für die Lagerungstemperatur 5°C wurden niedrigere Konzentrationen, nämlich 200,
600 und 3000 mg SO4-2/l gewählt, weil erfasst werden sollte, ob es bei der geringen
Konzentration von 200 mg SO4-2/l überhaupt zu einer Schädigung kommt, obwohl seitens F.
Bellmann auch Konzentrationen von ≤500 mg SO4-2/l für die Thaumasitbildung als ausreichend
angesehen werden [26].
Die Untersuchung des Einflusses schwankender Sulfatkonzentrationen auf die

17
Thaumasitbildung war nicht vorgesehen, weil im Rahmen des Antrags nur die Unterschiede
der Angriffscharakteristik von Ettringit und Thaumasit erfasst werden sollten. Um den Einfluss
der Temperatur zu dokumentieren wurden aber kalksteinhaltige Proben bei den genannten
Konzentrationen im Labor sowohl bei 20°C als auch bei 5°C ausgelagert.
Die Untersuchung war nach Lagerungszeiten von 6 Monaten, 1,5 Jahren, 2,5 und 3,5 Jahren
vorgesehen. Bei 4 Auslagerungsstellen im Kanalsystem und 4 Prüfterminen ergibt sich eine
Anzahl von 16 Prüfkörpern je Rezeptur (je 16 Prismen und Würfel). Hinzu kommt ein
Prüfkörper je Rezeptur zur Bestimmung des Sulfatgehaltes zu Beginn. In Linz sollte
hauptsächlich der Einfluss der Porosität (W/Z-Wert) und des C3A-Gehaltes des Zementes
erfasst werden. Daher wurden nur zwei Zementsorten (CEM I 42,5 R und CEM I 42,5 N C3A-
frei) vorgesehen und nur von einem Zement (CEM I 42,5 R) Prüfkörper mit verschiedenem
W/Z-Wert (0,70 und 0,45) hergestellt. Der relativ hohe W/Z-Wert von 0,70 wurde gewählt, um
die Eindiffusion von Sulfat zu erleichtern und innerhalb der Projektlaufzeit zu aussagekräftigen
Ergebnissen zu kommen. Zusätzlich sind Proben mit W/Z-0,45 - nur aus dem CEM I 42,5 R -
hergestellt worden, um auch den Einfluss der Porosität auf die Sulfataufnahme zu erfassen.
Insgesamt erforderte dies je 48 Würfel und 48 Prismen für die Auslagerung im Kanalsystem.
Da man nicht wissen konnte, ob die in Kanal ausgelagerten Proben nicht durch mitgeführte
Feststoffe (etwa Treibholz) beschädigt werden würden, sind sicherheitshalber je Rezeptur 4
zusätzliche Würfel bzw. Prismen hergestellt worden (je Lagerstelle eine zusätzliche Probe).
Für die Untersuchungen in Linz mussten somit 60 Würfel und Prismen hergestellt werden.
Hinzu kamen die für die Auslagerung im Labor bei 20°C (Ettringit) und 5°C (Thaumasit)
benötigten Proben. Bei der ersten gemeinsamen Besprechung mit den Projektpartnern nach
Bewilligung des Antrages wurden die vorgesehenen Zemente diskutiert und beschlossen,
dass zwei Zementsorten nicht ausreichen und dass zusätzlich ein CEM III/B 32,5 N und eine
Mischung aus 70% CEM I C3A-frei mit 30% Fluamix C einbezogen werden soll, um den
Einfluss von Hüttensand bzw. Flugasche zu erfassen. Die diesbezüglichen Proben hatten
einen W/Z-Wert von 0,70 und wurden nur bei 3000 mg SO4-2/l gelagert. Es wurden also für die
Laboruntersuchungen vier verschiedene Bindemittel verwendet. Weiter wurde beschlossen,
Proben nicht nur in Natriumsulfatlösungen auszulagern, sondern auch gesättigte
Gipslösungen einzubeziehen, da in natürlichen Wässern enthaltenes Sulfat hauptsächlich von
Gips stammt. Das ergab 7 Rezepturen (W/Z-0,70; bei CEM I 42,5 R auch 0,45; Verwendung
von Normensand als Zuschlag für die Lagerung bei 20°C und Kalkstein bzw. gefälltem CaCO3
anstelle des Normensandes fein für die Lagerung bei 5°C, weil davon ausgegangen werden
musste, dass Kalziumkarbonat in der Probe vorhanden sein muss). Damit hat sich die Zahl

18
der Prüfkörper für die Laboruntersuchungen auf rund 150 Würfel und Prismen erhöht.
Insgesamt wurden daher mehr als 200 Primen und Würfel, also rund 400 Prüfkörper
hergestellt.
Da den Autoren keine in einem Regelwerk verankerte nasschemische Methode zur exakten
Bestimmung der Verteilung von Schadstoffen in bestehendem Beton bekannt war, war die
Entwicklung eines geeigneten Prüfverfahrens ein weiteres Ziel des Forschungsprojektes.
Dazu sollten Betone unterschiedlicher Zusammensetzung verwendet und nach
unterschiedlich langen Lagerungszeiten in gesättigten Gipslösungen der Sulfatgehalt in
unterschiedlichen Tiefen der Betone bestimmt werden.
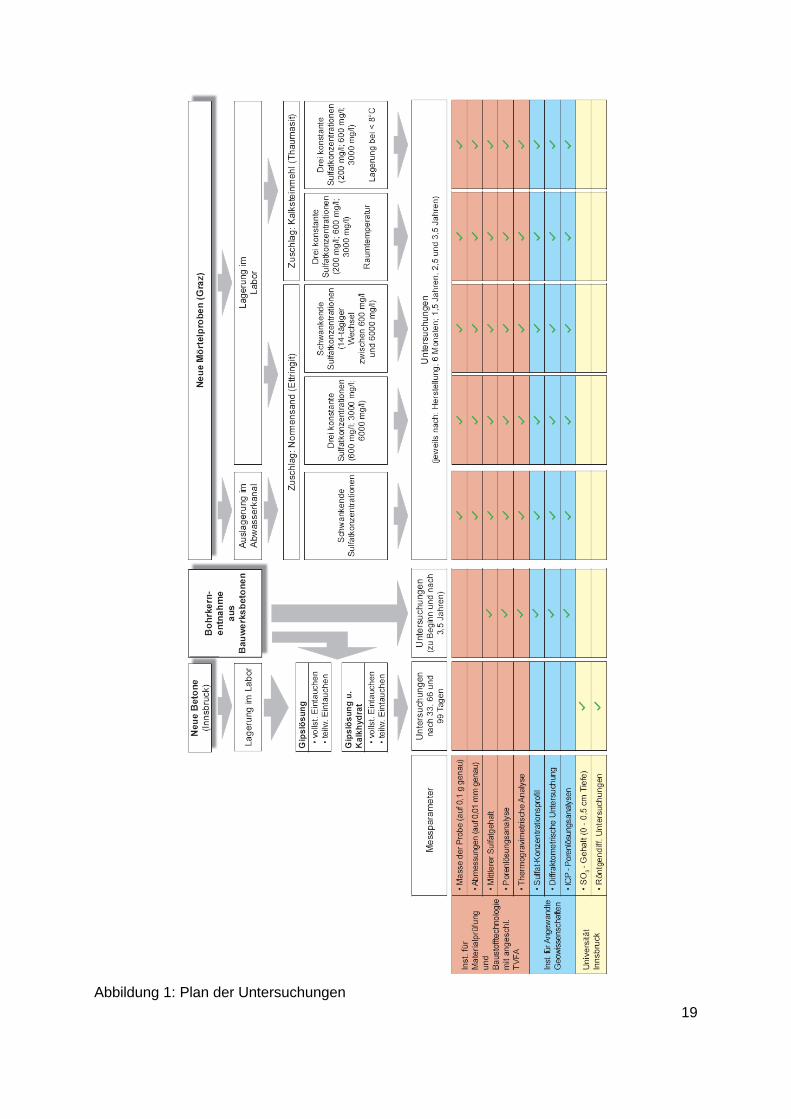
Abbildung 1 gibt eine Übersicht über die Untersuchungen nach deren Planung, aus der auch
ersichtlich ist, welche Untersuchungen von den beteiligten Forschungsstellen durchgeführt
werden sollten. Darin nicht enthalten sind die Untersuchungen der an Betonteilen in Tunneln
aufgetretenen Sulfat-Schädigungen, weil zum Zeitpunkt der Planung nicht bekannt war, dass
die dort aufgetretenen Schäden besichtigt und Proben entnommen werden konnten. Die zur
Klärung der dortigen Schadensursachen nötigen Untersuchungen sind nachträglich
mitaufgenommen worden. Dazu waren, wie sich erst im Verlauf der Untersuchungen
herausstellte, umfangreiche Messungen, u. a. des Gehaltes von Isotopen verschiedener
Elemente notwendig, um die Schadensmechanismen klären zu können. Die Klärung der
Mechanismen war aber notwendig, um angeben zu können, wie in Zukunft derlei Schäden
vermieden werden können.
19
Abbildung 1: Plan der Untersuchungen

20
6. Prüfkörperherstellung, -auslagerung und Probenentnahme aus
bestehenden Bauwerken
6.1 Herstellung und Auslagerung der Mörtelproben
Die Rezeptur der zur Prüfkörperherstellung benutzten Mörtel war ähnlich der, wie sie für die
Festigkeitsprüfung von Zement gemäß der ÖN EN 196-1 [59] vorgeschrieben ist. Abgesehen
von den davon abweichenden W/B-Werten wurde aus Restbeständen noch vorhandener
Normensand grob und fein verwendet, wie er früher gemäß der ÖNORM B 3310 für die
Zementnormenprüfung vorgeschrieben war. Eine Mischung bestand aus 225 g bzw. 350 g
Wasser (für W/Z-0,45 bzw. 0,70), 500 g Bindemittel, 400 g Normensand fein oder
Kalksteinmehl und 900 g Normensand grob. Das Mischen und die Herstellung der Prismen
erfolgten gemäß der gültigen Norm. Für die Herstellung der Würfel wurden ebenfalls die für
die Prismenherstellung vorhandenen Stahlformen benutzt, die 16 cm langen Fächer jedoch
mit 2 cm dicken Kunststofftafeln so geteilt, so dass je Kammer anstelle eines Primas drei
Würfel mit 4 cm Kantenlänge erhalten wurden. Abbildung 2 zeigt eine solche Form vor und
nach dem Einfüllen des Mörtels. Wie in der Norm vorgeschrieben, wurde der Mörtel auch hier
zunächst bis etwa zur halbe Höhe eingefüllt, am Schocktisch verdichtet und danach die Form
gefüllt und wieder verdichtet. Aus einer Mörtelmischung, die für die Herstellung von 3 Prismen
ausgelegt ist, wurden somit 9 Würfel hergestellt. Die Mörtel wurden im Einklang mit der Norm
nach 24 Stunden entformt und danach bis ins Alter von 28 Tagen unter Wasser gelagert.
Abbildung 2: Stahlform mit unterteilten Kammern

21
6.1.1 Laborlagerung
Die Prüfkörper für die Laboruntersuchungen wurden zunächst mit einem der jeweiligen
Rezeptur entsprechenden Code beschriftet und in einen Drahtkorb aus nichtrostendem Stahl
auf Kunststoff-Dreieckleisten gelegt. Der Korb war mittels Kunststoffteilern in Fächer zur
Probenaufnahme unterteilt. Abbildung 3 links zeigt einen solchen Korb mit Fachunterteilung.
Dann wurden die Körbe in Kunststoffboxen gestellt, die mit aufklappbaren Deckeln versehen
waren (Abbildung 3 rechts). Der Deckel der Boxen war an einer Längsseite fix mit der Box
verbunden und konnte nur aufgeklappt aber nicht abgenommen werden. Jede Box wurde mit
14 Liter Natriumsulfat-Lösung befüllt. Die Sulfatlösungen wurden hergestellt, indem zunächst
die, der jeweiligen Sulfatkonzentrationen entsprechende Menge an Natriumsulfat-Salz
(Reinheit: >99%-ig) in entkalktem Wasser gelöst und die Lösung dann in die Boxen
eingegossen wurden. Dann sind die mit den Proben bestückten Körbe in die Lösungen
vollständig eingetaucht, und die Boxen mittels der Deckel verschlossen worden. Die Deckel
wurden so auf die Box gedrückt, dass die seitlich vorhandenen Laschen einrasteten und die
Boxen somit weitgehend dicht verschlossen waren. Die Boxen mit den bei 20°C zu lagernden
Proben sind in einen Klimaraum mit 20±2°C und die bei 5°C gelagerten Boxen in eine
Kühltruhe dieser Temperatur gestellt worden.
Abbildung 3: Stahlkorb mit Probenfächern(links) und Lagerungsgefäß mit Proben (rechts)
6.1.2 Auslagerung im Kanalsystem von Linz
Für die Auslagerung im Kanalsystem wurden vier verschiedene Stellen ausgewählt, die
seitens der LINZ AG als aussagekräftig erachtet wurden. Davon befanden sich drei Stellen
innerhalb des Kanalnetzes und als vierte Stelle wurde das Nachklärbecken der
Hauptkläranlage Linz-Asten festgelegt. Die Lage der Auslagerungsstellen ist aus Abbildung
4 ersichtlich. Die an den Auslagerungsstellen vorhandenen Abwässer stammten aus a)

22
Haushalt (Messstelle 1), b) Industrie (Messstelle 2; in unmittelbarer Nähe einer
Einleitungsstelle), c) Haushalt und Industrie (Messstelle 3) sowie d) Nachklärbecken der
Kläranlage Asten bei Linz (Messstelle 4). Der Beton an den Auslagerungsstellen wurde nach
Angaben der LINZ AG in den Jahren 1977 bis 1979 (Auslagerungsstellen 1, 2 und 3) und
1980 bis 1982 (Auslagerungsstelle 4) errichtet, war also zu Beginn des
Forschungsvorhabens an den Auslagerungsstellen 1, 2 und 3 zwischen 30 und 32 Jahre,
an der Auslagerungsstelle 4 zwischen 27 und 29 Jahre alt.
Abbildung 4: Auslagerungsstellen im Linzer Abwasser-Kanalsystem
Für die Auslagerung im fließenden Wasser der Abwasserkanäle waren stabile Gefäße
erforderlich, die an der Kanalwand fest fixiert werden mussten. Dazu wurden seitens
der LINZ AG dickwandige Kunststoffrohre zur Verfügung gestellt, in die Löcher gebohrt
waren. Um zu verhindern, dass Proben unbeabsichtigt aus den Rohren fallen können,
wurden die Rohröffnungen an die Rohrenden mittels Schrauben gesichert (Abbildung
5, links). Zwischen die einzelnen Proben wurden Abstandhalter aus Kunststoff
gegeben und so sichergestellt, dass die Proben immer allseitig Wasserkontakt hatten.
Im rechten Bild der Abbildung 5 ist einer der beiden benötigten Koffer zu sehen, in
denen die für die Auslagerung im Nachklärbecken benötigten Proben enthalten waren.

23
Die Koffer enthielten Fächer aus Kunststoff zur Probenaufnahme (Abbildung 3) und
waren mit Bohrlöchern versehenen. Damit die Deckel nicht unbeabsichtigt aufgehen
konnten, wurden um die Koffer Stahlbügel angebracht, die mit Schraubkarabinern an
den Bügelenden verschraubt waren. Mittels an den Karabinern angebunden Seilen
konnten die Koffer so tief in das Klärbecken hinabgelassen werden, dass sie
vollständig im Wasser eintauchten und allseitig von Wasser umspült waren (kein
Berührungskontakt zu Beton oder anderen Bauteilen).
Abbildung 5: Mit Proben bestücktes Rohr (links) und Probenkoffer beim Eintauchen in das
Nachklärbecken (rechts)
6.1.3 Betonproben
Für die Untersuchungen zur nasschemischen Bestimmung des Sulfatprofils von Beton
wurden Betonbalken mit 70 x 15 x 15 cm hergestellt. Als Zementsorten wurden auch hier
„CEM I 52,5 R“, „CEM I 42,5 N C3A frei“ und eine Mischung von 70% des C3A-freien
Zementes mit 30% Fluamix verwendet. Der Bindemittelgehalt des Betons betrug 350 kg/m³
und der W/B-Wert 0,45, 0,50 bzw. 0,55. Insgesamt wurden also 9 Balken hergestellt. Sie
wurden nach 1 Tag entformt und 6 Tage unter Wasser gelagert. Danach sind Bohrkerne (Ø
70 mm) entnommen, von diesen 25 mm dicke Scheiben angeschnitten und planparallel

24
geschliffen worden (Abbildung 48 und 49). Diese Prüfkörper sind sodann in gesättigter
Gipslösung (Bodensatz vorhanden) bei Raumtemperatur gelagert worden.
6.2 Probenentnahme
6.2.1 Kanalsystem von Linz
An jenen vier unterschiedlichen Stellen des Kanalsystems, an denen die Mörtelproben
ausgelagert wurden, sind zu Beginn der Forschungsarbeiten Bohrkerne (BK) des
Kanalbetons mit 200 mm Durchmesser entnommen worden. Es war geplant 3,5 Jahre
später an denselben Stellen erneut Bohrkerne zu entnehmen, um die Veränderungen
zwischen Kanalbeton und Mörtelproben zu erfassen. Daraus und mit den anhand der
Laborproben gewonnenen Erkenntnissen - ab welchem Sulfatgehalt in Abhängigkeit von
der Zementsorte und Porosität der Prüfkörper (W/Z-Wert) Schäden auftreten - sollten
Rückschlüsse gezogen werden, wie lange es etwa dauern wird, bis der Sulfatgehalt des
Betons so stark angestiegen ist, dass mit dem Auftreten von Schäden gerechnet werden
muss. Wie sich später herausgestellt hat, ist der Sulfatgehalt der ausgelagerten
Mörtelproben im Laufe der Auslagerung nur so wenig angestiegen, dass die erhofften
Informationen leider nicht erhalten werden konnten. Da die Entnahme der Bohrkerne mit
nicht unerheblichen Schwierigkeiten verbunden war, weil die Entnahmestellen unterhalb
der Wasserlinie lagen und der Wasserspiegel daher vor Probenentnahme gesenkt werden
musste, sind nach 3,5 Jahren nur noch aus dem Nachklärbecken erneut Bohrkerne
entnommen worden.
Die Mörtelproben wurden zu den geplanten Terminen entnommen. Wegen der mit der
Montage und Demontage verbundenen Schwierigkeiten, war es nicht möglich, die Rohre
erst am Tag des Besuches der Sachbearbeiter der TVFA abzumontieren. Daher sind die
Rohre, in denen sich die Würfel und Prismen befanden, einen oder zwei Tage vorher
seitens der LINZ AG abmontiert und in Kunststoffplanen verpackt worden, damit sie nicht
austrocknen konnten (Abbildung 6, links). Wie sich zeigte, waren die Proben durch
Ablagerungen verschmutzt (Abbildung 6, rechts) und mussten durch Abspritzen gereinigt
werden.

25
Abbildung 6: Verpackte Probenrohre (links) und Aussehen der Proben bei Entnahme (rechts)
6.2.2 Tunnel mit Sulfatschäden am Beton
Im Zuge der ersten gemeinsamen Besprechung nach Bewilligung des
Forschungsvorhabens wurde einer Probenentnahme aus dem Bosruck-Eisenbahntunnel
seitens der ÖBB zugestimmt. Die Besichtigung des Tunnels und die Probenentnahmen
erfolgten im Oktober 2009. Dazu wurde eine Lokomotive mit offenem Aufbau und Kranarm
bereitgestellt. Der Tunnel ist ca. 100 Jahre alt. Wie mitgeteilt wurde, ist im Bereich instabiler
Gebirgsbereiche vor dem Felsen eine Steinwand errichtet worden. Der gesamte Tunnel ist
später - vor ca. 50 Jahren - mit Spritzbeton ausgekleidet worden. Bei der Besichtigung
wurden festgestellt, dass der Beton stellenweise seine Festigkeit über die gesamte Tiefe
verloren hatte. Diese Schäden gingen von Fugen bzw. anderen Stellen aus, an denen die
Betonoberfläche unterbrochen war (Abbildung 7). Auf der Betonoberfläche hafteten an
einigen Stellen nadelige Ausblühungen an (Abbildung 8). Proben des zerstörten
Spritzbetons und der Ausblühungen wurden entnommen.
Weiter ist an einer Stelle ein Bohrkern entnommen worden, an der der Spritzbeton
augenscheinlich nicht schadhaft war, an der aber kein Verbund zum darunterliegenden
Mauerwerk mehr gegeben war und der Spritzbeton daher hohl lag. Dieser Bereich befand

26
sich in Fortsetzung von einer Stelle mit zerstörtem Spritzbeton. Dies deutete darauf hin,
dass es zwischen Mauerwerk und Spritzbeton zu einer Betonschädigung gekommen ist,
was zu einer Lösung des Spritzbetons vom Untergrund geführt hat. Daher durfte
angenommen werden, dass die nicht sichtbare Rückseite des noch anhaftenden
Spritzbetons geschädigt war, weshalb von dort ein Bohrkern (Ø 200 mm) entnommen
worden ist (Abbildung 9). Wie sich nach der Entnahme zeigte, haftete an der rückwärtigen
Oberfläche eine weiße Substanz an, bei der es sich offensichtlich um Reaktionsprodukte
handelte (Abbildung 10).
Abbildung 8: Ausblühungen auf Spritzbeton
Abbildung 7: Schadhafter Spritzbeton (neben Fuge)

27
Abbildung 9: Bohrkernentnahme neben einem Bereich mit ab-gefallenem Spritzbeton
Abbildung 10: Reaktionsprodukte auf Rückseite des Bohr–kerns
Im Oktober 2012 wurde im Einvernehmen mit der ASFINAG auch der Bosruck-
Straßentunnel besichtigt und an mehreren Stellen Proben entnommen. Die sichtbaren
Schäden befanden sich vornehmlich in dem, unter dem Fahrbahnniveau liegenden
Belüftungsstollen, der auch als Fluchtstollen bezeichnet wurde sowie in Nischen, die von
der Fahrbahn aus zugänglich waren und normal zur Fahrbahn in das Gebirge ragten. Das
Schadensbild war nicht immer gleichartig sondern an verschiedenen Stellen beider Tunnel
sehr unterschiedlich. In den Nischen war der Spritzbeton teilweise abgefallen und die
Stücke lagen am Boden (Abbildung 11). Innerhalb des Belüftungsstollens war ein Sockel
einer Türe vollkommen zerstört (Abbildung 12) und der Beton konnte mittels Schaufel leicht
entnommen werden. Wie sich zeigte, verlief darunter ein Drainagerohr, das aber nicht mehr
funktionstüchtig war, denn es war keinerlei Fließbewegung des darin befindlichen Wassers
erkennbar. An wieder anderer Stelle war der Beton bei einer im Spritzbeton vorhandenen
Öffnung schadhaft, die feucht war (Abbildung 13).

28
Abbildung 11: Abgefallener Spritzbeton in einer Nische
Abbildung 12: Zerstörter Beton eines Türsockels
Abbildung 13: Schadhafter Beton bei feuchter Wandöffnung
Abbildung 14: Entnahme von Wasserproben

29
Von solchen Stellen sind Proben des schadhaften Betons entnommen worden. Zudem
wurden Proben des Gebirgswassers an unterschiedlichen Stellen innerhalb des Tunnels
entnommen und vor Ort Messungen des pH-Wertes, der Temperatur und der elektrischen
Leitfähigkeit durchgeführt. Die Wasserproben sind zunächst mittels einer Spritze
aufgesaugt worden. Dann wurde auf die Spitze der Spritze ein Filter gesteckt und die
Proben durch diesen Filter hindurch in Probengläser gedrückt (Abbildung 14), die sofort
nach Befüllung dicht verschraubt wurden.
Neben den beiden Bosruck-Tunneln (Eisenbahn- und Straßentunnel) sind auch Proben aus
dem in Reparatur befindlichen Tauerntunnel entnommen worden. Darin sind schon 2008
zur Beseitigung von Fahrbahnhebungen über die ganze Fahrbahnbreite reichende
Aushebungen durchgeführt worden, in die zur Stabilisierung Bewehrungskörbe eingehoben
und dann mit Beton verfüllt wurden. Innerhalb des Fundamentbetons (unter der Fahrbahn)
waren stellenweise Zonen mit zersetztem Beton ohne Festigkeit vorhanden. Vom solchen
Stellen wurden schon 2008 seitens des Instituts für Angewandte Geowissenschaften
Proben entnommen, die projektrelevant waren und daher hier mit erwähnt werden. Im
Rahmen des FFG-Projektes ist u. a. neben einer solchen Aushebung ein Bohrkern
entnommen worden, der von oben nach unten bis in die Tiefe einer schadhaften Zone
reichte (Bezeichnung: „Fundamentbeton“).
Ein weiterer Bohrkern (Ø 200 mm) ist bei einem Besuch im August 2010 vom
Konstruktionsbeton aus der Westulme im Bereich eines Risses entnommen worden. Der
Bohrkern bestand aus vier Stücken. Für die Untersuchungen im Labor wurde nur das dritte
Bohrkernstück (Bezeichnung: „Ulmenbeton/3“) verwendet, weil aus dieser Zone beim
Bohren weiße Substanzen mit dem Kühlwasser ausgeschwemmt wurden, was den
Verdacht auf eine Schädigung durch Thaumasit erweckte und der Abschnitt darüber hinaus
besonders porös aussah. Wie bereits bei einer vorhergehenden Besichtigung sind
zusätzlich Handstückproben sowohl vom nicht zersetzten als auch vom vollkommen
zersetzten Beton zu deren mineralogisch-petrologischen und chemischen
Charakterisierung gezogen worden. Ebenso sind Proben von Tunnelwässern entnommen
worden.

30
7. Beschreibung der Untersuchungen
7.1 In Labor und Kanalsystem ausgelagerte Proben
Die Würfelproben wurden zu jedem Prüftermin von oben (abgezogene Oberfläche) nach
unten durchtrennt. Ein Teil der Probe wurde für die Bestimmung des Sulfatprofils mittels
Elektronenstrahlmikrosonde (ESM) verwendet und die andere Hälfte wurde nach dem
Trocknen und anschließendem Zerkleinern der Proben auf Analysenfeinheit (<0,09 mm)
zur nass chemischen Analyse (Gesamtsulfatgehalt) sowie für Untersuchungen mittels
Röntgendiffraktometer und Röntgenfluoreszenz verwendet.
Zur Bestimmung des Gesamtsulfatgehalts wurden die Proben mit verdünnter Salzsäure
(1:1) aufgeschlossen, filtriert und danach am Sandbad über Nacht zur Trockene
eingedampft. Dabei ist die Partikelgröße der unmittelbar nach dem Aufschluss in kolloidaler
Form vorliegenden und daher filtrierbaren Kieselsäure (SiO2; sie wird deshalb auch
„lösliche“ SiO2 genannt) so vergrößert worden, dass sie filtriert werden konnte. Ihre Menge
ist nach Verglühen bei 1000°C gravimetrisch quantifiziert worden. Die SiO2-Bestimmung
hat sich als notwendig herausgestellt, weil sich die ursprüngliche Annahme, dass der
Zementgehalt von einer Hälfte eines Würfels dem der Rezeptur entspricht, als nicht richtig
herausstellte. Aus dem bestimmten Gehalt an Kieselsäure und dem (bekannten) Gehalt
des jeweiligen Zementes an löslicher Kieselsäure konnte dann der Zementgehalt jeder
einzelnen Probe berechnet werden. Im Filtrat der Kieselsäureabscheidung wurde das Sulfat
als Bariumsulfat gefällt und gravimetrisch bestimmt. Zudem wurden die Feststoffe auch
mittels Röntgenfluoreszenzanalyse zur Bestimmung weiterer Bestandteile analysiert.
Die Pulver der Mörtelproben sind zudem mittels Röntgendiffraktometrie hinsichtlich der
enthaltenen kristallinen Phasen untersucht worden. Diese Methode ist auch zum
qualitativen bzw. quantitativen Nachweis von Ettringit und Thaumasit benutzt worden. Dazu
musste eine Kalibrierung des Messgerätes mit Gemischen von Ettringit/Thaumasit in
zerkleinerten Betonproben durchgeführt werden. So konnte Ettringit und Thaumasit bei
gleichzeitigem Auftreten, trotz ihrer sehr ähnlichen Kristallstruktur, ab einem Gehalt jeder
Phase von ~2,5 M% eindeutig identifiziert werden.
Die ortsaufgelöste Sulfatverteilung ist mittels Elektronenstrahlmikrosonde bestimmt
worden. Dazu wurde aus dem nicht aufgemahlenen Stück jedes Prüfkörpers ein Dünnschliff

31
erzeugt. Der Probekörper wurde an der innenliegenden Fläche plan geschliffen und mittels
Epoxidharz auf einen Glasträger aufgeklebt. Im Anschluss wurde der überstehende Teil
des Klötzchens mittels Diamantsäge abgetrennt, mit diamantbesetzten Schleifscheiben bis
auf eine Dicke von 20-30 m abgetragen und mit Diamantsuspension poliert (Partikelgröße
3 m bzw. 1m). Der so erzeugte Dünnschliff wurde nach lichtmikroskopischer
Dokumentation mit Kohlenstoff bedampft, um eine elektrische Leitfähigkeit zu erzeugen und
an der Elektronenstrahlmikrosonde (Jeol JXA-8200 Superprobe) bei 15kV und 30nA
untersucht. Abbildung 15 zeigt die Schwefelverteilung innerhalb des Dünnschliffs nach 18
Monaten Lagerung bei 6000 mg SO4-2/l. Die hellblauen Zonen zeigen hohe, die
dunkelblauen Zonen niedrige Schwefelkonzentration an. Ein sogenanntes Sulfatprofil
(Abbildung 16) setzt sich aus einer Vielzahl von Einzelmessungen zusammen. Im
Normalfall wurde, beginnend an der Längsseite (Würfelaußenseite), ein ca. 2-3 cm langer
Bereich festgelegt. Von dort ausgehend wurde ein Rasterbereich (1,5 cm) in Richtung
Probenmitte aufgespannt. Bei Standarduntersuchungen wurden 150 Messpunkte in x-
Richtung (parallel zur Würfelaußenseite) und 150 Messpunkte in y-Richtung (in die Tiefe
des Probenkörpers) gesetzt. Ein Standardprofil setzt sich somit aus über 20.000
Einzelmesspunkten zusammen. An einigen Proben wurden zusätzlich über 106 Messpunkte
(1024x1024) aufgenommen, um noch detaillierte Profile aufzunehmen und zusätzlich
hochauflösende Elementverteilungsbilder zu erzeugen.
Nach der Kalibrierung des Röntgendiffraktometers wurden die Abschnitte der aus dem
Kanalsystem von Linz stammenden Bohrkerne untersucht. In den Randzonen (0-1 cm
Tiefe; siehe Kapitel 8.1) wurde mit Ausnahme von einem nicht näher charakterisierbaren
Ca-Fe-Sulfat-Hydrat im Bohrkern 1 (BK 1) - weder Ettringit noch Thaumasit nachgewiesen.

32
Abbildung 15: Die Schwefelverteilung einer Probe nach 18 Monaten Lagerung in Na2SO4 Lösung mit 6000 mg SO4
-2/l bei 20°C (CEM I/C3A-frei; W/Z 0.70)
Abbildung 16: SO3-Profil der Probe aus Abbildung 15

33
Anhand der Prismen wurde zunächst die Biegezug- und dann die Druckfestigkeit gemäß
der ÖN EN 196-1 [59] bestimmt. Die geprüften Proben sind sofort in Kunststoffsäcken
verpackt worden. Aus ihnen wurde danach die Porenlösung ausgepresst. Dazu ist ein
dickwandiger Stahlzylinder benutzt worden, der auf eine Bodenplatte gestellt war, in der
sich eine Ringnut befand, von der eine Bohrung nach außen führte. In die Bohrung wurde
eine Kunststoffspritze oder -rohr gesteckt. Die Porenlösung wurde entweder durch
Aufziehen des Kolbens in die Spritze gesaugt oder sie tropfte aus dem Kunststoffrohr in ein
Probenauffanggefäß, das mittels Klebeband am Rohr befestigt war. In das über die ganze
Höhe reichende mittige Bohrloch (Ø 70 mm) wurde die Probe gegeben, auf diese ein
Scheibe aus Teflon und auf sie eine Scheibe aus gehärtetem Stahl. Dann wurde ein
Stahlstempel in die Zylinderöffnung gesteckt und auf den Stempel eine Stahlplatte gelegt.
Die ganze Vorrichtung stand in einer Presse (300 kN-Presse; Abbildung 17) mittels der der
erforderliche Innendruck aufgebracht wurde (max. 7000 bar), der zum Auspressen der
Porenlösung aus Mörtel- bzw. Betonproben mit baupraktisch niedrigem W/Z-Wert benötigt
wird. Die Details zur Methode und dass sie zu richtigen Ergebnissen führt, sind in [54, 78-
81] beschrieben.
Abbildung 17: Presse zum Auspressen der Porenlösung

34
Die ausgepressten Porenlösungen wurden hinsichtlich der Konzentrationen der Ionen
Hydroxid (OH-), Chlorid (Cl-), Sulfat (SO42-), Natrium (Na+), Kalium (K+) und Calcium (Ca2+)
untersucht. Die Konzentration der OH--Ionen wurde durch Titration mit 0,1 oder 0,01
molarer Salzsäure (HCl) gegen m-Kresolpurpur bestimmt. Der Farbumschlag dieses
Indikators erfolgt von tiefblau (pH: ≥ 9,0) nach hellgelb (pH: 7,4) und ist äußerst scharf.
Allerdings muss zur Verdünnung ein CO2-freies dest. Wasser verwendet werden, das durch
Kochen am Rückfußkühler hergestellt wurde. Beim Abkühlen und der weiteren Lagerung
war am Vorratsgefäß ein CO2-Adsorptionsrohr angebracht (gefüllt mit NaOH auf Träger),
um jeden CO2 Zutritt zu unterbinden. Die Chloridbestimmung (Cl-) wurde fallweise durch
potentiometrische Titration mit einer 0,01 molaren Lösung von Silbernitrat (AgNO3) unter
Verwendung eines mikroprozessorgesteuerten Titrierautomaten durchgeführt (ORION 960;
Schreiberegistrierung), erfolgte im Regelfall aber mittels Ionenchromatographie. Weiter
wurden die Konzentrationen von Sulfat (SO4-2), Natrium (Na+), Kalium (K+) und Calcium
(Ca+2) mittels Ionenchromatographie (Dionex DX-120) sowie fallweise zusätzlich durch ICP-
OES (inductive coupled plasma-optical emmisions spectrscopy) quantifiziert.
7.2 Aus Kanalsystem und Tunneln entnommene Beton- und
Gebirgswasserproben
Von den aus dem Kanalsystem von Linz und den Tunneln entnommenen Bohrkernen sind
im Labor fünf, je 1 cm dicke Scheiben parallel zur Grundfläche der Bohrkerne abgeschnitten
worden. Da das Sägeblatt 5 mm dick war, betrug der Schnittverlust 5 mm und die Abschnitte
stammten somit aus 0-1 cm, 1,5-2,5 cm, 3-4 cm usw. Da es sich teilweise um lufttrockenen
Beton handelte, mussten die Abschnitte zunächst mit Wasser gefüllt werden. Dazu wurden
sie in einen Exsikkator gelegt und zunächst die Luft aus dem Exsikkator gesaugt. Bei
laufendem Vakuum wurden die Abschnitte dann mit Wasser überschichtet, danach das
Vakuum aufgehoben und über Nacht im Wasser belassen. Anschließend wurden sie in
Kunststoffsäcken verpackt und bei 20°C etwa 2 Monate lang gelagert. Die lange
Lagerungszeit war notwendig, um sicherzustellen, dass sich zwischen den Feststoffen und
der Porenlösung ein Gleichgewicht eingestellt hat, wofür nach früheren Untersuchungen
zumindest einige Wochen erforderlich sind [82, 83]. Die aus den Bohrkernabschnitten
ausgepressten Porenlösungen sind gleich untersucht worden, wie die Porenlösungen der
Mörtelproben.
Die aus den zersetzten Betonproben ausgepressten Lösungen und die Bergwasserproben

35
sind hinsichtlich ausgewählter hydrochemischer Kennwerte mittels a) ICP (Inductive
coupled plasma mass spectrometer) auf die Elementkonzentrationen und b) WS-CRDS
(wavelength-scanned cavity ring-down spetroscopy) auf die Isotopenwerte von Sauerstoff,
Wasserstoff, Kohlenstoff und fallweise Schwefel (δ2H, δ18O, δ13C, δ34S) untersucht worden.
7.3 Für die Entwicklung einer Prüfmethode zur
nasschemischen Bestimmung der Sulfatverteilung
verwendete Betonproben
Aus den, von den Bohrkernen aus den Betonbalken hergestellten Prüfkörpern (25 mm dicke
Scheiben) sind nach Lagerungszeiten von 33, 66, 99 und 152 Tagen in gesättigter
Gipslösung Bohrmehlproben entnommen worden. Dazu wurde mittels einer dafür
adaptierten Drehbank die jeweils eingespannte Betonscheibe mittels einer PKD-
Wendeschneidplatte (Polykristalliner Diamant mit Hartmetallunterlage) abgedreht und das
so entnommene Material in 0,2 mm Tiefenabstufungen gesammelt. Abbildung 18 zeigt das
Abdrehen einer Betonscheibe.
Abbildung 18: Abdrehen einer Betonscheibe
Das Betonmehl wurde in 100 ml Salzsäure (1 + 9) aufgeschlämmt, auf 70 °C erwärmt und
anschließend durch einen Glasfaserfilter filtriert. Im Filtrat ist der Sulfatgehalt photometrisch
bestimmt und auf den SO3-Gehalt in Masse-% umgerechnet worden.

36
8. Ergebnisse der im Kanalsystem von Linz durchgeführten
Untersuchungen
8.1 Betonproben
Die Betonproben (Bohrkerne) sind an denselben Stellen des Kanalsystems entnommen
worden, an denen die Mörtelproben ausgelagert waren. Sie wurden mit den in Abbildung 4
angegebenen Ziffern bezeichnet (1 bis 4). Aus Abbildung 19 ist ersichtlich, dass auf den
Bohrkernen von den Entnahmestellen 1 (HSM) und 3 (Weikerlsee) eine Schicht eines Mörtels
anhaftete, dessen Größtkorn deutlich kleiner war als das des Bauwerksbetons. Die
Schichtdicke des Mörtels betrug bei Entnahmestelle 1 ca. 2 cm und bei Entnahmestelle 3 ca.
7 cm. Weshalb der Mörtel aufgebracht wurde und warum die Schichtdicke sehr unterschiedlich
war, ist den Autoren nicht bekannt. Bei den Bohrkernen der anderen beiden Entnahmestellen
(ULK und Nachklärbecken) fehlte eine solche Schicht.
Abbildung 19: Bohrkerne von den Entnahmestellen 1 (links) und 3 (rechts)
Das Diagramm rechts in Abbildung 20 zeigt die Ergebnisse der Porenlösungsanalyse. Beim
Bohrkern 4 konnte nur aus dem ersten Abschnitt etwas Porenlösung ausgepresst werden, aus
den übrigen Abschnitten jedoch nicht, weshalb nur die bei den Bohrkernen 1-3 gemessenen
Konzentrationen dargestellt sind. Die Abbildung zeigt, dass die OH--Konzentration von außen
nach innen kontinuierlich, also von eher schwach alkalisch zu mittelstark alkalisch ansteigt (bei
BK 3 betrug der, aus der OH--Konzentration rechnerisch ermittelte pH-Wert in 0-1 cm Tiefe
11,86, bei BK 1 und bei BK 2 etwa 12,4; der höchste, bei BK 2 in 4,5-5,5 cm Tiefe gemessene
pH-Wert errechnete sich zu 12,89). Auch der letztere Wert ist noch relativ gering
(normalerweise liegt der pH-Wert der Porenlösung eines nicht karbonatisierten Betons über
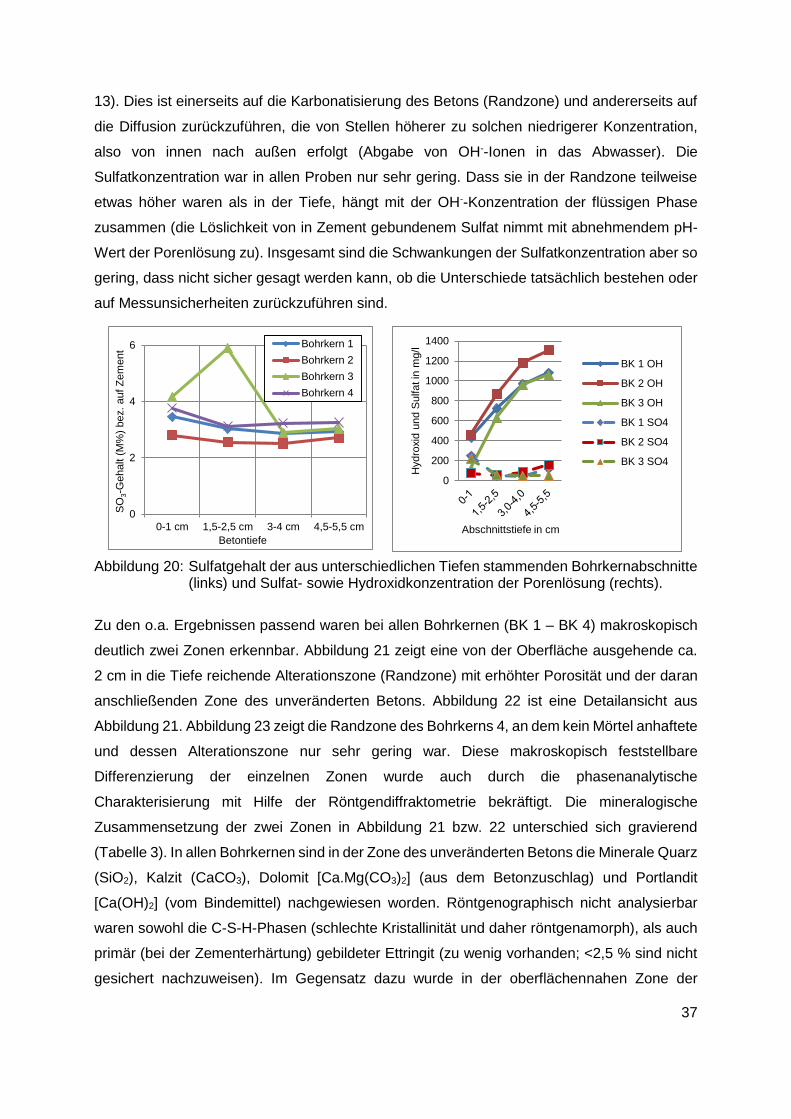
37
13). Dies ist einerseits auf die Karbonatisierung des Betons (Randzone) und andererseits auf
die Diffusion zurückzuführen, die von Stellen höherer zu solchen niedrigerer Konzentration,
also von innen nach außen erfolgt (Abgabe von OH--Ionen in das Abwasser). Die
Sulfatkonzentration war in allen Proben nur sehr gering. Dass sie in der Randzone teilweise
etwas höher waren als in der Tiefe, hängt mit der OH--Konzentration der flüssigen Phase
zusammen (die Löslichkeit von in Zement gebundenem Sulfat nimmt mit abnehmendem pH-
Wert der Porenlösung zu). Insgesamt sind die Schwankungen der Sulfatkonzentration aber so
gering, dass nicht sicher gesagt werden kann, ob die Unterschiede tatsächlich bestehen oder
auf Messunsicherheiten zurückzuführen sind.
Abbildung 20: Sulfatgehalt der aus unterschiedlichen Tiefen stammenden Bohrkernabschnitte (links) und Sulfat- sowie Hydroxidkonzentration der Porenlösung (rechts).
Zu den o.a. Ergebnissen passend waren bei allen Bohrkernen (BK 1 – BK 4) makroskopisch
deutlich zwei Zonen erkennbar. Abbildung 21 zeigt eine von der Oberfläche ausgehende ca.
2 cm in die Tiefe reichende Alterationszone (Randzone) mit erhöhter Porosität und der daran
anschließenden Zone des unveränderten Betons. Abbildung 22 ist eine Detailansicht aus
Abbildung 21. Abbildung 23 zeigt die Randzone des Bohrkerns 4, an dem kein Mörtel anhaftete
und dessen Alterationszone nur sehr gering war. Diese makroskopisch feststellbare
Differenzierung der einzelnen Zonen wurde auch durch die phasenanalytische
Charakterisierung mit Hilfe der Röntgendiffraktometrie bekräftigt. Die mineralogische
Zusammensetzung der zwei Zonen in Abbildung 21 bzw. 22 unterschied sich gravierend
(Tabelle 3). In allen Bohrkernen sind in der Zone des unveränderten Betons die Minerale Quarz
(SiO2), Kalzit (CaCO3), Dolomit [Ca.Mg(CO3)2] (aus dem Betonzuschlag) und Portlandit
[Ca(OH)2] (vom Bindemittel) nachgewiesen worden. Röntgenographisch nicht analysierbar
waren sowohl die C-S-H-Phasen (schlechte Kristallinität und daher röntgenamorph), als auch
primär (bei der Zementerhärtung) gebildeter Ettringit (zu wenig vorhanden; <2,5 % sind nicht
gesichert nachzuweisen). Im Gegensatz dazu wurde in der oberflächennahen Zone der
0
2
4
6
0-1 cm 1,5-2,5 cm 3-4 cm 4,5-5,5 cm
SO
3-G
ehalt (
M%
) bez.
auf Z
em
ent
Betontiefe
Bohrkern 1
Bohrkern 2
Bohrkern 3
Bohrkern 4
0
200
400
600
800
1000
1200
1400
Hydro
xid
und S
ulfat in
mg/l
Abschnittstiefe in cm
BK 1 OH
BK 2 OH
BK 3 OH
BK 1 SO4
BK 2 SO4
BK 3 SO4

38
Bohrkerne neben den erwähnten Mineralphasen des Betonzuschlages die Mineralneubildung
Vaterit (µ- CaCO3) und offensichtlich vom Sediment des Kanalsystems als Verunreinigung in
den Beton gelangter Glimmer nachgewiesen, aber kein Ca(OH)2. Die Bildung der CaCO3
Modifikation Vaterit erfolgte mit Sicherheit sekundär und steht im Zusammenhang mit der
Anwesenheit von organischen Substanzen. Es ist anzunehmen, dass auch ein Teil des
bestimmten Kalzits neu gebildet wurde. Dieser Anteil konnte aber nicht quantifiziert werden.
Abbildung 21: Bohrkern Linz (Ø 50 mm; Messstelle 1)
Abbildung 22: Bohrkern 1; links Dünnschliff, rechts Sulfatverteilung (helle Farben hoher Sulfatanteil, schwarz kein Sulfat vgl. Zuschlagskörner)
3,5cm
Ob
erflä
ch
e
Bohrkern 1 (BK 1)
Linz-Abwasserkanal
2 cm

39
Abbildung 23: Bohrkern 4; links Dünnschliff, rechts Sulfatverteilung
Tabelle 3: Qualitative Mineralogische Zusammensetzung der Bohrkernabschni
Bohrkern Nummer
(die Nr. gibt die Messstelle in Abb. 4 an)
durch Karbonatisierung veränderte Randzone
tiefer liegender, unveränderter Beton
1
Quarz; Dolomit; Kalzit bzw.
Vaterit; Glimmer;
Ca-Fe-Sulfat-Hydrat
Quarz
Dolomit
Kalzit
Portlandit
2 Quarz; Dolomit; Kalzit Vaterit;
Glimmer 3
4
Dass die Porosität im karbonatisierten Bereich sichtlich höher war als im nicht karbonatisierten
Beton, war überraschend, weil die Karbonatisierung von luftgelagerten Beton bekanntlich zu
einer Porenverengung führt. Als Ursache dafür ist primär eine fortgesetzte Auslaugung des
Ca(OH)2 zufolge des häufigen Kontaktes zum Abwasser zu sehen [die Löslichkeit von Ca(OH)2
beträgt immerhin 1,7 g/l]. Vermutlich hatte der Beton im Entnahmebereich abwechselnd Luft-
und Wasserkontakt (wechselnder Füllstand des Kanals).
3,5 Jahre später, also am Ende der Laufzeit des Forschungsprojektes wurde nur aus dem
Nachklärbecken eine neuerliche Bohrkernentnahme durchgeführt. Dies einerseits, weil die
Bohrkernentnahme bei laufendem Betrieb erhebliche Schwierigkeiten mit sich bringt und
andererseits, weil der Sulfatgehalt lediglich innerhalb der Schicht des ungewöhnlich porösen
Reparaturmörtels stärker überhöht war, ansonsten aber in dem alten Kanalbeton nicht bzw.
3,5cm
Ob
erflä
ch
e
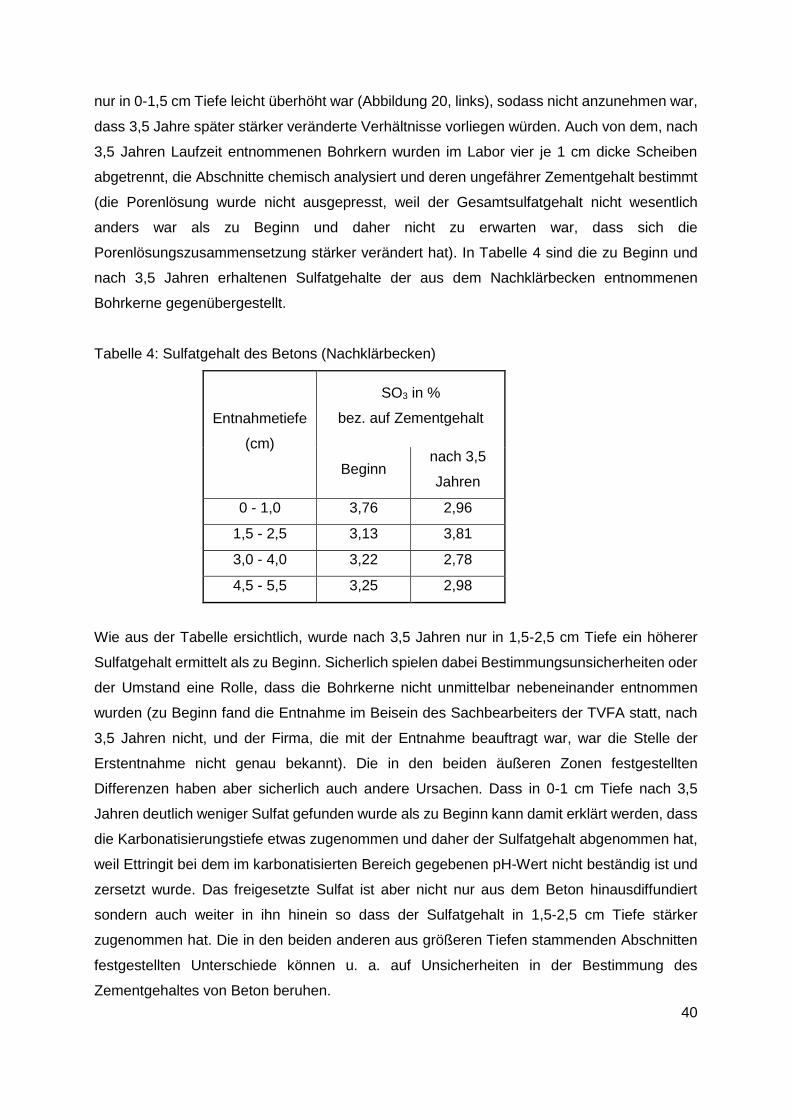
40
nur in 0-1,5 cm Tiefe leicht überhöht war (Abbildung 20, links), sodass nicht anzunehmen war,
dass 3,5 Jahre später stärker veränderte Verhältnisse vorliegen würden. Auch von dem, nach
3,5 Jahren Laufzeit entnommenen Bohrkern wurden im Labor vier je 1 cm dicke Scheiben
abgetrennt, die Abschnitte chemisch analysiert und deren ungefährer Zementgehalt bestimmt
(die Porenlösung wurde nicht ausgepresst, weil der Gesamtsulfatgehalt nicht wesentlich
anders war als zu Beginn und daher nicht zu erwarten war, dass sich die
Porenlösungszusammensetzung stärker verändert hat). In Tabelle 4 sind die zu Beginn und
nach 3,5 Jahren erhaltenen Sulfatgehalte der aus dem Nachklärbecken entnommenen
Bohrkerne gegenübergestellt.
Tabelle 4: Sulfatgehalt des Betons (Nachklärbecken)
Entnahmetiefe
(cm)
SO3 in %
bez. auf Zementgehalt
Beginn nach 3,5
Jahren
0 - 1,0 3,76 2,96
1,5 - 2,5 3,13 3,81
3,0 - 4,0 3,22 2,78
4,5 - 5,5 3,25 2,98
Wie aus der Tabelle ersichtlich, wurde nach 3,5 Jahren nur in 1,5-2,5 cm Tiefe ein höherer
Sulfatgehalt ermittelt als zu Beginn. Sicherlich spielen dabei Bestimmungsunsicherheiten oder
der Umstand eine Rolle, dass die Bohrkerne nicht unmittelbar nebeneinander entnommen
wurden (zu Beginn fand die Entnahme im Beisein des Sachbearbeiters der TVFA statt, nach
3,5 Jahren nicht, und der Firma, die mit der Entnahme beauftragt war, war die Stelle der
Erstentnahme nicht genau bekannt). Die in den beiden äußeren Zonen festgestellten
Differenzen haben aber sicherlich auch andere Ursachen. Dass in 0-1 cm Tiefe nach 3,5
Jahren deutlich weniger Sulfat gefunden wurde als zu Beginn kann damit erklärt werden, dass
die Karbonatisierungstiefe etwas zugenommen und daher der Sulfatgehalt abgenommen hat,
weil Ettringit bei dem im karbonatisierten Bereich gegebenen pH-Wert nicht beständig ist und
zersetzt wurde. Das freigesetzte Sulfat ist aber nicht nur aus dem Beton hinausdiffundiert
sondern auch weiter in ihn hinein so dass der Sulfatgehalt in 1,5-2,5 cm Tiefe stärker
zugenommen hat. Die in den beiden anderen aus größeren Tiefen stammenden Abschnitten
festgestellten Unterschiede können u. a. auf Unsicherheiten in der Bestimmung des
Zementgehaltes von Beton beruhen.

41
Auch nach 3,5 Jahren war der Sulfatgehalt des Betons nicht besonders hoch und ist somit
jedenfalls als eher unauffällig anzusehen. Leider konnte aber keine Information erhalten
werden ob der durchschnittliche Sulfatgehalt während des Beobachtungszeitraums überhaupt
zugenommen hat, oder ob die Sulfatzufuhr wegen der, verglichen mit den
Sulfatkonzentrationen der Laborlagerlösungen, geringen Sulfatkonzentration des Wassers im
Nachklärbecken so langsam erfolgte, dass sie nicht messbar war
8.2 Mörtelproben
8.2.1 Probenbeschaffenheit
Wie die in Abbildung 24 gezeigten Proben wiesen auch alle anderen Würfel und Prismen
oberflächlich eine dünne bräunliche Schicht auf, die nach der Entfernung der vom Klärwasser
stammenden Ablagerungen (Abbildung 6) sichtbar wurde und umso stärker ausgeprägt war,
je länger die Proben gelagert waren (Abbildung 24 links zeigt einen Würfel der im Labor
gespalten wurde). Diese, anfangs nur ca. 0,5 mm dicke Schicht war mineralogisch ident mit
der karbonatisierten Betonzone der Bohrkerne (Anwesenheit von Vaterit).
Abbildung 24: Braunfärbung der Probenoberfläche (links nach 6 Monaten und rechts nach 3,5 Jahren Lagerung)
8.2.2 Festigkeiten
In den Abbildungen 25 und 26 sind die Veränderungen der Druck- bzw. Biegezugfestigkeiten
dargestellt. Bei den Druckfestigkeiten der im Nachklärbecken gelagerten Proben ist auffällig,
dass sie nach 3,5 Jahren mit Ausnahme des mit C3A-freiem Zement hergestellten Mörtels
deutlich geringer war als nach 2,5 Jahren. Die Messwertdifferenzen zu den unterschiedlichen
Terminen sind allerdings teilweise sehr hoch, was eine Interpretation erschwert. Die
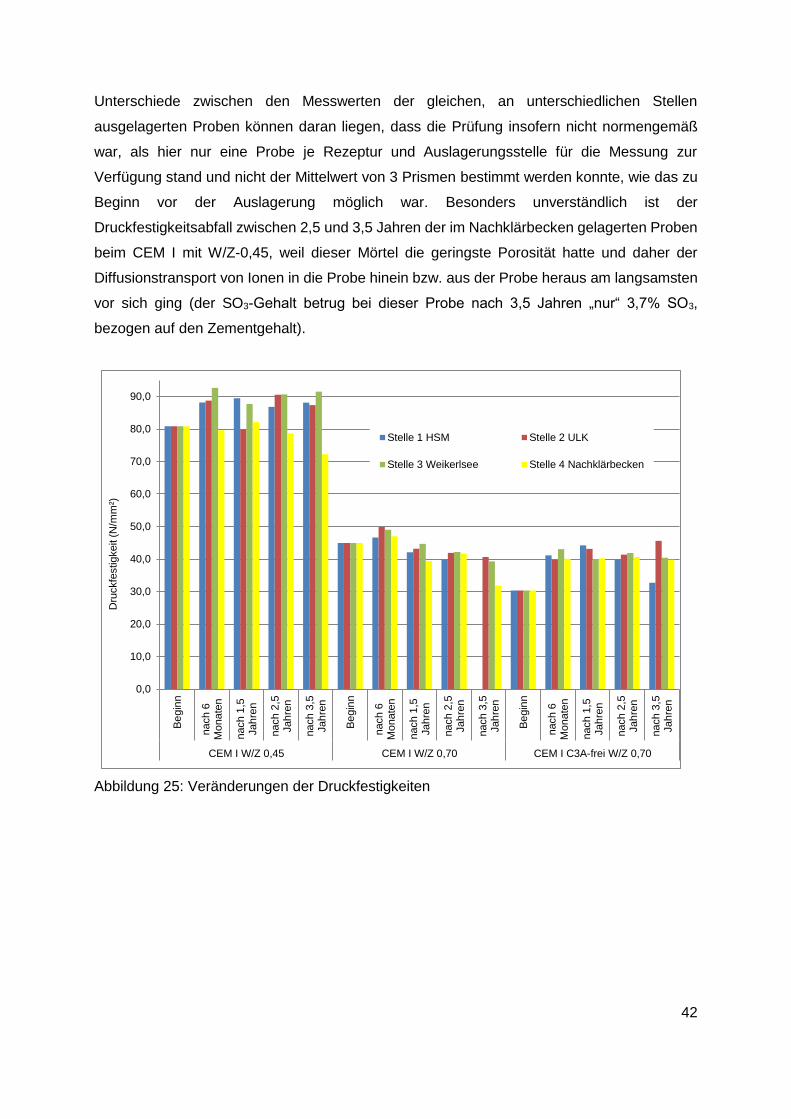
42
Unterschiede zwischen den Messwerten der gleichen, an unterschiedlichen Stellen
ausgelagerten Proben können daran liegen, dass die Prüfung insofern nicht normengemäß
war, als hier nur eine Probe je Rezeptur und Auslagerungsstelle für die Messung zur
Verfügung stand und nicht der Mittelwert von 3 Prismen bestimmt werden konnte, wie das zu
Beginn vor der Auslagerung möglich war. Besonders unverständlich ist der
Druckfestigkeitsabfall zwischen 2,5 und 3,5 Jahren der im Nachklärbecken gelagerten Proben
beim CEM I mit W/Z-0,45, weil dieser Mörtel die geringste Porosität hatte und daher der
Diffusionstransport von Ionen in die Probe hinein bzw. aus der Probe heraus am langsamsten
vor sich ging (der SO3-Gehalt betrug bei dieser Probe nach 3,5 Jahren „nur“ 3,7% SO3,
bezogen auf den Zementgehalt).
Abbildung 25: Veränderungen der Druckfestigkeiten
0,0
10,0
20,0
30,0
40,0
50,0
60,0
70,0
80,0
90,0
Be
gin
n
nach 6
Mo
nate
n
nach 1
,5Jah
ren
nach 2
,5Jah
ren
nach 3
,5Jah
ren
Be
gin
n
nach 6
Mo
nate
n
nach 1
,5Jah
ren
nach 2
,5Jah
ren
nach 3
,5Jah
ren
Be
gin
n
nach 6
Mo
nate
n
nach 1
,5Jah
ren
nach 2
,5Jah
ren
nach 3
,5Jah
ren
CEM I W/Z 0,45 CEM I W/Z 0,70 CEM I C3A-frei W/Z 0,70
Dru
ckfe
stig
keit (
N/m
m²)
Stelle 1 HSM Stelle 2 ULK
Stelle 3 Weikerlsee Stelle 4 Nachklärbecken
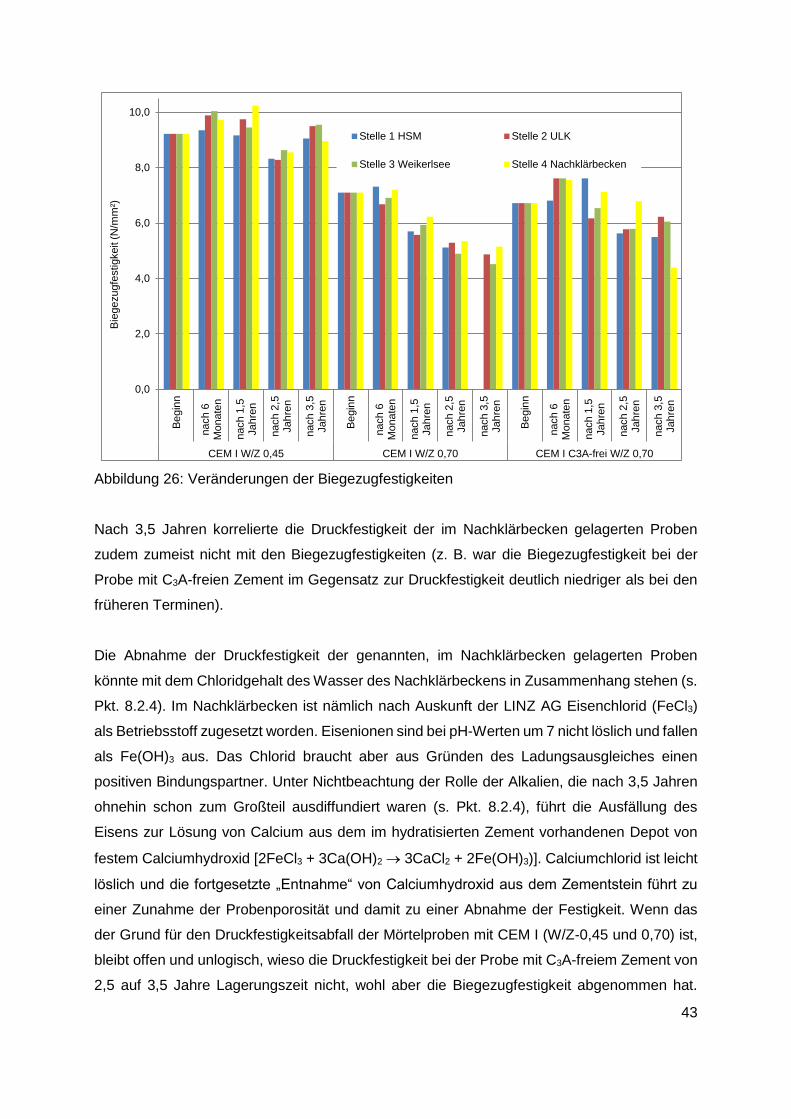
43
Abbildung 26: Veränderungen der Biegezugfestigkeiten
Nach 3,5 Jahren korrelierte die Druckfestigkeit der im Nachklärbecken gelagerten Proben
zudem zumeist nicht mit den Biegezugfestigkeiten (z. B. war die Biegezugfestigkeit bei der
Probe mit C3A-freien Zement im Gegensatz zur Druckfestigkeit deutlich niedriger als bei den
früheren Terminen).
Die Abnahme der Druckfestigkeit der genannten, im Nachklärbecken gelagerten Proben
könnte mit dem Chloridgehalt des Wasser des Nachklärbeckens in Zusammenhang stehen (s.
Pkt. 8.2.4). Im Nachklärbecken ist nämlich nach Auskunft der LINZ AG Eisenchlorid (FeCl3)
als Betriebsstoff zugesetzt worden. Eisenionen sind bei pH-Werten um 7 nicht löslich und fallen
als Fe(OH)3 aus. Das Chlorid braucht aber aus Gründen des Ladungsausgleiches einen
positiven Bindungspartner. Unter Nichtbeachtung der Rolle der Alkalien, die nach 3,5 Jahren
ohnehin schon zum Großteil ausdiffundiert waren (s. Pkt. 8.2.4), führt die Ausfällung des
Eisens zur Lösung von Calcium aus dem im hydratisierten Zement vorhandenen Depot von
festem Calciumhydroxid [2FeCl3 + 3Ca(OH)2 3CaCl2 + 2Fe(OH)3)]. Calciumchlorid ist leicht
löslich und die fortgesetzte „Entnahme“ von Calciumhydroxid aus dem Zementstein führt zu
einer Zunahme der Probenporosität und damit zu einer Abnahme der Festigkeit. Wenn das
der Grund für den Druckfestigkeitsabfall der Mörtelproben mit CEM I (W/Z-0,45 und 0,70) ist,
bleibt offen und unlogisch, wieso die Druckfestigkeit bei der Probe mit C3A-freiem Zement von
2,5 auf 3,5 Jahre Lagerungszeit nicht, wohl aber die Biegezugfestigkeit abgenommen hat.
0,0
2,0
4,0
6,0
8,0
10,0
Be
gin
n
nach 6
Mo
nate
n
nach 1
,5Jah
ren
nach 2
,5Jah
ren
nach 3
,5Jah
ren
Be
gin
n
nach 6
Mo
nate
n
nach 1
,5Jah
ren
nach 2
,5Jah
ren
nach 3
,5Jah
ren
Be
gin
n
nach 6
Mo
nate
n
nach 1
,5Jah
ren
nach 2
,5Jah
ren
nach 3
,5Jah
ren
CEM I W/Z 0,45 CEM I W/Z 0,70 CEM I C3A-frei W/Z 0,70
Bie
gezugfe
stigkeit (
N/m
m²)
Stelle 1 HSM Stelle 2 ULK
Stelle 3 Weikerlsee Stelle 4 Nachklärbecken
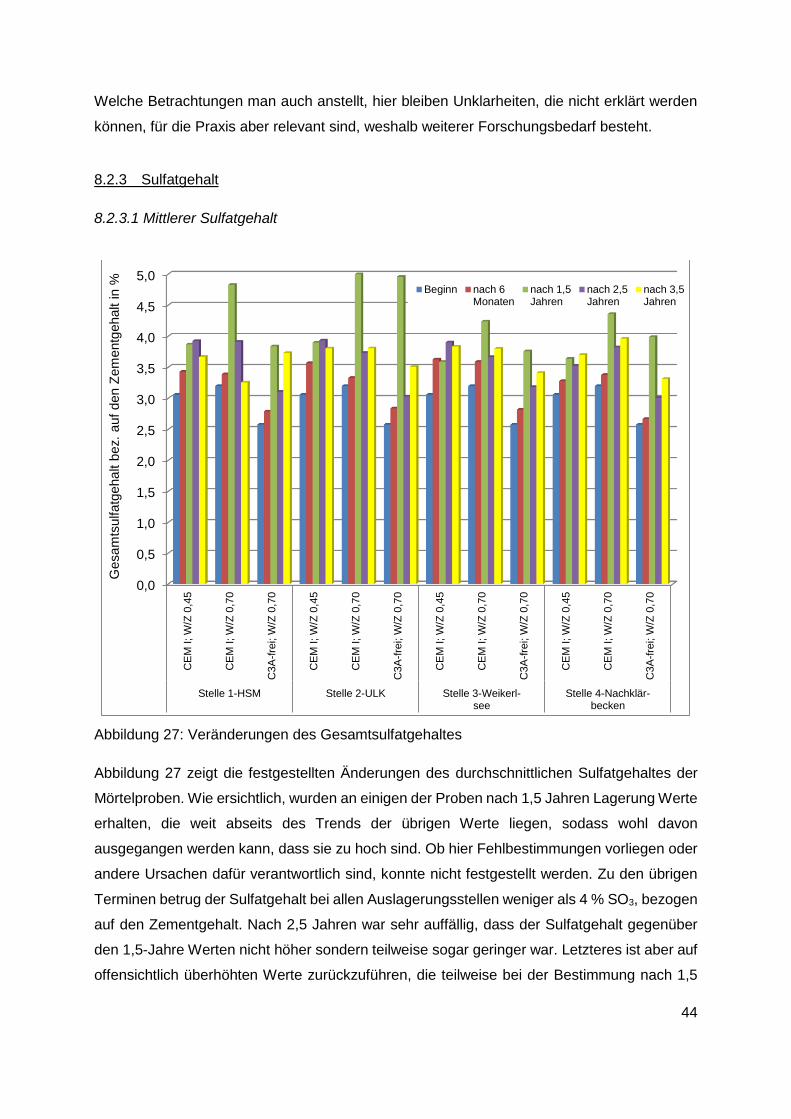
44
Welche Betrachtungen man auch anstellt, hier bleiben Unklarheiten, die nicht erklärt werden
können, für die Praxis aber relevant sind, weshalb weiterer Forschungsbedarf besteht.
8.2.3 Sulfatgehalt
8.2.3.1 Mittlerer Sulfatgehalt
Abbildung 27: Veränderungen des Gesamtsulfatgehaltes
Abbildung 27 zeigt die festgestellten Änderungen des durchschnittlichen Sulfatgehaltes der
Mörtelproben. Wie ersichtlich, wurden an einigen der Proben nach 1,5 Jahren Lagerung Werte
erhalten, die weit abseits des Trends der übrigen Werte liegen, sodass wohl davon
ausgegangen werden kann, dass sie zu hoch sind. Ob hier Fehlbestimmungen vorliegen oder
andere Ursachen dafür verantwortlich sind, konnte nicht festgestellt werden. Zu den übrigen
Terminen betrug der Sulfatgehalt bei allen Auslagerungsstellen weniger als 4 % SO3, bezogen
auf den Zementgehalt. Nach 2,5 Jahren war sehr auffällig, dass der Sulfatgehalt gegenüber
den 1,5-Jahre Werten nicht höher sondern teilweise sogar geringer war. Letzteres ist aber auf
offensichtlich überhöhten Werte zurückzuführen, die teilweise bei der Bestimmung nach 1,5
0,0
0,5
1,0
1,5
2,0
2,5
3,0
3,5
4,0
4,5
5,0
CE
M I; W
/Z 0
,45
CE
M I; W
/Z 0
,70
C3A
-fre
i; W
/Z 0
,70
CE
M I; W
/Z 0
,45
CE
M I; W
/Z 0
,70
C3A
-fre
i; W
/Z 0
,70
CE
M I; W
/Z 0
,45
CE
M I; W
/Z 0
,70
C3A
-fre
i; W
/Z 0
,70
CE
M I; W
/Z 0
,45
CE
M I; W
/Z 0
,70
C3A
-fre
i; W
/Z 0
,70
Stelle 1-HSM Stelle 2-ULK Stelle 3-Weikerl-see
Stelle 4-Nachklär-becken
Gesam
tsulfatg
ehalt
bez.
auf
den Z
em
entg
ehalt in % Beginn nach 6
Monatennach 1,5Jahren
nach 2,5Jahren
nach 3,5Jahren

45
Jahren erhalten wurden. Die Untersuchungen hierzu haben ergeben, dass innerhalb dieser
Proben nahe der Oberfläche eine Karbonatisierung stattgefunden hat (Abbildung 28). Dies
führte zu einer Porenverengung und somit zu einer Abnahme der Geschwindigkeit, mit der
Substanzen von außen in die Probe eindringen können. Offensichtlich wurde die Randzone
oberhalb der Karbonatabscheidung verstärkt ausgelaugt, was erklären könnte, dass bei den
elektronenmikroskopischen Untersuchungen eine stärker poröse Randzone vorgefunden
wurde.
Abbildung 28: Karbonatisierte Randzone einer Mörtelprobe [helle Bereiche stammen von Kalzit (CaCO3)]
Die Karbonatisierung ist entstanden, weil im Abwasser Kohlendioxid (CO2) gelöst war und mit
den Proben in Kontakt kam (stammt aus der Luft bzw. aus Zersetzungsvorgängen organischer
Stoffe). Unter Wasseraufnahme hat sich daraus teilweise Kohlensäure (H2CO3) gebildet (CO2
+ H2O H2CO3), die bei Kontakt zu Natrium bzw. Kaliumhydroxid (NaOH bzw. KOH; ist in der
Porenlösung der Proben enthalten) zu Natrium- bzw. Kaliumkarbonat reagiert (Na2CO3 bzw.
K2CO3). Diese Salze sind leicht wasserlöslich und liegen in gelöstem Zustand dissoziiert vor
(Na2CO3 2Na+ + CO3-2). Die so gebildeten Karbonat-Ionen (CO3
-2) können mit den vom
Zement stammenden Calciumionen zu unlöslichem CaCO3 reagieren. Aus dem unmittelbaren
Oberflächenbereich wurde das Ca(OH)2 des Zementsteins mit dem fließendem Wasser
offenbar vollständig abtransportiert, so dass nur die in die Proben hinein diffundierende
Kohlensäure bzw. CO3-2-Ionen eine unterhalb der Oberfläche beginnende Karbonatisierung
bewirken konnten und sich die Kalkschicht nicht - wie bei den im Labor gelagerten Proben (s.
Kapitel 9.1.2) an der Probenoberfläche ablagern konnte.
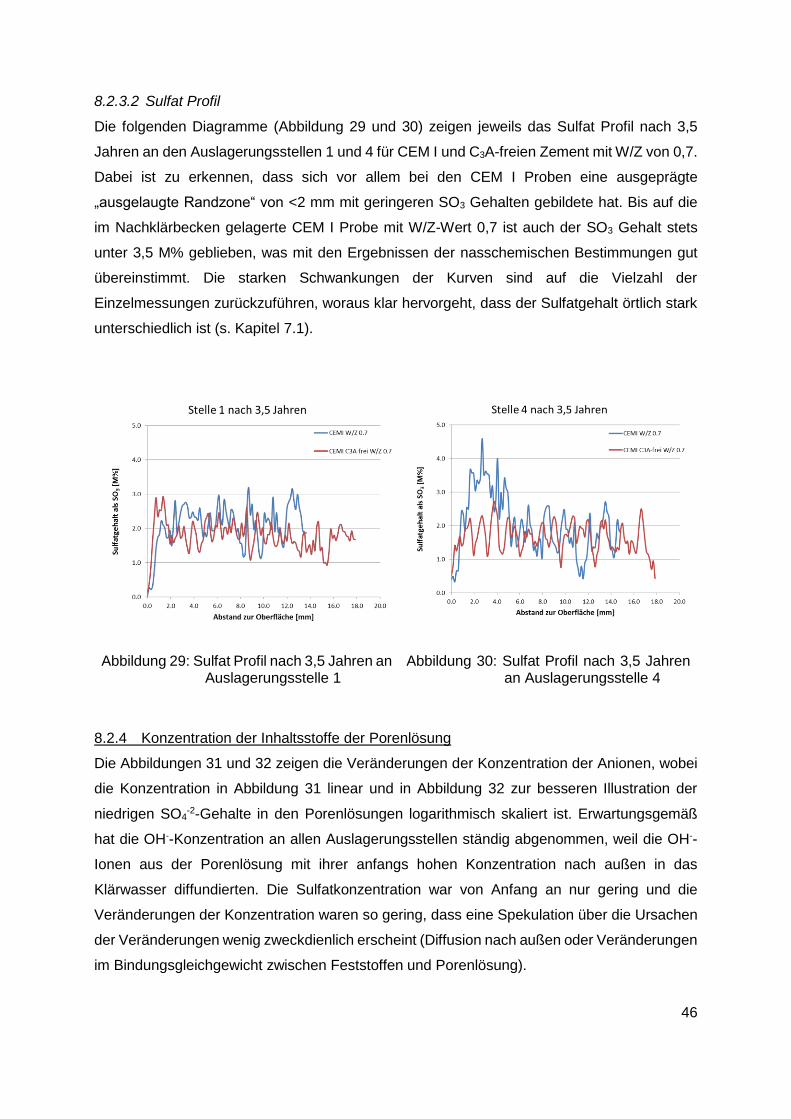
46
8.2.3.2 Sulfat Profil
Die folgenden Diagramme (Abbildung 29 und 30) zeigen jeweils das Sulfat Profil nach 3,5
Jahren an den Auslagerungsstellen 1 und 4 für CEM I und C3A-freien Zement mit W/Z von 0,7.
Dabei ist zu erkennen, dass sich vor allem bei den CEM I Proben eine ausgeprägte
„ausgelaugte Randzone“ von <2 mm mit geringeren SO3 Gehalten gebildete hat. Bis auf die
im Nachklärbecken gelagerte CEM I Probe mit W/Z-Wert 0,7 ist auch der SO3 Gehalt stets
unter 3,5 M% geblieben, was mit den Ergebnissen der nasschemischen Bestimmungen gut
übereinstimmt. Die starken Schwankungen der Kurven sind auf die Vielzahl der
Einzelmessungen zurückzuführen, woraus klar hervorgeht, dass der Sulfatgehalt örtlich stark
unterschiedlich ist (s. Kapitel 7.1).
Abbildung 29: Sulfat Profil nach 3,5 Jahren an Auslagerungsstelle 1
Abbildung 30: Sulfat Profil nach 3,5 Jahren an Auslagerungsstelle 4
8.2.4 Konzentration der Inhaltsstoffe der Porenlösung
Die Abbildungen 31 und 32 zeigen die Veränderungen der Konzentration der Anionen, wobei
die Konzentration in Abbildung 31 linear und in Abbildung 32 zur besseren Illustration der
niedrigen SO4-2-Gehalte in den Porenlösungen logarithmisch skaliert ist. Erwartungsgemäß
hat die OH--Konzentration an allen Auslagerungsstellen ständig abgenommen, weil die OH--
Ionen aus der Porenlösung mit ihrer anfangs hohen Konzentration nach außen in das
Klärwasser diffundierten. Die Sulfatkonzentration war von Anfang an nur gering und die
Veränderungen der Konzentration waren so gering, dass eine Spekulation über die Ursachen
der Veränderungen wenig zweckdienlich erscheint (Diffusion nach außen oder Veränderungen
im Bindungsgleichgewicht zwischen Feststoffen und Porenlösung).

47
Abbildung 31: Veränderung der Konzentration der Anionen der Porenlösung
Abbildung 32: Konzentration aus Abbildung 31 in logarithmischer Ordinatenskalierung
0
1000
2000
3000
4000
5000
6000
7000
8000
zu…
nac
h 6
…
nac
h 1
,5…
nac
h 2
,5…
nac
h 3
,5…
zu…
nac
h 6
…
nac
h 1
,5…
nac
h 2
,5…
nac
h 3
,5…
zu…
nac
h 6
…
nac
h 1
,5…
nac
h 2
,5…
nac
h 3
,5…
OH-mg/l
SO4 --mg/l
Cl-mg/l
Ko
nze
ntr
atio
n in
mg/
L Stelle 2-ULK CEM I W/Z 0,45
Stelle 2-ULK CEM I W/Z 0,70
Stelle 2-ULK C3A-frei W/Z 0,70
Stelle 4-Nachklär-becken CEM I W/Z 0,45

48
Überrascht hat aber die stetige Zunahme der Chloridkonzentration der im Nachklärbecken
gelagerten Proben, die nur an dieser Auslagerungsstelle derart ausgeprägt war. Da dem
Abwasser des Nachklärbeckens Eisenchlorid (FeCl3) als Betriebsstoff zugesetzt worden ist,
konnte Chlorid von außen in die Proben hineindiffundierten. Wegen des dichten Gefüges der
Probe mit W/Z-0,45 stieg der Chloridgehalt der Porenlösung langsamer und insgesamt
deutlich weniger stark an als bei W/Z-0,70, bei dem wiederum beim Mörtel mit C3A-freien
Zement die Zunahme stärker war als beim „normalen“ CEM I (mit C3A), weil letzterer Zement
etwas Chlorid als Friedel´sches Salz (3CaO.Al2O3.CaCl2.10H2O) binden kann, C3A-freier
Zement aber nicht. Interessant und bislang ungeklärt sind die Ursachen der hohen
Chloridkonzentrationen, denn die Cl--Konzentration im Nachklärbecken war nach Angabe der
LINZ AG nie höher als etwa 500 mg Cl-/l. Da die hohen Cl--Konzentration nur im
Nachklärbecken festgestellt wurden, hier aber bei den Proben aller Zemente, handelt es sich
um einen Effekt. In dieselbe Richtung weist auch die Cl--Konzentration von 1643 mg/l, die im
Bohrkernabschnitt aus 0-1 cm Tiefe gefunden wurde, der von dem aus dem Nachklärbecken
stammenden Bohrkern abgetrennt worden ist und auch wesentlich höher war als im Wasser
des Nachklärbeckens. Wegen dieser Ergebnisse sind an den Mörtelproben zusätzlich
Gesamtchloridgehaltsbestimmungen durchgeführt worden Die Ergebnisse sind in Tabelle 5
enthalten. Darin ist auch der Zementgehalt angegeben, der an diesen Proben im Zuge der
Sulfatgehaltsbestimmungen ermittelt worden ist.
Die ermittelten Werte des Gesamtchloridgehaltes passen gut zu den Cl--Konzentrationen der
Porenlösung. Die oben beschriebenen Ursachen für die Unterschiede gelten auch hier. Das
unterschiedliche Bindungsverhalten zwischen dem C3A-haltigen und –freien Zement (W/Z-
0,70) sind auch beim Gesamtchloridgehalt abgebildet. Da der C3A-freie Zement Chlorid nur
physikalisch durch Adsorption binden kann, konnte der Gesamtchloridgehalt nicht so hoch
anstiegen wie bei der Probe mit CEM I und demselben W/Z-Wert, aber die Cl--Konzentration
der Porenlösung war dennoch höher. Aufgrund welcher Ursachen es dazu kommen konnte,
dass entgegen dem zwischen innen und außen gegebenen Konzentrationsgefälles Chlorid in
die Proben hineindiffundieren und der Chloridgehalt auf so hohe Werte ansteigen konnte, kann
nicht gesagt werden.
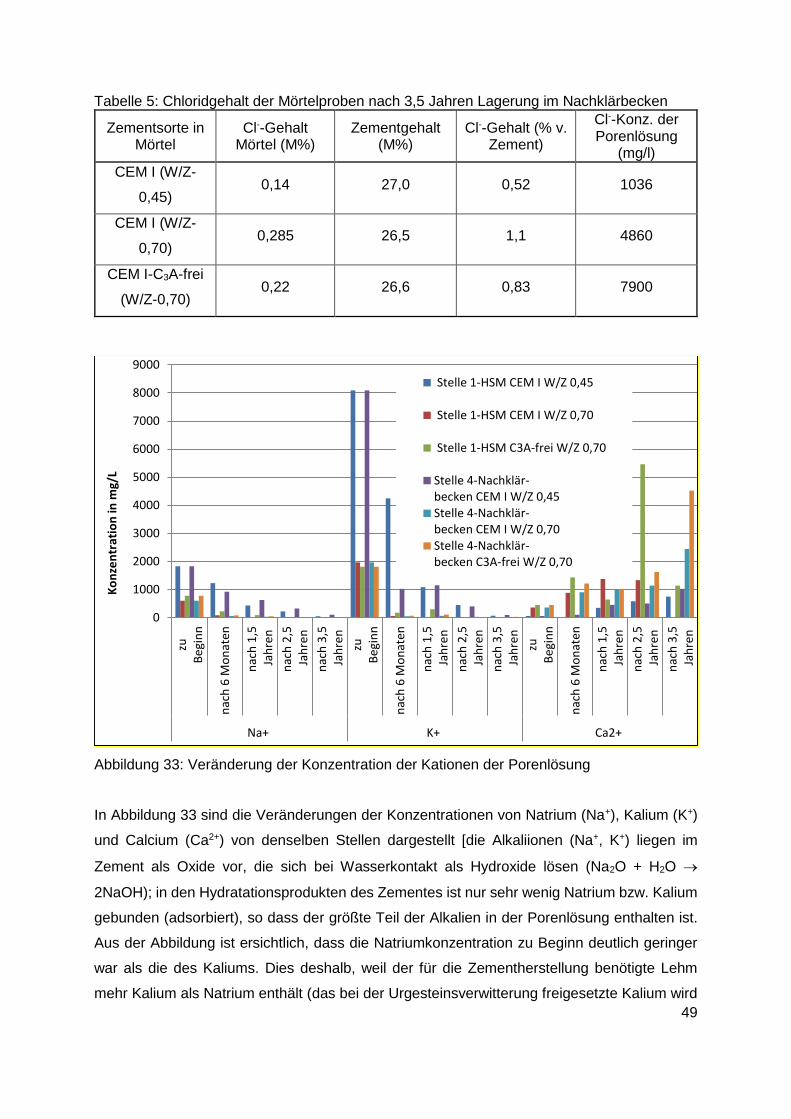
49
Tabelle 5: Chloridgehalt der Mörtelproben nach 3,5 Jahren Lagerung im Nachklärbecken
Zementsorte in Mörtel
Cl--Gehalt Mörtel (M%)
Zementgehalt (M%)
Cl--Gehalt (% v. Zement)
Cl--Konz. der Porenlösung
(mg/l)
CEM I (W/Z-
0,45) 0,14 27,0 0,52 1036
CEM I (W/Z-
0,70) 0,285 26,5 1,1 4860
CEM I-C3A-frei
(W/Z-0,70) 0,22 26,6 0,83 7900
Abbildung 33: Veränderung der Konzentration der Kationen der Porenlösung
In Abbildung 33 sind die Veränderungen der Konzentrationen von Natrium (Na+), Kalium (K+)
und Calcium (Ca2+) von denselben Stellen dargestellt [die Alkaliionen (Na+, K+) liegen im
Zement als Oxide vor, die sich bei Wasserkontakt als Hydroxide lösen (Na2O + H2O
2NaOH); in den Hydratationsprodukten des Zementes ist nur sehr wenig Natrium bzw. Kalium
gebunden (adsorbiert), so dass der größte Teil der Alkalien in der Porenlösung enthalten ist.
Aus der Abbildung ist ersichtlich, dass die Natriumkonzentration zu Beginn deutlich geringer
war als die des Kaliums. Dies deshalb, weil der für die Zementherstellung benötigte Lehm
mehr Kalium als Natrium enthält (das bei der Urgesteinsverwitterung freigesetzte Kalium wird
0
1000
2000
3000
4000
5000
6000
7000
8000
9000
zuB
egi
nn
nac
h 6
Mo
nat
en
nac
h 1
,5 J
ahre
n
nac
h 2
,5 J
ahre
n
nac
h 3
,5 J
ahre
n
zuB
egi
nn
nac
h 6
Mo
nat
en
nac
h 1
,5 J
ahre
n
nac
h 2
,5 J
ahre
n
nac
h 3
,5 J
ahre
n
zuB
egi
nn
nac
h 6
Mo
nat
en
nac
h 1
,5 J
ahre
n
nac
h 2
,5 J
ahre
n
nac
h 3
,5 J
ahre
n
Na+ K+ Ca2+
Ko
nze
ntr
atio
n in
mg/
L
Stelle 1-HSM CEM I W/Z 0,45
Stelle 1-HSM CEM I W/Z 0,70
Stelle 1-HSM C3A-frei W/Z 0,70
Stelle 4-Nachklär-becken CEM I W/Z 0,45Stelle 4-Nachklär-becken CEM I W/Z 0,70Stelle 4-Nachklär-becken C3A-frei W/Z 0,70

50
vom Boden adsorbiert, Natrium nicht). Mit zunehmender Lagerungsdauer hat die
Konzentration der Alkalien kontinuierlich abgenommen, weil sie aus den Proben in das
Abwasser hinausdiffundierten. Da sie als Hydroxide vorlagen, korreliert die Abnahme der
Alkalikonzentration mit der des Hydroxids. Anders sind die Verhältnisse beim Calcium dessen
Konzentration zu Beginn von allen Kationen am geringsten war und mit zunehmender
Lagerungsdauer angestiegen ist. Dies ist mit dem Absinken der OH--Konzentration zu
erklären. Je höher die OH--Konzentration, umso geringer ist die Löslichkeit von Calcium.
Allerdings erscheinen einzelne Ca+2-Werte, wie die nach 2,5 Jahren der bei Stelle 1
ausgelagerten Probe (C3A-freier Zement; grüner Balken) fragwürdig, denn nach 3,5 Jahren
wurde an dieser Stelle eine wesentlich geringere Ca+2-Konzentration gemessen (vermutlich
liegt hier ein Bestimmungsfehler vor).
Insgesamt ergibt sich aus den Untersuchungen, dass es sehr unwahrscheinlich erscheint,
dass es im Kanalsystem in überschaubarer Zukunft zu einer gefährlichen Sulfat-Anreicherung
des Betons kommen wird. Dies zumal die Ergebnisse der Laboruntersuchungen zeigten, dass
bei wechselnden Sulfat-Konzentrationen im angreifenden Wasser die mittlere Konzentration
relevant ist (s. Kapitel 9.1.2) und dass an Proben mit CEM I als Bindemittel und einem W/Z-
Wert von 0,70, die im Labor in einer Sulfatlösung mit 6.000 mg SO4-2/l ausgelagert waren -
trotz eines Anstieges des Sulfatgehaltes auf ~10% SO3, bezogen auf Zementgehalt - lediglich
erste Anzeichen beginnender Schädigung durch Ettringitbildung festzustellen waren. Es muss
jedoch darauf hingewiesen werden, dass die Laborexperimente nicht direkt auf die Praxis
übertragen werden können, da in Abwasserkanälen immer komplexe Mischlösungen z.B.
Sulfat mit hohen Chloridwerten und hohen Gehalten an organischen Substanzen auftreten.
Weiter muss erwähnt werden, dass es vor allem im Nachklärbecken zu einer signifikanten
Reduzierung der mechanischen Kennwerte der Mörtelproben (Druck- und Biegezugfestigkeit)
gekommen ist. Zusätzlich zeigten die CEM I Proben mit W/Z Wert 0,7 auch an den anderen
Auslagerungsstellen einen deutlich abfallenden Trend bei der Biegezugfestigkeit. Wie bereits
zuvor ausgeführt, dürfte dafür hauptsächlich die Auslaugung von Portlandit aus dem
Zementstein verantwortlich sein. Begünstigt durch hohe Chloridwerte, wie sie im
Nachklärbecken auftraten, wird Portlandit gelöst und im Gegensatz zu den im Labor
durchgeführten Experimenten nicht immer durch Kalzit ersetzt. Die daraus resultierende
Erhöhung der Porosität begünstigt in weitere Folge etwaige chemische Angriffe. Da die
Temperatur im Kanalsystem so hoch ist, dass es zu keinem Angriff durch Thaumasitbildung
kommen wird, wird darauf nicht näher eingegangen.

51
9. Ergebnisse der im Labor ausgelagerten Mörtelproben
9.1 Experimente zur Ettringitbildung
(mit Normensand hergestellte Mörtelproben, ausgelagert bei 20°C)
9.1.1 Festigkeiten
Abbildung 34: Druckfestigkeiten bei Lagerung in Na2SO4-Lösungen mit unterschiedlichen SO4
-2-Konzentrationen
Abbildung 34 zeigt die Ergebnisse der Druckfestigkeitsprüfungen der bei unterschiedlichen
Sulfatkonzentrationen im Labor bei ~20°C gelagerten Proben, die keinen Kalkstein sondern
Normensand enthielten (bei den Proben, die Kalksteinmehl anstelle des Normen-Feinsandes
enthielten, waren die Ergebnisse erwartungsgemäß vergleichbar, weshalb darauf nicht näher
eingegangen wird). Nach 3,5 Jahren fand keine Prüfung statt, weil die Versuchsdauer bei
diesen Proben auf 4,5 Jahre ausgedehnt werden musste und daher nach 3,5 Jahren keine
Festigkeits- und Porenlösungsuntersuchungen durchgeführt werden konnten (s. Kapitel 9.1.2).
Wie ersichtlich, war die Druckfestigkeit nach 6 Monaten bei allen Proben höher als zu Beginn,
was vermutlich nur auf die Nacherhärtung zurückzuführen ist. Es ist aber auch erkennbar,
dass die Vertrauenswürdigkeit in die Messwerte als nicht besonders hoch einzustufen ist. Dies
deshalb, weil etwa die bei 600 mg SO4-2/l ausgelagerte Probe mit W/Z-0,45 nach 6 Monaten
0,00
10,00
20,00
30,00
40,00
50,00
60,00
70,00
80,00
90,00
100,00
CE
M I W
/Z 0
,45
CE
M I W
/Z 0
,70
C3A
-fre
i W
/Z 0
,70
CE
M I W
/Z 0
,45
CE
M I W
/Z 0
,70
C3A
-fre
i W
/Z 0
,70
CE
M I W
/Z 0
,45
CE
M I W
/Z 0
,70
C3A
-fre
i W
/Z 0
,70
CE
M I W
/Z 0
,45
CE
M I W
/Z 0
,70
C3A
-fre
i W
/Z 0
,70
wechselnde Konz. zw.600 und 6000 mg/l
600 mg/l 3000 mg/l 6000 mg/l
Dru
ckfe
stig
keiten
in N
/mm
2
zu Beginn nach 6 Monaten nach 1,5 Jahren
nach 2,5 Jahren nach 4,5 Jahren

52
die höchste Druckfestigkeit von allen Prüfterminen hatte. Auch war die Druckfestigkeit dieser
Probe nach 2,5 Jahren deutlich geringer war als die des bei 6000 mgSO4-2/l gelagerten Probe.
Grund dafür könnte sein, dass die Poren bei der hohen Sulfatkonzentration vermutlich schon
stärker mit neu gebildeten Sulfatphasen gefüllt waren und daher die Porosität geringer war als
bei der niedrigen Sulfatkonzentration der Lagerlösung. Wenn darin die Erklärung läge, müsste
aber die Druckfestigkeit der abwechselnd bei 600 und 3000 mg SO4-2/l gelagerten Probe mit
W/Z-0,45 niedriger sein als die der Probe, die bei 6000 mg SO4-2/l gelagert war. Weiter muss
angemerkt werden, dass die Verwendung von Druckfestigkeitsuntersuchungen zur
Beurteilung des Schädigungsgrades als nicht besonders gut geeignet angesehen wird.
Abbildung 35: Druckfestigkeit der Prismen mit W/Z-0,70 bei Lagerung in Na2SO4-Lösung mit 3000 mg SO4
-2/l
Abbildung 35, in der die Druckfestigkeiten der bei Raumtemperatur in der Lösung mit 3000
mg/SO4-2/l gelagerten Proben (W/Z-Wert: 0,70) dargestellt sind, gibt einen Überblick über die
Druckfestigkeit der Mörtel mit verschiedenem Bindemittel (die Ergebnisse des CEM I und des
C3A-freien Zementes sind auch enthalten, weil die Ordinate anders skaliert ist als in Abbildung
34 und die Messwerte so leichter verglichen werden können).
Die Balken in der Spalte „nach 4,5/3,5 Jahren“ enthält beim CEM I und CEM I/C3A-frei die
Werte nach 4,5 Jahren Lagerung, bei CEM III und Fluamix (MIX 70/30) aber die nach 3,5
Jahren. Die Druckfestigkeit war bis nach 2,5 Jahren bei dem Mörtel mit CEM I am höchsten,
0,00
10,00
20,00
30,00
40,00
50,00
60,00
zu Beginn nach 6Monaten
nach 1,5Jahren
nach 2,5Jahren
nach 4,5/3,5Jahren
Dru
ckfe
stig
keit (
N/m
m²)
CEM I W/Z 0,70 C3A-frei W/Z 0,70
CEM III MIX 70/30

53
gefolgt von der des Mörtels mit C3A-freien Zement. Nach 3,5 Jahren hatte jedoch der Mörtel
mit CEM III die höchste Druckfestigkeit und die des MIX 70/30 war etwa gleich hoch wie die
des Mörtels mit C3A-freien Zement nach 4,5 Jahren. Offenbar kam es bei den Mörteln mit CEM
III und Fluamix zu einer langandauernden Nacherhärtung ohne dass sich die Einwirkung der
Lagerlösung mindernd auf die Druckfestigkeit auswirkte. Im Gegensatz dazu kam es aber bei
dem Mörteln mit CEM I sowie dem C3A-freien Zement zwischen 2,5 und 4,5 Jahren Lagerung
zu einer deutlichen Reduktion der Druckfestigkeit, was auf eine Schädigung durch die
entstandenen neuen Sulfatphasen hindeutet.
festgestellt werden konnten.
Abbildung 36: Biegezugfestigkeiten bei Lagerung in Na2SO4-Lösungen mit unterschiedlichen SO4
-2–Konzentrationen
Aus Abbildung 36 sind die Veränderungen der Biegezugfestigkeiten ersichtlich. Auch hier hat
man es mit ähnlich hohen Prüfstreuungen wie bei den Druckfestigkeiten zu tun, was eine
Interpretation sehr erschwert. Immerhin weist aber der Umstand, dass die Biegezugfestigkeit
der bei 6000 mg SO4-2/l gelagerte Proben des Mörtels mit W/Z-0,70 und CEM I als Bindemittel
zwischen 6 Monaten und Versuchsende ständig geringer wurden und dass sie nach 2,5 Jahren
schon unter 5 N/mm² lag (die mit Abstand bis dahin niedrigste Biegezugfestigkeit), sehr auf
eine Schädigung durch Sulfat hin. Der Gesamtsulfatgehalt dieses Mörtels betrug nach 2,5
Jahren schon über 7 % und im inneren des Mörtels waren bereits Risse vorhanden (s. Pkt.
0,00
1,00
2,00
3,00
4,00
5,00
6,00
7,00
8,00
9,00
10,00
11,00
12,00
13,00
14,00
CEM IW/Z 0,45
CEM IW/Z 0,70
C3A-freiW/Z 0,70
CEM IW/Z 0,45
CEM IW/Z 0,70
C3A-freiW/Z 0,70
CEM IW/Z 0,45
CEM IW/Z 0,70
C3A-freiW/Z 0,70
CEM IW/Z 0,45
CEM IW/Z 0,70
C3A-freiW/Z 0,70
wechselnde Konz. zw. 600und 6000 mg/l
600 mg/l 3000 mg/l 6000 mg/l
Bie
ge
zu
ge
sti
gk
eit
in
N/m
m2
Beginn
nach 6 Monaten
nach 1,5 Jahren
nach 2,5 Jahren
nach 4,5 Jahren
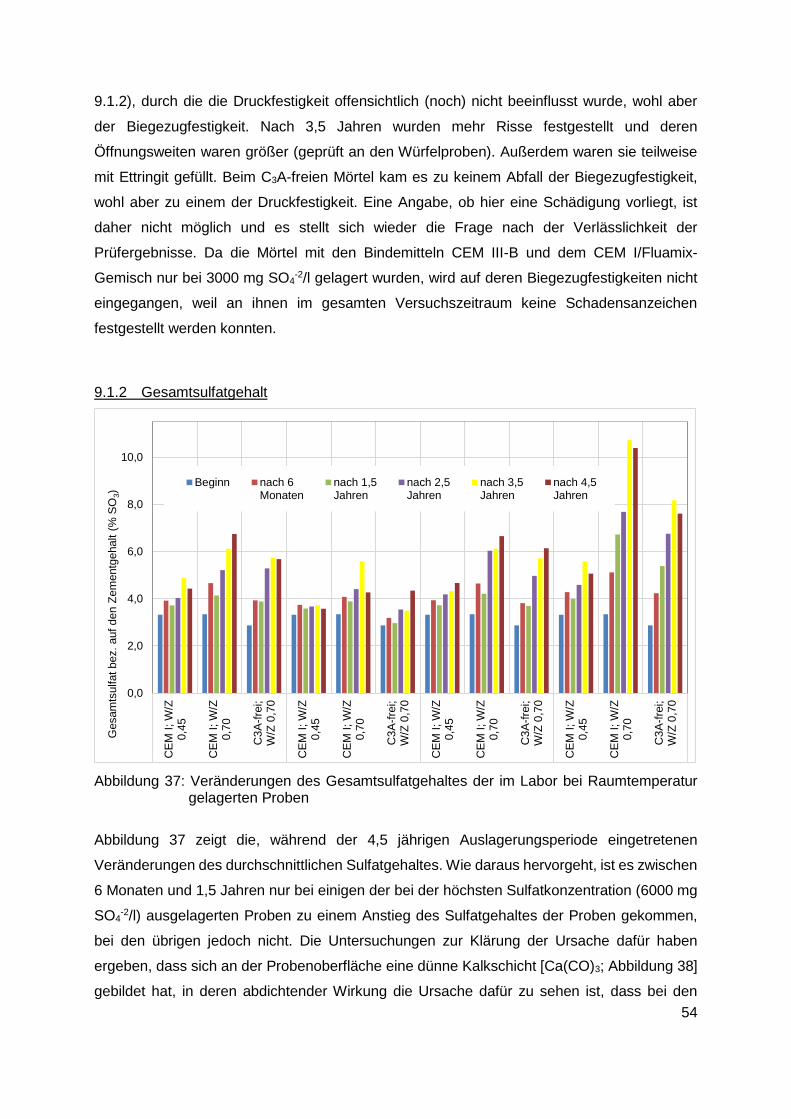
54
9.1.2), durch die die Druckfestigkeit offensichtlich (noch) nicht beeinflusst wurde, wohl aber
der Biegezugfestigkeit. Nach 3,5 Jahren wurden mehr Risse festgestellt und deren
Öffnungsweiten waren größer (geprüft an den Würfelproben). Außerdem waren sie teilweise
mit Ettringit gefüllt. Beim C3A-freien Mörtel kam es zu keinem Abfall der Biegezugfestigkeit,
wohl aber zu einem der Druckfestigkeit. Eine Angabe, ob hier eine Schädigung vorliegt, ist
daher nicht möglich und es stellt sich wieder die Frage nach der Verlässlichkeit der
Prüfergebnisse. Da die Mörtel mit den Bindemitteln CEM III-B und dem CEM I/Fluamix-
Gemisch nur bei 3000 mg SO4-2/l gelagert wurden, wird auf deren Biegezugfestigkeiten nicht
eingegangen, weil an ihnen im gesamten Versuchszeitraum keine Schadensanzeichen
festgestellt werden konnten.
9.1.2 Gesamtsulfatgehalt
Abbildung 37: Veränderungen des Gesamtsulfatgehaltes der im Labor bei Raumtemperatur gelagerten Proben
Abbildung 37 zeigt die, während der 4,5 jährigen Auslagerungsperiode eingetretenen
Veränderungen des durchschnittlichen Sulfatgehaltes. Wie daraus hervorgeht, ist es zwischen
6 Monaten und 1,5 Jahren nur bei einigen der bei der höchsten Sulfatkonzentration (6000 mg
SO4-2/l) ausgelagerten Proben zu einem Anstieg des Sulfatgehaltes der Proben gekommen,
bei den übrigen jedoch nicht. Die Untersuchungen zur Klärung der Ursache dafür haben
ergeben, dass sich an der Probenoberfläche eine dünne Kalkschicht [Ca(CO)3; Abbildung 38]
gebildet hat, in deren abdichtender Wirkung die Ursache dafür zu sehen ist, dass bei den
0,0
2,0
4,0
6,0
8,0
10,0
CE
M I
; W
/Z0,4
5
CE
M I
; W
/Z0,7
0
C3
A-f
rei;
W/Z
0,7
0
CE
M I
; W
/Z0,4
5
CE
M I
; W
/Z0,7
0
C3
A-f
rei;
W/Z
0,7
0
CE
M I
; W
/Z0,4
5
CE
M I
; W
/Z0,7
0
C3
A-f
rei;
W/Z
0,7
0
CE
M I
; W
/Z0,4
5
CE
M I
; W
/Z0,7
0
C3
A-f
rei;
W/Z
0,7
0
Gesam
tsulfat bez.
auf
den Z
em
entg
ehalt (
% S
O3)
Beginn nach 6Monaten
nach 1,5Jahren
nach 2,5Jahren
nach 3,5Jahren
nach 4,5Jahren

55
meisten Proben keine messbare Zunahme des Sulfatgehaltes stattgefunden hat. Wie die
Untersuchungen der Porenlösung der Proben ergeben haben, hat das aber keine vollständige
Abdichtung bewirkt, denn die Hydroxid- und Alkali-Ionen konnten weiterhin aus der Probe in
die Lagerlösung diffundieren, was an der Abnahme der Konzentration dieser Ionen in der
Porenlösung zwischen 6 Monaten und 1,5 Jahren kenntlich ist (Abbildung 44 bzw. 45).
Möglicherweise konnten nur Sulfat-Ionen die Schicht nicht bzw. nur äußerst langsam
durchdringen, weil sie ein viel größeres Volumen einnehmen als die OH--bzw. Alkali Ionen. Im
Gegensatz zu den im Kanal ausgelagerten Proben hat sich bei der Laborlagerung die
Kalkschicht an der Probenoberfläche ausgebildet, weil das ausdiffundierte Calciumhydroxid
nicht abtransportiert wurde wie im fließenden Wasser und daher direkt mit dem in der
Lagerlösung vorhandenen CO2 zu Calciumkarbonat reagieren konnten [Ca(OH)2 + CO2 →
CaCO3 + H2O].
Abbildung 38: Kalkschicht auf Mörtelprobe (ca. 0,01 mm dick; heller Streifen am linken Bildrand)
Um die Eindiffusion von SO4-2-Ionen wieder zu ermöglichen, wurde die Kalkschicht in der Folge
alle 6 Monate durch Absäuern der Proben mit Ameisensäure entfernt. Zwischen 1,5 und 2,5
Jahren ist es dann wieder bei allen Proben zu einer Sulfataufnahme gekommen, wobei die
Zunahme in allen Lagerlösungen erwartungsgemäß beim CEM I mit W/Z-Wert 0,70 am
stärksten war. Bei dieser Probe betrug der auf den Zementgehalt bezogene Sulfatgehalt nach
3,5 bzw. 4,5 Jahren Lagerung in der Lösung mit 6000 mg SO4-2/l über 10 % SO3.
Aus Abbildung 37 ist auch ersichtlich, dass der Anstieg des Sulfatgehaltes bei
Wechsellagerung zwischen 600 und 6000 mg SO4-2/l (Mittelwert 3300 mg SO4
-2/l) vergleichbar
war mit dem bei konstant 3000 mg SO4-2/l gelagerten Proben. Auf die Menge an Sulfat, die
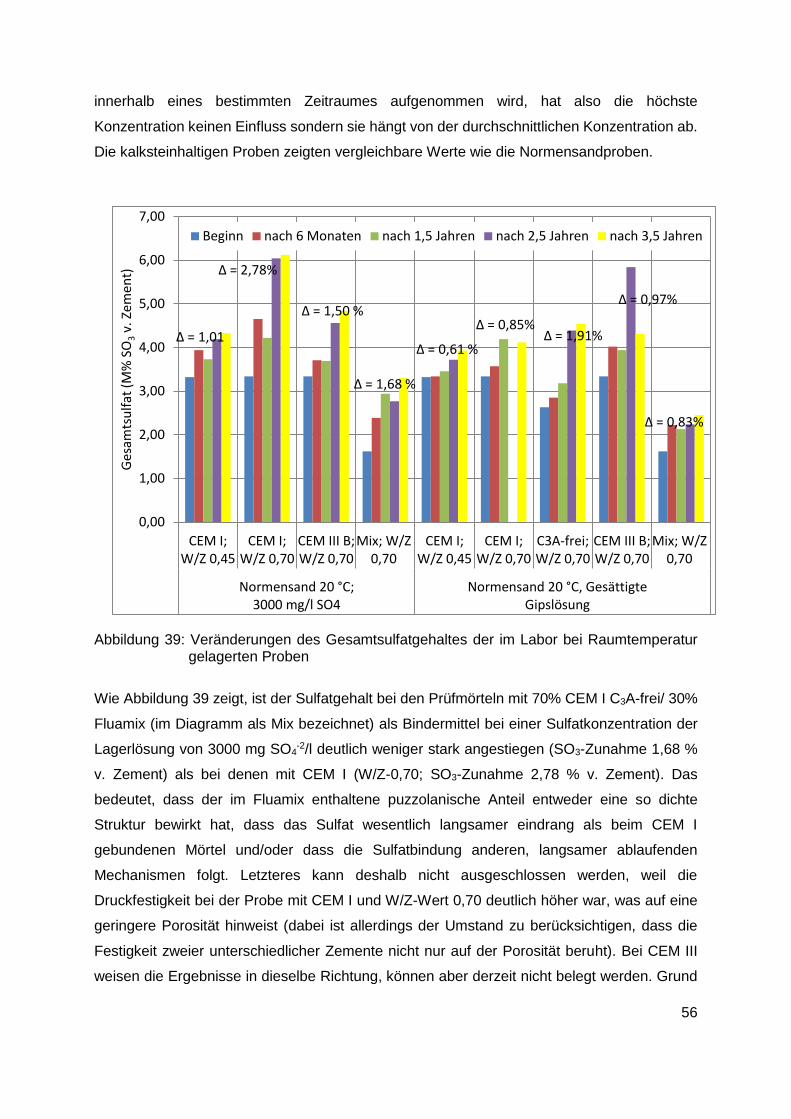
56
innerhalb eines bestimmten Zeitraumes aufgenommen wird, hat also die höchste
Konzentration keinen Einfluss sondern sie hängt von der durchschnittlichen Konzentration ab.
Die kalksteinhaltigen Proben zeigten vergleichbare Werte wie die Normensandproben.
Abbildung 39: Veränderungen des Gesamtsulfatgehaltes der im Labor bei Raumtemperatur
gelagerten Proben
Wie Abbildung 39 zeigt, ist der Sulfatgehalt bei den Prüfmörteln mit 70% CEM I C3A-frei/ 30%
Fluamix (im Diagramm als Mix bezeichnet) als Bindermittel bei einer Sulfatkonzentration der
Lagerlösung von 3000 mg SO4-2/l deutlich weniger stark angestiegen (SO3-Zunahme 1,68 %
v. Zement) als bei denen mit CEM I (W/Z-0,70; SO3-Zunahme 2,78 % v. Zement). Das
bedeutet, dass der im Fluamix enthaltene puzzolanische Anteil entweder eine so dichte
Struktur bewirkt hat, dass das Sulfat wesentlich langsamer eindrang als beim CEM I
gebundenen Mörtel und/oder dass die Sulfatbindung anderen, langsamer ablaufenden
Mechanismen folgt. Letzteres kann deshalb nicht ausgeschlossen werden, weil die
Druckfestigkeit bei der Probe mit CEM I und W/Z-Wert 0,70 deutlich höher war, was auf eine
geringere Porosität hinweist (dabei ist allerdings der Umstand zu berücksichtigen, dass die
Festigkeit zweier unterschiedlicher Zemente nicht nur auf der Porosität beruht). Bei CEM III
weisen die Ergebnisse in dieselbe Richtung, können aber derzeit nicht belegt werden. Grund
0,00
1,00
2,00
3,00
4,00
5,00
6,00
7,00
CEM I;W/Z 0,45
CEM I;W/Z 0,70
CEM III B;W/Z 0,70
Mix; W/Z0,70
CEM I;W/Z 0,45
CEM I;W/Z 0,70
C3A-frei;W/Z 0,70
CEM III B;W/Z 0,70
Mix; W/Z0,70
Normensand 20 °C;3000 mg/l SO4
Normensand 20 °C, GesättigteGipslösung
Ges
amts
ulf
at (M
% S
O3
v. Z
emen
t)
Beginn nach 6 Monaten nach 1,5 Jahren nach 2,5 Jahren nach 3,5 Jahren
Δ = 1,01
Δ = 2,78%
Δ = 1,50 %
Δ = 1,68 %
Δ = 0,61 %
Δ = 0,85%Δ = 1,91%
Δ = 0,97%
Δ = 0,83%

57
dafür könnte sein, dass der Ca(OH)2-Anteil in den Hydratationsprodukten die Sulfatreaktion
stärker beeinflusst hat. Zudem hat hier offenbar die Bestimmung des Zementgehaltes über die
lösl. SiO2 als Leitelement nicht funktioniert, weil der SiO2-Gehalt hoch war und offenbar
deshalb nicht mehr die gesamte lösl. SiO2 filtrierbar war, sich somit ein unkorrekter
Zementgehalt und damit ein unkorrekter Gehalt des auf den Zementgehalt bezogenen
Sulfatgehaltes ergeben hat. Zusätzliche Untersuchungen haben nämlich gezeigt, dass der
Gehalt des säureunlöslichen Anteils mitunter stark schwankte. Aus Abbildung 39 ist auch
ersichtlich, dass die Ergebnisse bei Lagerung in gesättigter Gipslösung
(Sättigungskonzentration: ~1200 mg SO4-2/l) dieselbe Tendenz zeigen, dass der Sulfatgehalt
aber deutlich weniger stark angestiegen ist als bei 3000 mg SO4-2/l.
Obwohl der Versuchszeitraum auch der Laboruntersuchungen mit 3,5 Jahren festgesetzt war,
erschien es zweckdienlich die Auslagerung noch nicht zu beenden und die in jeder
Lagerlösung noch vorhandenen letzten Proben nicht zu prüfen, weil dann mit Sicherheit keine
Aussagen hinsichtlich des kritischen Sulfatgehaltes getroffen hätten werden können. Daher
wurde eine Verlängerung der Projektlaufzeit um ein Jahr beantragt und nach 3,5 Jahren
lediglich der mittlere Sulfatgehalt anhand der Würfelproben bestimmt. Die Verlängerung der
Auslagerung erschien deshalb vielversprechend, weil beim Prisma mit W/Z-0,70 und CEM I
als Bindemittel nach 3,5 Jahren Lagerung bei 6000 mg SO4-2/l andeutungsweise eine
Längenänderung zu bemerken war (~0,05 mm; entspricht 0,3 – 0,6 mm/m), die
Biegezugfestigkeit deutlich abgenommen hat sowie innerhalb der Proben unter dem
Elektronenmikroskop feststellbare Risse vorhanden waren (Abbildung 40), weshalb
anzunehmen war, dass in nicht mehr ferner Zukunft erste sichtbare Schäden entstehen
würden. Von den nach der Prüfung der Biegezugfestigkeit verblieben zwei Prismenhälften
wurde dann aus einer Hälfte die Porenlösung ausgepresst und danach der Gesamtsulfatgehalt
der Feststoffe bestimmt. An der anderen Hälfte wurden die übrigen Untersuchungen
(Röngendiffraktometrie, Elektronenmikroskop) durchgeführt. Daher konnten alle Prüfungen
mit Ausnahme von Festigkeit und Porenlösung sowohl nach 3,5 als auch nach 4,5 Jahren
durchgeführt werden.

58
Abbildung 40: Risse innerhalb einer Probe (CEM I; W/Z 0.70) nach ca. 2,5 Jahren (links) und nach 3,5 Jahren (rechts)
Abbildung 41: Ablagerungen von Ettringit in Poren (links) bzw. Gips (rechts) und entlang der Übergangszone Zuschlagskorn/Bindemittelmatrix (links CEM I - W/Z-0,70 nach 3,5 Jahren; rechts C3A-freier Zement nach 4,5 Jahren)
Es hat sehr überrascht, dass es nach 3,5 Jahren Lagerung auch an den bei der höchsten
Sulfatkonzentration gelagerten Proben zu keinen für das unbewaffnete Auge erkennbaren
Anzeichen von Schadensbildung gekommen ist. Bei Vergrößerungen im Elektronenmikroskop
waren allerdings schon viel früher Anzeichen von Schäden zu erkennen. Abbildung 40 zeigt
Bilder der Probe mit der Rezeptur W/Z-0,70; CEM I. Nach 2,5 Jahren waren bei der mittleren
Sulfatkonzentration der Lagerlösung von 3000 mg SO4-2/l Ettringitablagerungen in Poren bzw.
anderen Hohlräumen zu beobachten, von denen feine Haarrisse ausgingen (Abbildung 40,
links). Das rechte Bild zeigt die Struktur des gleichen Mörtels nach 3,5 Jahren Lagerung bei
6000 mg SO4-2/l und 20°C. Risse waren vermehrt festzustellen und ihre Öffnungsweite war
größer. Auch wurde festgestellt, dass in der Übergangszone Gesteinskorn/Bindemittelmatrix

59
teilweise Ablagerungen von Ettringit bzw. Gips vorhanden waren (Abbildung 41). Dies steht im
Einklang mit der Literatur, wonach die Übergangszone poröser ist als die abseits von
Gesteinskörnern befindliche Bindemittelmatrix und dass der Transport eindringender Stoffe
bevorzugt entlang von Gesteinskörnern stattfindet [84].
9.1.3 Sulfatverteilung innerhalb der Proben
Abbildung 42: Sulfatverteilung innerhalb der Proben
Abbildung 43: Schwefelverteilung innerhalb der Proben mit CEM I; W/Z-0,70; nach 6 Monaten (links) und 1,5 Jahren (rechts) Lagerung bei 6000 mg SO4
-2/l

60
Wie aus den Diagrammen in Abbildungen 42 hervorgeht, hat sich das aufgenommene Sulfat
innerhalb der Proben nicht gleichmäßig sondern sehr ungleichmäßig verteilt. Die Erstellung
der Sulfatprofile erfolgte wie in Kapitel 7.1 beschrieben. Die ungleichmäßige Verteilung
innerhalb der Proben kommt auch aus den Bildern in Abbildung 43 zum Ausdruck, in denen
die Schwefelkonzentration innerhalb einer Probe (ca. 2 cm² große Fläche zwischen
Probenrand und Probenmitte) in unterschiedlichen Farben dargestellt ist [dunkelblau <1%
Schwefel (S), hellgrün ~5% S; s. Farbenskala zwischen den Bildern; 1 % S entspricht etwa 3
% SO4-2, 5% S etwa 15 % SO4
-2]. Daraus ist klar ersichtlich, dass die Farben unter der
Probenoberfläche (linker Bildrand) schon nach 6 Monaten Lagerung in der Lösung mit 6000
mg SO4-2/l unterschiedlich sind (die hellblaue Zone links unten reicht tiefer in die Probe als
darüber). Der Verteilungsgradient wurde bei den Proben mit W/Z-0,70 im Verlauf der Lagerung
schwächer, weil der Sulfatgehalt ab dem Erreichen der Probenmitte im Zentrum weiter anstieg,
nicht jedoch in den äußeren Probezonen. Bei der C3A-freien Probe beruht die Zunahme des
Sulfatgehaltes vornehmlich auf der Bildung der AfT-Phase und von Gips.
9.1.4 Porenlösung
Abbildung 44: Kationenkonzentration der Porenlösung
Abbildung 44 zeigt die während der Lagerung in den Lösungen mit 600 und 6000 mg SO4-2/l
entstandenen Veränderungen der Konzentration der in der Porenlösung enthaltenen Kationen.
0
2000
4000
6000
8000
10000
12000
14000
16000
18000
20000
CEM I W/Z0,45
CEM I W/Z0,70
C3A-frei W/Z0,70
CEM I W/Z0,45
CEM I W/Z0,70
C3A-frei W/Z0,70
600 mg/l 6000 mg/l
Konzentr
ation in m
g/L
Na K Ca
Werte zu Beginn, nach 6 Monaten, 1,5; 2,5 und 4,5 Jahren

61
Bei den übrigen Lagerlösungen waren die Veränderungen vergleichbar. Sie sind hier nicht
dargestellt, weil die Veränderungen bei weniger Einzelwerten besser erkannt werden können.
Die Balken derselben Farbe zeigen die Konzentration zu Beginn, nach 6 Monaten, 1,5 Jahren,
2,5 Jahren und zu Versuchsende an (von links nach rechts). Wie ersichtlich, war die
Konzentration der Alkaliionen beim niedrigeren W/Z-Wert (CEM I) höher, weil weniger
Lösungsmittel (Porenwasser) vorhanden war als beim höheren W/Z-Wert. Die
Natriumkonzentration war anfänglich deutlich geringer als die des Kaliums (s. Kapitel 8.2.4).
Weiter ist ersichtlich, dass die Ausgangskonzentration der Alkalien (vornehmlich die von
Kalium) in der Porenlösung des C3A-freien Zementes deutlich niedriger war als beim CEM I
mit W/Z-0,70. Dies deshalb, weil im C3A freien Zement weniger Alkalien enthalten waren. Wie
bei den im Kanalsystem von Linz ausgelagerten Mörtelproben hat die Konzentration der
Alkalien ständig ab- und die des Calciums zugenommen (s. Kapitel 8.2.4). Auch bei den
Anionen bestand derselbe Trend wie bei den im Kanalsystem ausgelagerten Proben, weshalb
auf die Änderungen und deren Ursachen hier nicht erneut näher eingegangen wird.
Abbildung 45: Anionenkonzentration der Porenlösung
In Abbildung 45 sind die Konzentrationsverschiebungen der OH- und SO4-2-Ionen dargestellt.
Die Konzentration der OH--Ionen hat wie bei den im Abwasserkanal ausgelagerten Proben
0
2000
4000
6000
8000
10000
12000
14000
CEM I W/Z0,45
CEM I W/Z0,70
C3A-frei W/Z0,70
CEM I W/Z0,45
CEM I W/Z0,70
C3A-frei W/Z0,70
600 mg/l 6000 mg/l
Konzentr
ation in m
g/L
OH SO4
Werte zu Beginn, nach 6 Monaten, 1,5; 2,5 und 4,5 Jahren

62
laufend abgenommen, was beim Sulfat nicht der Fall war. Die SO4-2-Konzentration hat bei
W/Z-0,45 bis zu 1,5 Jahren ab-, danach zugenommen, bei den Proben mit W/Z-0,70 immer
leicht zugenommen. Das steht in direktem Zusammenhang mit der OH--Konzentration, denn
mit abnehmender OH--Konzentration steigt die Löslichkeit von Calcium und Gips
(Calciumsulfat). Dies ist natürlich nur qualitativ zu sehen, denn die Gesamtverhältnisse des
sich zwischen den Feststoffen und der Porenlösung einstellenden Verteilungsgleichgewichtes
sind bei der Vielzahl an Hydratationsprodukten, die in einem hydratisierten Zement vorhanden
sind, nicht überschaubar. Die Sulfatkonzentration der Porenlösung blieb aber bei 6000 mg
SO4-2/l immer weit unterhalb der Sulfatkonzentration der zugehörigen Lagerlösung, so dass
davon ausgegangen werden kann, dass eindiffundiertes Sulfat Großteils gebunden wurde und
daher die Sulfataufnahmefähigkeit der Proben auch zu Versuchsende noch nicht erschöpft
war. Dass dies bei 600 mg SO4-2/l bei den Mörteln mit W/Z-0,70 nicht der Fall war, könnte ein
Hinweis dafür sein, dass der zu dieser Lagerlösungskonzentration gehörige Sulfat-
Maximalgehalt erreicht war. Aus theoretischen Überlegungen müsste dies der Fall sein (die
Sulfakonzentration der Porenlösung hängt von der Löslichkeit der Sulfatverbindungen ab),
kann aber nicht bewiesen werden, weil dazu durch länger andauernde (über 4,5 Jahre
hinausgehende) Untersuchungen festgestellt werden müsste ob der Gesamtsulfatgehalt bei
fortgesetzter Lagerung konstant bleibt

63
9.2 Thaumasitbildung
(mit Kalksteinmehl statt Normensand-fein hergestellte Mörtelproben, ausgelagert bei 5°C)
Abbildung 46: Veränderungen des Gesamtsulfatgehaltes während der Lagerung bei 5 °C
Der Bestimmung der Biegezug- und Druckfestigkeit (Prismen) wurde nur bis zum Prüftermin
2,5 Jahre durchgeführt, weil die bei 3000 mg SO4-2/l gelagerten Proben nach 3,5 Jahren schon
so stark geschädigt waren, dass die Prüfung nicht mehr möglich war bzw. nicht sinnvoll
erschien. Wohl aber wurden von den Würfelproben die lose anhaftenden Reaktionsprodukte
abgebürstet und der verbliebene Probenteil hinsichtlich des Sulfatgehaltes untersucht.
Abbildung 46 zeigt die Veränderungen des Sulfatgehaltes der bei 5°C gelagerten Proben.
Interessant war, dass die Zunahme des Sulfatgehaltes bei 3000 mg SO4-2/l recht gut mit den
bei Raumtemperatur eingetretenen Veränderungen (Abbildung 37) korrelierte. Die
Lagerungstemperatur oder die Art des Zuschlags hatten somit auf die Geschwindigkeit der
Sulfataufnahme (erwartungsgemäß) keinen starken Einfluss.
0,00
1,00
2,00
3,00
4,00
5,00
6,00
CEM I;W/Z 0,45
CEM I;W/Z 0,70
C3A-frei;W/Z 0,70
CEM I;W/Z 0,45
CEM I;W/Z 0,70
C3A-frei;W/Z 0,70
CEM I;W/Z 0,45
CEM I;W/Z 0,70
C3A-frei;W/Z 0,70
200 mg/l 600 mg/l 3000 mg/l
Gesam
tsulfatg
ehalt b
ez.
auf den Z
em
entg
ehalt in
%
Rezeptur und Sulfatkonzentration
zu Beginn nach 6 Monaten nach 1,5 Jahren nach 2,5 Jahren nach 3,5 Jahren

64
CEM I: Prismen mit Normensand - W/Z-0,45 (unten) bzw. 0,70 (oben) nach ca. 2 Jahren Lagerung in Na2SO4-
Lösung mit 3000 mg SO4-2/l
CEM I: Kalkstein -Zuschlag; W/Z-0,50 (unten) und 0,70 (oben) nach ca.3,5 Jahren Lagerung in Na2SO4-Lösung
mit 3000 mg SO4-2/l
CEM I: Kalkstein-Zuschlag; W/Z-0,50 (unten) und 0,70 (oben) nach ca.3,5
Jahren Lagerung in gesättigter Gipslösung
CEM I C3A-frei: W/Z-0,70 nach ca.3,5 Jahren (oben) und 4,5 Jahren (unten) Lagerung in gesättigter
Gipslösung
FluaMix: Kalkstein-Zuschlag; W/Z-0,70 nach ca.3,5 Jahren Lagerung in
gesättigter Gipslösung
CEM III-B: Kalkstein-Zuschlag; W/Z-0,70 nach ca.3,5 Jahren Lagerung in
gesättigter Gipslösung
FluaMix: Kalkstein-Zuschlag; W/Z-0,70 nach ca. 4,5 Jahren in gesättigter
Gipslösung
CEM III-B: Kalkstein-Zuschlag; W/Z-0,70 nach ca.4,5 Jahren Lagerung in
gesättigter Gipslösung
Abbildung 47: Thaumasitschäden an den Prismen (Lagerung im Labor bei 5°C)

65
Die an den Proben in der Lagerlösung mit 3000 mg SO4-2/l und in der gesättigten Gipslösung
entstandenen Schäden sind aus Abbildung 47 ersichtlich (bei 200 und 600 mg SO4-2/l sind
keine Schäden entstanden). Bei den Proben mit Normensand und Kalkstein als Zuschlag
(oberste Bilder) wirkt die Probe mit Kalkstein etwas stärker geschädigt. Diese Probe lagerte
aber länger, weshalb eine direkte Vergleichbarkeit nicht gegeben ist. Auch aus den übrigen
Ergebnissen konnte nicht abelessen werden, dass kalksteinhaltiger Zuschlag eine wesentlich
stärkere Schädigung bewirkt. Daraus geht hervor, dass die Anwesenheit einer karbonatischen
Gesteinskörnung weder eine notwendige Voraussetzung für einen Thaumasitangriff ist noch
dass der Angriff bei karbonatfreier Gesteinskörnung eindeutig schwächerer ist. Offenbar
stammt das benötigte Karbonat hauptsächlich aus dem CO2 der Luft und wurde nach
Verbrauch durch Diffusion in die Lagerlösung immer wieder nachtransportiert, obwohl die
Lagerungsboxen mittels Klappdeckel abgedeckt waren. Interessant war auch der Umstand,
dass bei Lagerung in gesättigter Gipslösung eine deutlich stärkere Schädigung eintrat als bei
3000 mg SO4-2/l obwohl die Sulfatkonzentration in der gesättigten Gipslösung wesentlich
geringer war (max. ~1200 mg SO4-2/l). Offensichtlich hat die Anwesenheit des für die
Thaumasitbildung auch benötigten Calciums im Sulfat-Salz (Gips: CaSO4 ∙2H2O) einen
wesentlichen Einfluss auf die Geschwindigkeit der Schädigung. Aus der Abbildung geht auch
klar hervor, dass C3A-freier Zement keinen Schutz gegen diese Art der Sulfatangriffs darstellt.
(dabei kommt es zu keiner Instabilität des Volumens (Ettringit) sondern zu einer Umwandlung
der C-S-H Phase in Thaumasit, verbunden mit einem Verlust der Festigkeit). Weiter zeigt die
Abbildung, dass die Proben mit Fluamix bzw. CEM III-B als Bindemittel unvergleichlich weniger
angegriffen waren, wobei an der Probe mit CEM III-B überhaupt keine Angriff stattgefunden
hat. Diese Bindemittel zeichnen sich somit nicht nur durch eine wesentlich langsamere
Sulfataufnahme als auch durch eine hohe Resistenz gegen Thaumasitschädigung aus. Durch
ein geeignetes Bindemittel kann also die Thaumasitschädigung vermieden bzw. stark reduziert
werden.
Abbildung 47 zeigt weiter, dass die Thaumasitschädigung wie ein lösender Angriff an der
Probenoberfläche beginnt und sich ins -innere fortsetzt, wobei die Zersetzungsprodukte
teilweise lose haften bleiben. Zuerst wurde immer die Oberfläche mit der höchsten Porosität
(abgezogene Oberfläche der Prismen) angegriffen. Die Porenlösungsuntersuchungen
ergaben, dass die Veränderungen der Porenlösungszusammensetzung mit den bei 20°C
festgestellten Veränderungen vergleichbar waren. Insbesondere die Sulfatkonzentration blieb
im nicht geschädigten Probenteil relativ gering (max. ca. 1000 mg SO4-2/l).
Zu erwähnen ist noch, dass auch an der Oberfläche aller Mörtelproben (mit Kalkstein oder

66
ohne), die bei 20°C und 6000 mg SO4-2/l ausgelagert waren, stellenweise Thaumasit
entstanden ist. Die gebildete Menge war aber nur sehr gering und hat die Festigkeit nicht
nachteilig verändert. Immerhin zeigen diese Ergebnisse aber, dass sich Thaumasit auch bei
Temperaturen über 15°C bilden kann. Die Frage welche Sulfatkonzentration eines
angreifenden Wassers unbedingt nötig ist, damit es überhaupt zur Bildung von Thaumasit
kommen kann, kann aus den Ergebnissen nur so beantwortet werden, dass es bei 5°C nur bei
der Sulfatkonzentration von 3000 mg/l zu einem Angriff gekommen ist, nicht aber bei 600 mg
SO4-2/l oder darunter. Aus dem Ergebnis, dass es bei Lagerung in gesättigter Gipslösung
(Sättigungskonzentration in neutraler Lösung ca. 1200 mg SO4-2/l; bei höheren pH-Werten
geringer) zu starken Schäden gekommen ist, ergibt sich, dass eine solche Konzentration
schädlich ist. Die Grenzkonzentration liegt also irgendwo zwischen 600 und 1200 mg SO4-2/l.
Daraus ergibt sich nachstehendes Scenario für die Thaumasitschädigung:
Im inneren der Proben, wo kein Berührungskontakt zur Lagerlösung bestand, blieb die
Sulfatkonzentration der Porenlösung offensichtlich zufolge fortgesetzter Bildung von
schwerlöslichen Sulfatverbindungen (Ettringit, Gips) ständig gering, so dass sich dort kein
Thaumasit bilden konnte. Die Sulfatkonzentration der Lagerlösung war aber so hoch, dass
Thaumasitbildung möglich war und deshalb begann der Angriff an der Probenoberfläche. Je
poröser der Mörtel war, umso schneller stieg der Sulfatgehalt, zuerst im oberflächennahen
Mörtelbereich und konnte eine Vorschädigung (Bildung von mit freiem Auge nicht erkennbaren
Rissen, etc.) bewirken. Die OH--Konzentration (der pH-Wert) der Porenlösung nahm nahe der
Oberfläche am schnellsten ab, weil OH--Ionen umso leichter aus der Probe diffundieren
können, je poröser die Bindemittelmatrix ist. Zudem reagiert der gebildete Gips geringfügig
sauer, sodass leicht vorstellbar ist, dass der pH-Wert der flüssigen Phase unter der Oberfläche
relativ rasch so stark abfiel, dass die zur Aufrechterhaltung der Stabilität von Ettringit benötigte
Alkalität der Porenlösung in der Nähe der Oberfläche bald verloren ging, daher hier die
Sulfatkonzentration der Porenlösung höhere Werte annehmen und sich Thaumasit bilden
konnte. Darin liegt wohl auch die Erklärung für die festgestellte starke Abhängigkeit der
Angriffsstärke vom W/Z-Wert. Berücksichtig man den offensichtlich starken Einfluss des
Kations in der Sulfatlösung (Vergleich Calcium- und Natriumsulfat) gibt es hier weiteren
Forschungsbedarf. Des Weiteren kommen in der Natur (siehe auch Kapitel 10) selten reine
Na+- bzw. Ca+2-Sulfatlösungen vor, sondern es handelt sich zumeist um komplexe
Mischlösungen, welche auch hohe Gehalte an z.B. Mg+2, Cl- und HCO3- beinhalten können.

67
10. Nasschemische Bestimmung der Sulfatverteilung in Betonproben
10.1 Allgemeines
Ausgangspunkt war ein in den Jahren 1980-1982 durchgeführtes Forschungsvorhaben
„Sulfatbeständiger Spritzbeton für Straßentunnel-Auskleidungen“ [77]. Dabei wurde eine
Prüfmethode für die Bewertung der Sulfatbeständigkeit auf Basis der Sulfataufnahme in
Zementsteinproben nach 99-tägiger Lagerung in gesättigter Gipslösung ausgearbeitet. Dabei
wurde aus einer Probentiefe von 0,5 mm Zementstein durch Abfräsen entnommen und die
Zunahme des Sulfatgehalts (als SO3) nach 33, 66 und 99 Tagen Lagerung in Bezug zum
Ausgangsgehalt an SO3 bestimmt.
Diese Prüfmethode wurde im gegenständlichen Forschungsvorhaben angewendet, wobei an
Stelle von Zementleimproben geschnittene Betonproben herangezogen werden. Durch eine
spezielle Abtragung wurde ebenfalls die Zunahme an SO3 im Verlauf der Lagerung in
gesättigter Gipslösung in unterschiedlichen Tiefenbereichen des Betons gemessen.
10.2 Betone für die Untersuchungen
Für die Untersuchungen wurden im Labor Betone mit 3 unterschiedlichen Bindemitteln und
W/B-Werten von 0,45, 0,50 und 0,55 hergestellt. Dabei wurde der Bindemittelgehalt konstant
mit 350 kg/m³ angesetzt. Als Bindemittel wurden folgende Zemente und ein AHWZ
(aufbereitetes hydraulisch wirksames Zusatzmittel) verwendet:
1.: CEM I 52,5 R
2.: Contragress CEM I 42,5 N C3A-frei
3.: 70 % Contragress + 30 % AHWZ (Fluamix C)
Fluamix C ist ein Kombinationsprodukt GC (Mischung aus gemahlenem Hüttensand und
gemahlener Flugasche mit max. 20 % Kalksteinmehl) gemäß ÖNorm B 3309 [61]. Als
Fließmittel wurde Glenium Sky 582 von BASF verwendet.
In den folgenden Tabellen sind die Mischungszusammensetzung sowie die
Frischbetoneigenschaften und die Druckfestigkeiten der Laborbetone zusammengestellt:
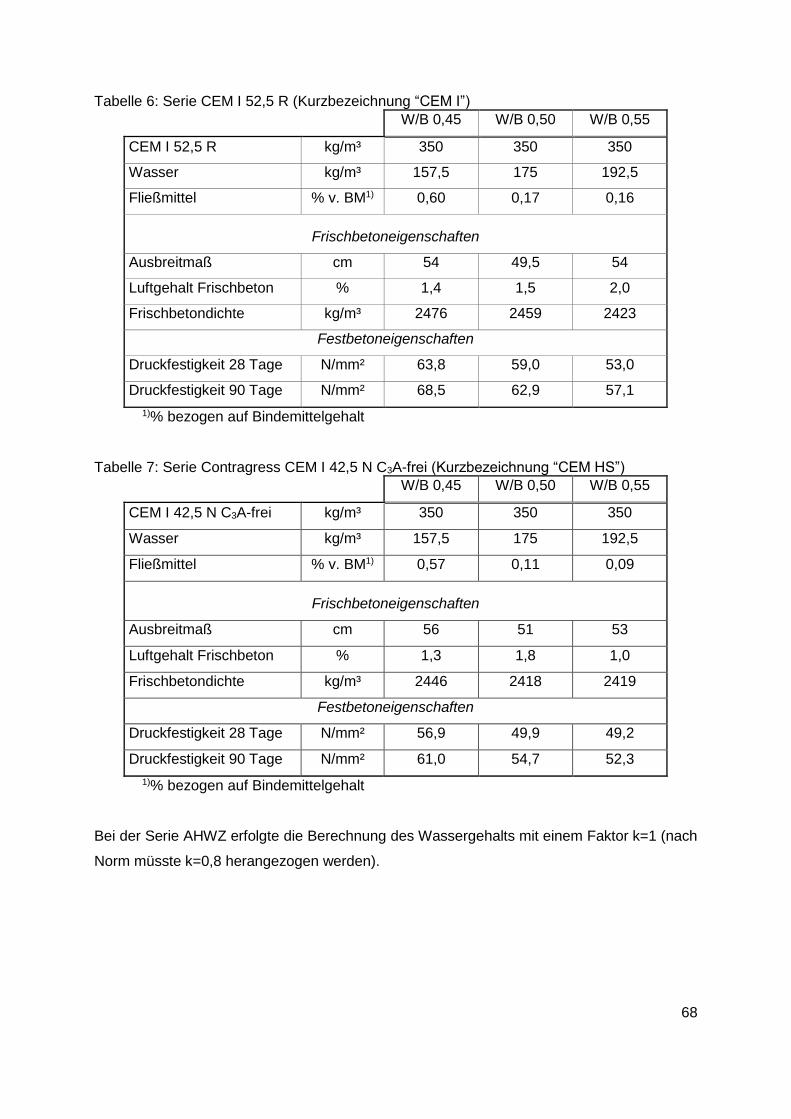
68
Tabelle 6: Serie CEM I 52,5 R (Kurzbezeichnung “CEM I”)
W/B 0,45 W/B 0,50 W/B 0,55
CEM I 52,5 R kg/m³ 350 350 350
Wasser kg/m³ 157,5 175 192,5
Fließmittel % v. BM1) 0,60 0,17 0,16
Frischbetoneigenschaften
Ausbreitmaß cm 54 49,5 54
Luftgehalt Frischbeton % 1,4 1,5 2,0
Frischbetondichte kg/m³ 2476 2459 2423
Festbetoneigenschaften
Druckfestigkeit 28 Tage N/mm² 63,8 59,0 53,0
Druckfestigkeit 90 Tage N/mm² 68,5 62,9 57,1
1)% bezogen auf Bindemittelgehalt
Tabelle 7: Serie Contragress CEM I 42,5 N C3A-frei (Kurzbezeichnung “CEM HS”)
W/B 0,45 W/B 0,50 W/B 0,55
CEM I 42,5 N C3A-frei kg/m³ 350 350 350
Wasser kg/m³ 157,5 175 192,5
Fließmittel % v. BM1) 0,57 0,11 0,09
Frischbetoneigenschaften
Ausbreitmaß cm 56 51 53
Luftgehalt Frischbeton % 1,3 1,8 1,0
Frischbetondichte kg/m³ 2446 2418 2419
Festbetoneigenschaften
Druckfestigkeit 28 Tage N/mm² 56,9 49,9 49,2
Druckfestigkeit 90 Tage N/mm² 61,0 54,7 52,3
1)% bezogen auf Bindemittelgehalt
Bei der Serie AHWZ erfolgte die Berechnung des Wassergehalts mit einem Faktor k=1 (nach
Norm müsste k=0,8 herangezogen werden).

69
Tabelle 8: Serie AHWZ (70 % Contragress + 30 % Fluamix C) (Kurzbezeichnung “HS+AHWZ”)
W/B 0,45 W/B 0,50 W/B 0,55
CEM I 42,5 N C3A-frei kg/m³ 245 245 245
Fluamix C kg/m³ 105 105 105
Wasser kg/m³ 157,5 175 192,5
Fließmittel % v. BM1) 0,38 0,14 0
Frischbetoneigenschaften
Ausbreitmaß cm 55 50 51,5
Luftgehalt Frischbeton % 1,0 1,1 0,6
Frischbetondichte kg/m³ 2466 2456 2444
Festbetoneigenschaften
Druckfestigkeit 28 Tage N/mm² 51,9 44,6 41,2
Druckfestigkeit 90 Tage N/mm² 55,2 48,1 43,9
1)% bezogen auf Bindemittelgehalt
10.3 Herstellung von Betonbalken und Probenentnahme
Abbildung 48: Betonprismen nach Bohrkern- entnahme
Abbildung 49: Betonscheiben
Für die Untersuchungen wurden Betonbalken (700 x 150 x 150 mm) hergestellt und nach
Ausschalen im Alter von 1 Tag bis zum 7. Tag unter Wasser gelagert. Die weitere Lagerung
erfolgte bei Raumtemperatur. Aus den Betonbalken wurden Bohrkerne mit Durchmesser 70
mm entnommen (Abbildung 48). Von den Bohrkernen wurden ca. 25 mm dicke Scheiben
abgetrennt (Abbildung 49). Die Betonscheiben wurden in der Folge mittels Drehbank

70
planparallel geschliffen (siehe Kapitel 7.3).
10.4 Probenlagerung, Untersuchung und deren Ergebnisse
Die Einlagerung der Betonscheiben erfolgte in gesättigter Gipslösung mit Bodensatz (6 g Gips
pro Liter). Nach Lagerungsdauern von 33, 66, 99 und 152 Tagen wurden jeweils 2
Betonscheiben aus der Lösung entnommen, mit Wasser abgewaschen und anschließend bei
80 °C getrocknet. An den getrockneten Betonscheiben wurde das Betonmehl aus den
Tiefenstufen 0,4 – 0,6 mm, 1,0 – 1,2 mm und 1,4 – 1,6 mm entnommen und der Gehalt an
SO3 bestimmt. Betonmehl aus diesen Tiefenstufen wurde an 2 Betonscheiben entnommen,
d.h. dass insgesamt 4 Betonmehlproben pro Tiefenstufe zur Verfügung standen (jeweils
Vorder- und Rückseite der Betonscheiben). Mengenmäßig ergab das Betonmehl aus der
jeweiligen Tiefenstufe ca. 2 bis 2,5 Gramm.
Nach 99 Tagen Lagerung wurden die Oberflächen der Betonscheiben in verdünnte Salzsäure
(1 + 9) für 30 sec eingelegt. Anschließend wurde die Betonprobe abgewaschen und wiederum
in die Gipslösung eingelegt.
10.4.1 Bestimmung des Sulfatgehalts
An den, aus den Betonscheiben entnommenen Betonmehlproben wurde der Sulfatgehalt
nasschemisch nach einem Aufschluss mit verdünnter Salzsäure bestimmt. Dazu wurde das
Betonmehl mit 100 ml Salzsäure (1:9) übergossen, auf 70 °C erwärmt und danach durch einen
Glasfaserfilter filtriert. In der erhaltenen Lösung wurde der Sulfatgehalt photometrisch
bestimmt und auf den SO3-Gehalt in Masse-% umgerechnet.
10.4.2 Ergebnisse
Wie bereits erwähnt, wurde für jede Tiefenstufe insgesamt 4 Probenentnahmen durchgeführt
(je Rezeptur zwei 2 Betonscheiben; Abdrehen des Betons auf der Vorder- und Hinterseite).
Das Betonmehl aus jeder Tiefenstufe beiden Scheiben wurde zu einer Probe vereint und der
SO3-Gehalt bestimmt. Der erhaltene Messwert ist somit der durchschnittliche SO3-Gehalt
beider Betonscheiben.
In der folgenden Tabelle sind die SO3-Gehalte der Betone mit W/B-Werten von 0,45 und 0,55
im Ausgangszustand und nach 33, 66, 99 und 152 Tagen Lagerung in der gesättigten
Gipslösung zusammengestellt. Es ist zu beachten, dass nach 99 Tagen die Oberfläche der
Betonscheiben mit verdünnter Salzsäure (1 + 9) für jeweils 30 sec behandelt wurde.
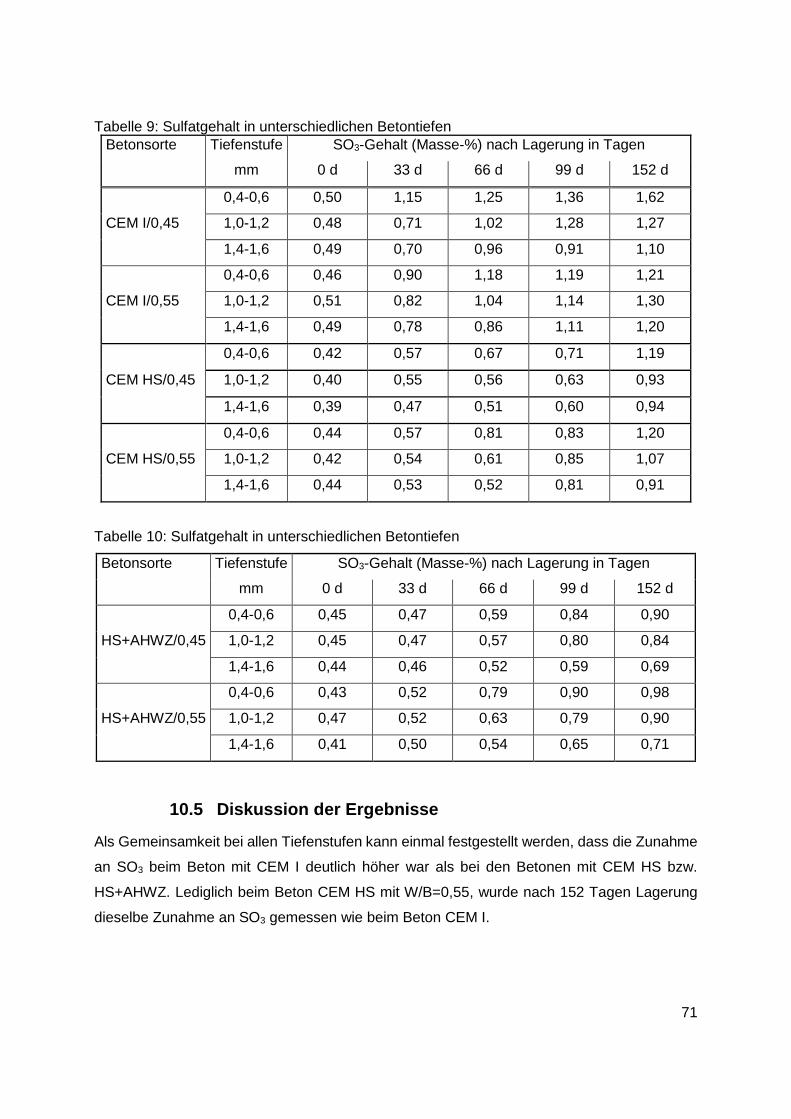
71
Tabelle 9: Sulfatgehalt in unterschiedlichen Betontiefen
Betonsorte Tiefenstufe SO3-Gehalt (Masse-%) nach Lagerung in Tagen
mm 0 d 33 d 66 d 99 d 152 d
CEM I/0,45
0,4-0,6 0,50 1,15 1,25 1,36 1,62
1,0-1,2 0,48 0,71 1,02 1,28 1,27
1,4-1,6 0,49 0,70 0,96 0,91 1,10
CEM I/0,55
0,4-0,6 0,46 0,90 1,18 1,19 1,21
1,0-1,2 0,51 0,82 1,04 1,14 1,30
1,4-1,6 0,49 0,78 0,86 1,11 1,20
CEM HS/0,45
0,4-0,6 0,42 0,57 0,67 0,71 1,19
1,0-1,2 0,40 0,55 0,56 0,63 0,93
1,4-1,6 0,39 0,47 0,51 0,60 0,94
CEM HS/0,55
0,4-0,6 0,44 0,57 0,81 0,83 1,20
1,0-1,2 0,42 0,54 0,61 0,85 1,07
1,4-1,6 0,44 0,53 0,52 0,81 0,91
Tabelle 10: Sulfatgehalt in unterschiedlichen Betontiefen
Betonsorte Tiefenstufe SO3-Gehalt (Masse-%) nach Lagerung in Tagen
mm 0 d 33 d 66 d 99 d 152 d
HS+AHWZ/0,45
0,4-0,6 0,45 0,47 0,59 0,84 0,90
1,0-1,2 0,45 0,47 0,57 0,80 0,84
1,4-1,6 0,44 0,46 0,52 0,59 0,69
HS+AHWZ/0,55
0,4-0,6 0,43 0,52 0,79 0,90 0,98
1,0-1,2 0,47 0,52 0,63 0,79 0,90
1,4-1,6 0,41 0,50 0,54 0,65 0,71
10.5 Diskussion der Ergebnisse
Als Gemeinsamkeit bei allen Tiefenstufen kann einmal festgestellt werden, dass die Zunahme
an SO3 beim Beton mit CEM I deutlich höher war als bei den Betonen mit CEM HS bzw.
HS+AHWZ. Lediglich beim Beton CEM HS mit W/B=0,55, wurde nach 152 Tagen Lagerung
dieselbe Zunahme an SO3 gemessen wie beim Beton CEM I.

72
Abbildung 50: SO3-Gehalt der untersuchten Betone in der Tiefenstufe 0,4 bis 0,6 mm nach Lagerungsdauer 99 und 152 Tage
Abbildung 50 zeigt die SO3-Zunahme der Betone in der Tiefenstufe 0,4 bis 0,6 mm zwischen
99 und 152 Tagen Lagerung in der gesättigten Gipslösung. Wie ersichtlich, bedeutet der
höhere W/B-Wert nicht immer auch eine höhere Zunahme an SO3. Auffallend ist weiter, dass
der Beton HS+AHWZ die geringste SO3-Zunahme aufwies.
Der Beton CEM I/0,55 zeigte nach 99 Tagen (genauer bereits nach 66 Tagen) praktisch
keine weitere Zunahme an SO3 mehr. Bei den Betonen HS+AHWZ war der SO3-Gehalt nach
152 Tagen nur geringfügig höher als nach 99 Tagen. Hingegen wiesen die Betone CEM HS
noch sehr deutliche SO3-Zunahmen zwischen 99 und 152 Tagen Lagerung auf.
Obwohl sich bei der Sulfataufnahme der untersuchten Betone deutliche Unterschiede ergeben
haben, muss angemerkt werden, dass die Proben der Betone mit CEM I auch nach 152 Tagen
Lagerungsdauer trotz stark überhöhter Sulfatwerte noch keine Hinweise auf
Treiberscheinungen erkennen ließen. Auf die Frage, warum die Betone mit CEM I noch keine
Treiberscheinungen zeigen, fehlt derzeit noch eine Antwort. Um Klarheit über die
diesbezüglichen Mechanismen zu erhalten, bedarf es noch weiterer Untersuchungen.
0,00
0,10
0,20
0,30
0,40
0,50
0,60
0,70
0,80
0,90
1,00
1,10
1,20
CEM I/0,45 CEM HS/0,45 HS+AHWZ/0,45 CEM I/0,55 CEM HS/0,55 HS+AHWZ/0,55
Zu
na
hm
e S
O3
(Ma
ss
e-%
)99 Tage
152 Tage
Tiefenstufe 0,4 - 0,6 mm

73
11. Untersuchung von Bauwerksschäden
Den Projektbeteiligten waren zu Beginn des Forschungsvorhabens hauptsächlich nur solche,
durch Sulfatkorrosion verursachte Schäden gravierenden Ausmaßes bekannt, die in
österreichischen Tunneln entstanden sind. Daher wurden nur die dort vorhandenen Schäden
untersucht, deren Erscheinungsbild, Probenentnahmen, etc. im Kapitel 6.2.2 beschrieben ist,
sodass in diesem Kapitel nur noch auf die durchgeführten Untersuchungen und deren
Ergebnisse eingegangen wird. Da sowohl die Schadensursache als auch die zugrunde
liegenden Schadensmechanismen überall dieselben waren, werden nur die zur Klärung der
Ursache und des Mechanismus der Schadensbildung durchgeführten Untersuchungen
exemplarisch beschrieben. Weiter Details sind folgenden Arbeiten zu entnehmen [54-58, 75,
76, 78, 84-86].
Die mittels Röntgendiffraktometrie, Polarisationsmikroskopie, Rasterelektronenmikroskopie
und Mikrosonde durchgeführten Untersuchungen haben ergeben, dass die Schädigung des
Betons hauptsächlich nur durch Thaumasitbildung verursacht wurde. Die typisch stängelige
bis faserige Morphologie des Thaumasits ist aus Abbildung 51 ersichtlich, die eine
rasterelektronenmikroskopische Aufnahme von Thaumasitkristallen einer, einem Tunnel
entnommenen Probe zersetzten Betons zeigt.
Abbildung 51: Thaumasitkristalle
Abbildung 52: In Thaumasitmatrix inkongruent
aufgelöstes Dolomitkorn [(Kalcit (Cal); Brucit (Brc)]
Neben dem Beton schädigenden Mineral Thaumasit ist meistens zusätzlich als zweites
schädigendes Sulfat-Mineral Gips (CaSO4.2H2O) festgestellt worden (Gips war allerdings nur
in geringen Mengen enthalten). Daneben sind in den zerstörten Betonproben noch Quarz,
Dolomit, Calzit, Vaterit (eine besondere Form von CaCO3) und in einigen Fällen Brucit
5 m
Dol
Tha
Brc
Cal
100 m

74
[Mg(OH)2; s. Abbildung 52] festgestellt worden. Dolomit und Quarz stammen sicher vom
verwendeten Zuschlag. Ob Kalzit ebenfalls ausschließlich vom Zuschlag stammt oder
sekundär im Zuge des Zersetzungsprozesses neu gebildet wurde, kann nicht eindeutig
beantwortet werden. Beide Varianten sind möglich (siehe dazu auch die Abbildungen 52 und
53). Sicher ist, dass neben dem Thaumasit auch das Mineral Brucit eine Neubildung darstellt
und auf die Zersetzung des Betons zurückzuführen ist. Besonders bemerkenswert dabei ist
das gemeinsame Auftreten von Dolomit, Calcit und Brucit. Dies lässt sich nur durch die
Auflösung des Dolomits (stammt aus der Gesteinskörnung) und der daran anschließenden
Neubildung von Calcit und Brucit erklären, was ein unter den gegebenen Bedingungen (hohe
Alkalinität) ungewöhnlicher Vorgang ist. Diese außergewöhnliche Paragenese ist durch
Abbildung 53 belegt, in der die Calcium und Magnesiumverteilung aus einer
Mikrosondenuntersuchung abgebildet ist.
Abbildung 53: Mikrosondenbilder der Phasenumwandlung von Dolomit nach Calcit und Brucit; Elementverteilungen von Calcium (mittleres Bild) und Magnesium (Bild rechts): Rote Farben bedeuten hohe Gehalte dunkle bzw. blaue Farben niedrige Gehalte des jeweiligen Elements
Hinsichtlich der Klärung der Mechanismen der Thaumasitbildung kommt der
Zusammensetzung der mit dem Beton in Kontakt stehenden Gebirgswässer und der im
geschädigten und nicht geschädigten Beton enthaltenen flüssigen Phase besonderes
Interesse zu, weil die relevanten chemischen Reaktionen nur über die flüssige Phase ablaufen
konnten. Die Untersuchung der aus den Tunneln entnommenen Gebirgswässer hat einen
mittleren Sulfatgehalt von etwa 400 – 500 mg SO4-2/l ergeben. Ansonsten enthielt das Wasser
hauptsächlich Ionen von Calcium und Hydrogenkarbonat, aber praktisch keine Alkalien
(Natrium, Kalium: <10 mg/l). Die aus den zersetzten Betonproben ausgepressten Wässer
hingegen enthielten relativ konstant 500 - 600 mg Calcium/l aber sehr viel Natrium, Sulfat und
Chlorid (je nach Feuchtigkeitsgehalt der entnommenen Proben mehr oder weniger). Die
Sulfatkonzentration lag in allen Fällen weit über der Sättigungskonzentration von Gips (etwa
1200 mg SO4-2/l) und reichte bis etwa 30.000 mg SO4
-2/l, was schon nahe an der
Sättigungskonzentration von Natriumsulfat liegt. Da das Sulfat nur aus dem Gebirgswasser in
100 m
Dol
Brc
Cal
100 m
Ca
100 m
Mg
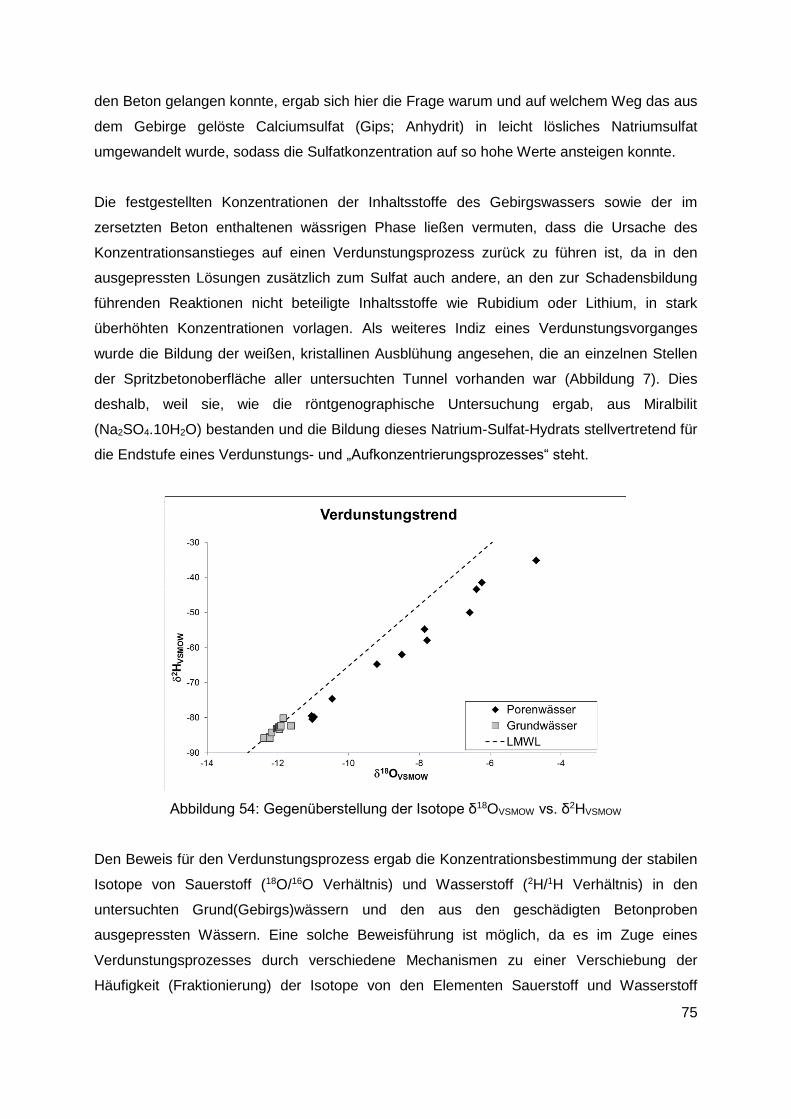
75
den Beton gelangen konnte, ergab sich hier die Frage warum und auf welchem Weg das aus
dem Gebirge gelöste Calciumsulfat (Gips; Anhydrit) in leicht lösliches Natriumsulfat
umgewandelt wurde, sodass die Sulfatkonzentration auf so hohe Werte ansteigen konnte.
Die festgestellten Konzentrationen der Inhaltsstoffe des Gebirgswassers sowie der im
zersetzten Beton enthaltenen wässrigen Phase ließen vermuten, dass die Ursache des
Konzentrationsanstieges auf einen Verdunstungsprozess zurück zu führen ist, da in den
ausgepressten Lösungen zusätzlich zum Sulfat auch andere, an den zur Schadensbildung
führenden Reaktionen nicht beteiligte Inhaltsstoffe wie Rubidium oder Lithium, in stark
überhöhten Konzentrationen vorlagen. Als weiteres Indiz eines Verdunstungsvorganges
wurde die Bildung der weißen, kristallinen Ausblühung angesehen, die an einzelnen Stellen
der Spritzbetonoberfläche aller untersuchten Tunnel vorhanden war (Abbildung 7). Dies
deshalb, weil sie, wie die röntgenographische Untersuchung ergab, aus Miralbilit
(Na2SO4.10H2O) bestanden und die Bildung dieses Natrium-Sulfat-Hydrats stellvertretend für
die Endstufe eines Verdunstungs- und „Aufkonzentrierungsprozesses“ steht.
Abbildung 54: Gegenüberstellung der Isotope δ18OVSMOW vs. δ2HVSMOW
Den Beweis für den Verdunstungsprozess ergab die Konzentrationsbestimmung der stabilen
Isotope von Sauerstoff (18O/16O Verhältnis) und Wasserstoff (2H/1H Verhältnis) in den
untersuchten Grund(Gebirgs)wässern und den aus den geschädigten Betonproben
ausgepressten Wässern. Eine solche Beweisführung ist möglich, da es im Zuge eines
Verdunstungsprozesses durch verschiedene Mechanismen zu einer Verschiebung der
Häufigkeit (Fraktionierung) der Isotope von den Elementen Sauerstoff und Wasserstoff

76
kommen kann. Zum Beispiel werden durch einen Verdunstungsprozess die schwereren
Isotope 18O und 2H angereichert (der Dampfdruck der leichten Wassermoleküle ist höher als
der der schwereren). In Abbildung 54 ist ihre Verteilung als δ18O und δ2H Werte in o/oo, VSMOW
(Vienna Standard Mean Ocean Water) dargestellt. Die strichlierte Gerade stellt frisch
gefallenes, lokales Niederschlagswasser aus dem Bereich des Feuerkogels dar (LMWL; Local
Meteoric Water Line). Die grauen Quadrate liegen auf der LMWL und zeigen die gemessenen
Werte aller Grundwasserproben der Tunnel dieses Gebirgsmassivs. Die schwarzen Quadrate
zeigen die Werte, die in den Porenwässern gemessen wurden, die aus den entnommenen
Proben des geschädigten Betons stammen. In ihnen war der Wassergehalt
(Austrocknungsgrad) je nach Entnahmestelle sehr verschieden. Deutlich ist zu erkennen, dass
es zu einer Anreicherung der schwereren Isotope gekommen ist, was nur durch Verdunstung
erklärbar ist. Aus solchen Messungen kann auch auf die an den Entnahmestellen
herrschenden rel. Luftfeuchtigkeit rückgeschlossen werden.
Da nicht verdampfbare und an der Betonschädigung nicht beteiligte Inhaltsstoffen in den aus
den zerstörten Betonproben ausgepressten wässrigen Lösung (Rubidium, Lithium, etc.)
wesentlich stärker konzentriert vorlagen als dem Verdunstungsgrad entspricht, der sich aus
der Abweichung der jeweiligen δ18O/ δ2H-Werte von der strichlierten Linie ergibt, muss Wasser
wiederholt verdampft und durch neu hinzutretendes Wasser wieder ergänzt worden sein.
Weitere Informationen sind in [54] enthalten.
Tabelle 11 enthält die Ergebnisse der nasschemischen Untersuchung der Abschnitte des in
Abbildung 10 gezeigten Spritzbeton-Bohrkerns mit 200 mm Durchmesser. Die Werte von CaO,
MgO und CO2 zeigen in Übereinstimmung mit den diffraktometrischen Untersuchungen, dass
der Betonzuschlag karbonatischer Natur war (Kalkstein und Dolomit). Bei einem (geschätzten)
Zementgehalt des Betons von ca. 15 M% (ca. 360 kg Zement pro m³ Beton mit einer Rohdichte
von 2400 kg/m³) und einem zulässigen SO3-Gehalt des Zementes von 3,5 M%, darf der Beton
max. 0,53 M% SO3 enthalten. In der dem Gebirge zugewandten Randzone (~0-1 cm Tiefe)
war der Sufatgehalt somit weit überhöht. Mit zunehmendem Abstand von der Randzone
wurden die SO3-Werte rasch kleiner, aber vermutlich war der SO3-Gehalt auch in einem
Abstand von ca. 5,5 cm noch überhöht. Aus der Tabelle ergibt sich weiter, dass der Gehalt an
säurelöslicher Kieselsäure (SiO2) unverhältnismäßig hoch ist. Portlandzement enthält etwa 20
M% lösl. SiO2, und bei 360 kg Zement pro m³ Beton müsste der Gehalt an lösl. SiO2 somit im
Bereich von 3 M% liegen (Gesteinskörnungen aus natürlichen Vorkommen enthalten nahezu
keine lösl. SiO2). Außerdem war der Gehalt an Natrium höher als der von Kalium. Da Zement
und natürliche Zuschläge jedoch gewöhnlich mehr Kalium als Natrium enthalten, wurde
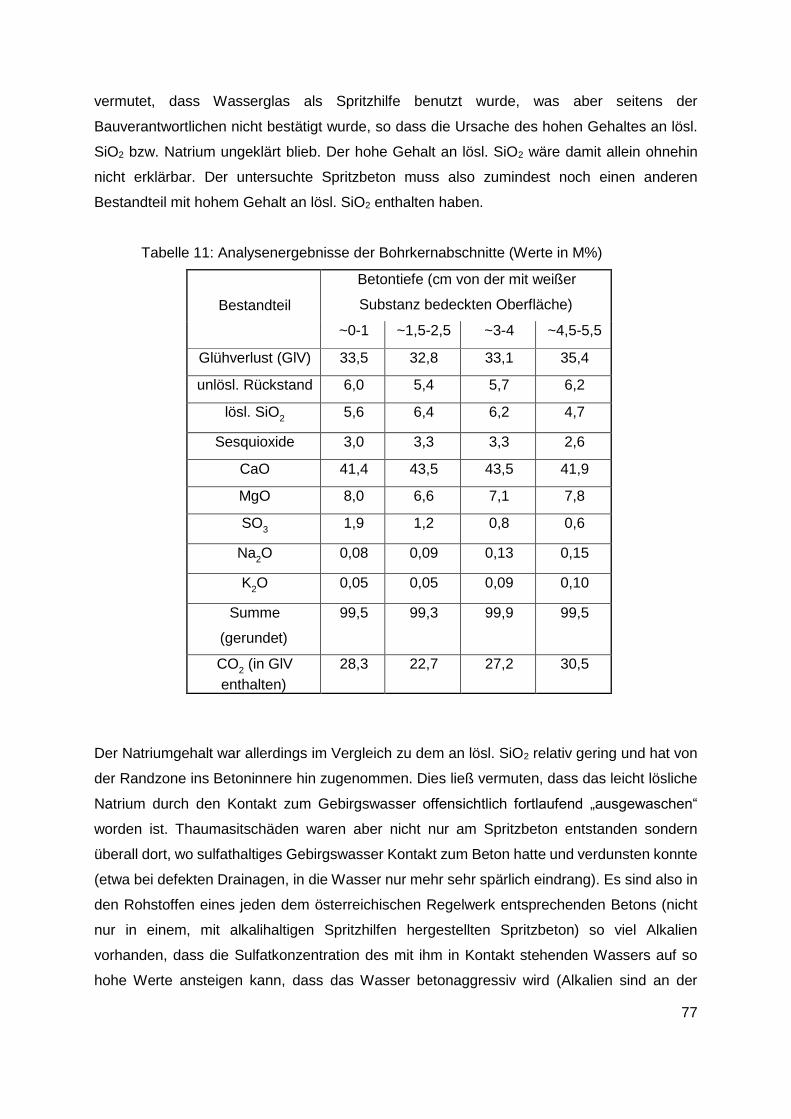
77
vermutet, dass Wasserglas als Spritzhilfe benutzt wurde, was aber seitens der
Bauverantwortlichen nicht bestätigt wurde, so dass die Ursache des hohen Gehaltes an lösl.
SiO2 bzw. Natrium ungeklärt blieb. Der hohe Gehalt an lösl. SiO2 wäre damit allein ohnehin
nicht erklärbar. Der untersuchte Spritzbeton muss also zumindest noch einen anderen
Bestandteil mit hohem Gehalt an lösl. SiO2 enthalten haben.
Tabelle 11: Analysenergebnisse der Bohrkernabschnitte (Werte in M%)
Bestandteil
Betontiefe (cm von der mit weißer
Substanz bedeckten Oberfläche)
~0-1 ~1,5-2,5 ~3-4 ~4,5-5,5
Glühverlust (GlV) 33,5 32,8 33,1 35,4
unlösl. Rückstand 6,0 5,4 5,7 6,2
lösl. SiO2 5,6 6,4 6,2 4,7
Sesquioxide 3,0 3,3 3,3 2,6
CaO 41,4 43,5 43,5 41,9
MgO 8,0 6,6 7,1 7,8
SO3 1,9 1,2 0,8 0,6
Na2O 0,08 0,09 0,13 0,15
K2O 0,05 0,05 0,09 0,10
Summe
(gerundet)
99,5 99,3 99,9 99,5
CO2 (in GlV
enthalten)
28,3 22,7 27,2 30,5
Der Natriumgehalt war allerdings im Vergleich zu dem an lösl. SiO2 relativ gering und hat von
der Randzone ins Betoninnere hin zugenommen. Dies ließ vermuten, dass das leicht lösliche
Natrium durch den Kontakt zum Gebirgswasser offensichtlich fortlaufend „ausgewaschen“
worden ist. Thaumasitschäden waren aber nicht nur am Spritzbeton entstanden sondern
überall dort, wo sulfathaltiges Gebirgswasser Kontakt zum Beton hatte und verdunsten konnte
(etwa bei defekten Drainagen, in die Wasser nur mehr sehr spärlich eindrang). Es sind also in
den Rohstoffen eines jeden dem österreichischen Regelwerk entsprechenden Betons (nicht
nur in einem, mit alkalihaltigen Spritzhilfen hergestellten Spritzbeton) so viel Alkalien
vorhanden, dass die Sulfatkonzentration des mit ihm in Kontakt stehenden Wassers auf so
hohe Werte ansteigen kann, dass das Wasser betonaggressiv wird (Alkalien sind an der
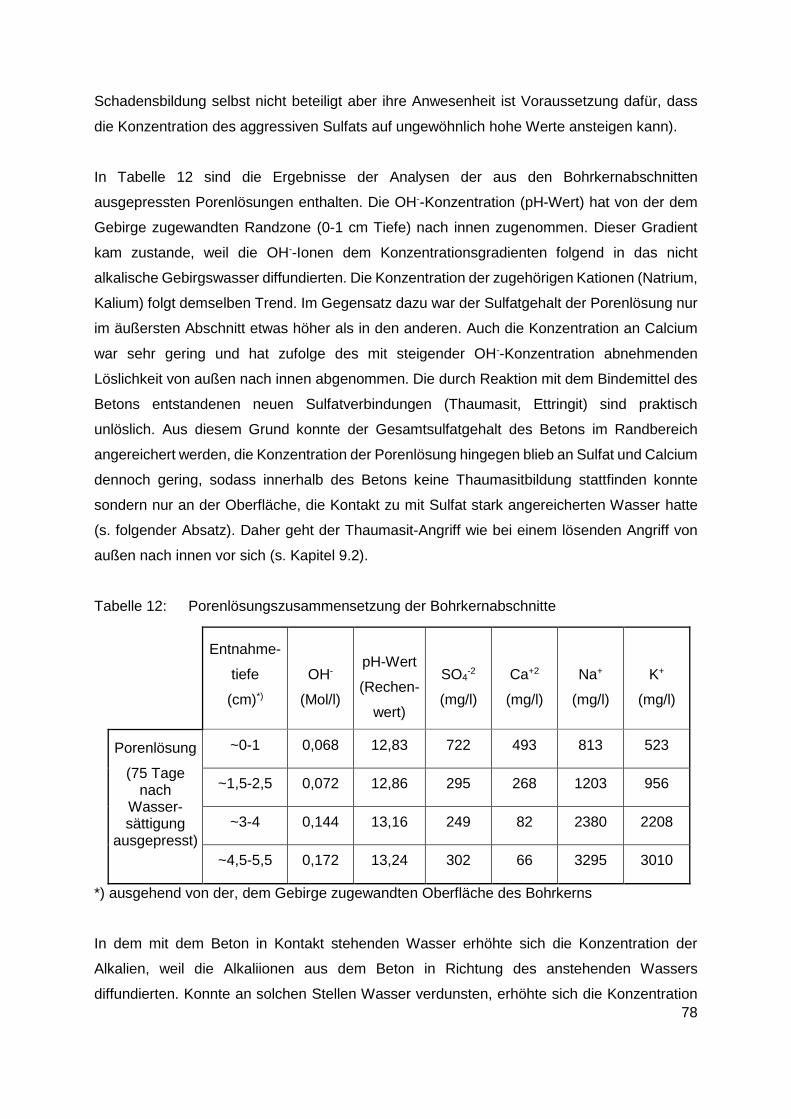
78
Schadensbildung selbst nicht beteiligt aber ihre Anwesenheit ist Voraussetzung dafür, dass
die Konzentration des aggressiven Sulfats auf ungewöhnlich hohe Werte ansteigen kann).
In Tabelle 12 sind die Ergebnisse der Analysen der aus den Bohrkernabschnitten
ausgepressten Porenlösungen enthalten. Die OH--Konzentration (pH-Wert) hat von der dem
Gebirge zugewandten Randzone (0-1 cm Tiefe) nach innen zugenommen. Dieser Gradient
kam zustande, weil die OH--Ionen dem Konzentrationsgradienten folgend in das nicht
alkalische Gebirgswasser diffundierten. Die Konzentration der zugehörigen Kationen (Natrium,
Kalium) folgt demselben Trend. Im Gegensatz dazu war der Sulfatgehalt der Porenlösung nur
im äußersten Abschnitt etwas höher als in den anderen. Auch die Konzentration an Calcium
war sehr gering und hat zufolge des mit steigender OH--Konzentration abnehmenden
Löslichkeit von außen nach innen abgenommen. Die durch Reaktion mit dem Bindemittel des
Betons entstandenen neuen Sulfatverbindungen (Thaumasit, Ettringit) sind praktisch
unlöslich. Aus diesem Grund konnte der Gesamtsulfatgehalt des Betons im Randbereich
angereichert werden, die Konzentration der Porenlösung hingegen blieb an Sulfat und Calcium
dennoch gering, sodass innerhalb des Betons keine Thaumasitbildung stattfinden konnte
sondern nur an der Oberfläche, die Kontakt zu mit Sulfat stark angereicherten Wasser hatte
(s. folgender Absatz). Daher geht der Thaumasit-Angriff wie bei einem lösenden Angriff von
außen nach innen vor sich (s. Kapitel 9.2).
Tabelle 12: Porenlösungszusammensetzung der Bohrkernabschnitte
Entnahme-
tiefe
(cm)*)
OH-
(Mol/l)
pH-Wert
(Rechen-
wert)
SO4-2
(mg/l)
Ca+2
(mg/l)
Na+
(mg/l)
K+
(mg/l)
Porenlösung
(75 Tage nach
Wasser-sättigung
ausgepresst)
~0-1 0,068 12,83 722 493 813 523
~1,5-2,5 0,072 12,86 295 268 1203 956
~3-4 0,144 13,16 249 82 2380 2208
~4,5-5,5 0,172 13,24 302 66 3295 3010
*) ausgehend von der, dem Gebirge zugewandten Oberfläche des Bohrkerns
In dem mit dem Beton in Kontakt stehenden Wasser erhöhte sich die Konzentration der
Alkalien, weil die Alkaliionen aus dem Beton in Richtung des anstehenden Wassers
diffundierten. Konnte an solchen Stellen Wasser verdunsten, erhöhte sich die Konzentration

79
weiter. Im Gegensatz dazu konnte sich die Calciumkonzentration nicht erhöhen. Calcium
wurde entweder durch die chemischen Reaktionen mit dem Beton verbraucht oder reagierte
mit CO2 bzw. HCO3-, das etwa aus der Luft oder aus dem Gebirgswasser stammen kann, zu
unlöslichem CaCO3 und ist so dem Wasser entzogen worden. Dadurch wurden die
Calciumionen mit der Zeit durch Natriumionen als Bindungspartner des Sulfats ersetzt,
weshalb das anstehende Wasser zufolge Verdunstung betonaggressiv werden konnte. Die
Sulfatkonzentration des im einmal geschädigten Beton vorhandenen Wassers konnte in
weiterer Folge durch Verdunstung solange zunehmen, bis die Sättigungskonzentration von
Natriumsulfat erreicht war. Natürlich stieg in den Bereichen, wo Wasser verdunsten konnte,
mit dem ansteigenden Sulfatgehalt auch seine Aggressivität stark an. Dies macht verständlich,
warum der Beton stellenweise von hinten bis an die sichtbare Innenoberfläche des
Spritzbetons seine Festigkeit vollkommen verlieren konnte. Dieses stark sulfathaltige Wasser
wurde kapillar entlang von Rissen an die sichtbare Innenoberfläche des Spritzbetons
befördert. Dort kam es nach der Verdunstung des Wassers zur z.T. flächenhaften
Mirabilitbildung (Abbildung 8) [11, 53, 87, 88].
Wie in Kapitel 6.2.2 beschrieben, war das Schadensbild sehr unterschiedlich. Für die
Schädigung war jedoch immer notwendige Voraussetzung, dass der Sulfatgehalt des
Gebirgswassers durch Verdunstung ansteigen konnte, weil die Sulfatkonzentration des
Gebirgswassers nur 400 – 500 mg SO4-2/l betrug, für eine Schädigung aber höhere
Konzentrationen erforderlich sind (siehe Kapitel 9.2). Die in Abbildung 7 sichtbare Zone befand
sich im Anschluss an eine Betonfuge. Durch die Fuge konnte Wasser verdunsten, so dass es
von dort ausgehend zur Betonschädigung kam, wobei die Schadensbildung sicherlich auch
hier von der Gebirgsseite ausging und sich nach innen fortsetzte, bis der gesamte Spritzbeton
dieses Bereiches zerstört war. Der Bereich des Spritzbetons, aus dem der Bohrkern
entnommen wurde (Abbildung 9), befand sich in Fortsetzung einer, auch an eine Betonfuge
grenzenden Zone mit bereits abgefallenen Spritzbeton. Offensichtlich hat die Schädigung auch
hier an der Fuge ihren Ausgang genommen und vermutlich war der Verbund zwischen
Spritzbeton und Gebirge von Anfang an nicht besonders gut, so dass Wasser mit bereits
konzentrierten Inhaltsstoffen eingesaugt werden und/oder von der Spritzbetonrückseite durch
den zerstörten Bereich des Spritzbetons nach außen verdunsten konnte. Wohl nur deswegen
wurde der Spritzeton hier von der Rückseite her angegriffen und es kam zu dessen
flächenhafter Ablösung vom Fels. Der Angriff des Spritzbetons innerhalb der Nische
(Abbildung 11) erfolgte ebenfalls von der Rückseite her und bewirkte dessen Abfallen, obwohl
dort keine Fugen vorhanden waren. Der Spritzbeton war hier aber sehr dünnschichtig, so dass
Wasser sicherlich durch den Spritzbeton hindurch verdampfen und die Schädigung bewirken

80
konnte. Der in Abbildung 12 gezeigte zerstörte Türsockelbeton befand sich oberhalb eines
Drainagerohrs, das nicht mehr funktionstüchtig war (kein fließendes Wasser), in dem aber
noch etwas Wasser stand. Da das Rohr weitgehend leer war, konnte Wasserdampf entlang
des Rohres abtransportiert werden und der Angriff beginnen. Die Ursache der
Betonzerstörung bei der in Abbildung 13 gezeigten Wandöffnung bedarf keiner näheren
Erklärung, weil die Möglichkeit der Wasserverdunstung offensichtlich ist.
12. Zusammenfassung
12.1 Allgemeines
Wie einleitend beschrieben, wird im europäischen Regelwerk (EN 206-1) hinsichtlich des
Angriffs von Sulfaten auf Beton nur auf die Gefahr von Treiberscheinungen durch
Ettringitbildung Bezug genommen. Die darin enthaltene Unterteilung des Schadensrisikos in
Expositionsklassen, die durch verschieden hohe Grenzwerte des Sulfatgehaltes von
angreifenden Wässern bzw. Böden gekennzeichnet sind, ist auf Neubauten ausgelegt, vor
deren Errichtung die mit dem Beton in Kontakt kommenden Umwelteinflüsse untersucht
werden sollen, sodass die notwendigen betontechnischen bzw. sonstigen Maßnahmen
getroffen werden können, um Schäden zu vermeiden. Warum die Grenzwerte gerade die
angegebenen Zahlenwerte haben (s. Tabelle 1), ist nicht leicht nachzuvollziehen und sind
vermutlich auf Grund langjähriger Erfahrungen mit einem entsprechenden Sicherheitspolster
festgelegt worden. Sie liegen daher weit auf der sicheren Seite und bedeuten nicht, dass es
bei deren Überschreitung jedenfalls zu einem Schaden kommen wird. Da vor der
Bauausführung die erforderlichen Untersuchungen, die eine Beurteilung der Expositionsklasse
ermöglichen, nicht immer durchgeführt werden und weil es bei älteren Bauwerken zur Zeit
derer Errichtung keine oder andere Sulfat-Grenzwerte gegeben hat, interessiert vornehmlich,
wie hoch der Sulfatgehalt des Betons ansteigen darf, ohne dass es zu einem Schaden kommt.
Da außerdem die Sulfatkonzentration angreifender Wässer in der Praxis kaum jemals eine
konstant bleibende Höhe hat (Voraussetzung zur Beurteilung der Expositionsklasse), wurde
das Forschungsvorhaben beantragt. Zu den hauptsächlichen Zielen (siehe Kapitel 1) des
Vorhabens gehörte, herauszufinden ob es bei Ettringitbildung einen kritischen Sulfatgehalt von
Beton gibt, bei dessen Überschreitung Schutzmaßnahmen getroffen werden müssen und wie
die Beurteilung der Expositionsklasse bei schwankenden Sulfatkonzentrationen des mit dem
Beton in Kontakt stehenden Wassers erfolgen soll (mittlere oder die höchste Konzentration).

81
Da im Regelwerk zudem keinerlei Hinweis enthalten ist, wie im Fall eines Sulfatangriffs
vorgegangen werden soll, bei dem die Ursache der Schädigung nicht die Bildung von Ettringit
(3CaO.Al2O3.3CaSO4.32H2O) sondern von Thaumasit (CaSiO3.CaCO3.CaSO4. 12H2O) ist,
war es notwendig, zu untersuchen nach welchen Mechanismen die Thaumasit-Schädigung
abläuft. Es waren keiner der am Forschungsprojekt beteiligten Personen Schäden bekannt,
die in Österreich an einem Konstruktionsbeton durch Ettringitbildung aufgetreten sind sondern
nur solche, die aber durch Thaumasitbildung verursacht wurden. Letztere sind in Tunneln
aufgetreten, die durch Gebirgsstöcke mit gipshaltigen Bereichen führten (bei
Thaumasitbildung kommt es zu keinen Treiberscheinungen sondern zu einer Aufweichung des
Zementsteins, weil die C-S-H-Phase unter Einwirkung von Karbonat bzw. CO2 und Sulfat in
Thaumasit umgewandelt wird [15, 43, 54-58, 64, 75, 76, 78, 84-86, 89-91].
12.2 Durchgeführte Untersuchungen
Der Versuchsplan war in Labor- und Praxisuntersuchungen unterteilt. Die Laborversuche
wurden an Mörtel- und Betonproben durchgeführt. Zunächst zu den mit den Mörtelproben
durchgeführten Untersuchungen:
Es handelte sich um Mörtel, die in Anlehnung an die Zementnormenprüfung hergestellt
wurden. Sie sind mit Quarzsand (Normensand) oder Kalksteinmehl anstelle des
Normensandes-fein, unterschiedlichen Zementen und W/Z-Werten hergestellt worden und
hatten die Form von a) Würfeln mit 4 cm Kantenlänge und b) von Prismen mit den
Abmessungen 4 x 4 x 16 cm und wurden sowohl für das Studium der Ettringitbildung als auch
der Thaumasitbildung verwendet. Der Unterschied lag hauptsächlich darin, dass für die
Thaumasitbildung die Lagerungstemperatur 5°C (± 3°C) betrug, für die Ettringitbildung
hingegen Raumtemperatur (20°C ± 2°C), weil sich Thaumasit laut Literatur bevorzugt bei
Temperaturen unterhalb von 15°C bildet.
Für das Studium der Schädigung durch Ettringitbildung wurden Mörtelproben mit CEM I 52,5-
R Zement mit W/Z-Wert 0,45 und 0,70 sowie CEM I 42,5 R/C3A-frei (HS-Zement) einerseits
im Labor in einem Klimaraum mit 20°C in Lösungen mit Sulfatkonzentrationen ausgelagert,
die den Grenzkonzentrationen der Expositionsklassen XA1, XA2 und XA3 entsprachen,
nämlich 600, 3000 und 6000 mg SO4-2/l sowie wechselweise bei 600 und 6000 mg SO4
-2/l. Die
Lagerlösungen wurden durch Lösen von Natriumsulfat in entkalktem Leitungswasser
hergestellt. Außerdem wurden Proben in einer gesättigten Gipslösung ausgelagert, weil der
Sulfatgehalt natürlicher österreichischer Wässer praktisch immer von Gips stammt. Zudem

82
wurden Proben mit CEM III/B 32,5 N und einer Mischung aus 70% HS-Zement mit 30%
Fluamix C einbezogen, um den Einfluss von Hüttensand bzw. Flugasche zu erfassen, die aber
nur bei 3000 mg SO4-2/l und in gesättigter Gipslösung ausgelagert wurden. Des Weiteren sind
Proben mit den Zementen CEM I 52,5-R (W/Z-Wert 0,45 und 0,70) und HS-Zement (W/Z-0,70)
an vier verschiedenen Stellen des Abwasserkanalsystems von Linz ausgelagert worden, wo
schwankende Sulfatkonzentrationen gegeben waren, die an zumindest einer Stelle kurzzeitig
die Grenzkonzentration der Expositionsklasse XA3 überschritten haben, im Mittel aber im
Bereich von XA1 bzw. darunter lagen. Außerdem wurde zu Beginn an den vier
Auslagerungsstellen je ein Bohrkern des Kanalbetons und am Ende des Versuchszeitraumes
von 3,5 Jahren an einer der Auslagerungsstellen erneut ein Bohrkern entnommen, um
Veränderungen des Sulfatgehaltes zu ermitteln. Die Untersuchung der im Kanal ausgelagerten
Mörtelproben hatten den Vorteil, dass die Lagerung unter praktischen Bedingungen erfolgte,
was für die Deutung der Ergebnisse vorteilhaft erschien, weil Ergebnisse aus
Laborbedingungen gewöhnlich nur mit Vorbehalt auf die Praxis übertragbar sind.
Für das Studium der Schädigung durch Thaumasit wurden Mörtelproben mit allen vier
Zementsorten und Kalksteinmehl anstelle des Normensandes fein verwendet, die in einer
Kühltruhe bei 5°C gelagert wurden. Die Sulfatkonzentration der Lagerlösung betrug 200, 600
und 3000 mg SO4-2/l. Die Konzentration von 200 mg SO4
-2/l (anstelle der bei 20°C
einbezogenen 6000 mg SO4-2/l) wurde deshalb verwendet, weil erfasst werden sollte, ob es
auch bei einer so geringen Konzentration zu einer Schädigung kommt. Dies deshalb, weil die
Sulfatkonzentration von Wässern in gipshaltigen Gebirgsbereichen häufig unter 500 mg
SO4-2/l liegt. Hinzu kam noch eine gesättigte Gipslösung. Einbezogen wurden auch einige
Mörtelproben ohne Kalkstein, um festzustellen, ob eine kalkhaltige Gesteinskörnung als Quelle
für das zur Thaumasitbildung benötigte Karbonat notwendig ist bzw. ob sie eine
aggressivitätssteigernde Wirkung hat.
Von den im Labor bei 20°C gelagerten Würfelproben wurde planmäßig nach 6 Monaten, 1,5
Jahren, 2,5 Jahren und 3,5 Jahren je Rezeptur und Lagerlösung ein Würfel entnommen und
mittig durchtrennt. Eine der beiden Hälften wurde a) zur Bestimmung enthaltener
Mineralphasen (Ettringit/Thaumasit) und b) hinsichtlich der Sulfatverteilung verwendet. An der
anderen Hälfte ist der Gesamtsulfatgehalt sowie der Gehalt an löslicher SiO2 nach dem
Trocknen und anschließendem Zerkleinern der Proben auf Analysenfeinheit (<0,09 mm)
nasschemisch bestimmt worden. Die Bestimmung des löslichen SiO2 war notwendig, weil aus
ihrem Gehalt der ungefähre Zementgehalt der jeweiligen Probe bestimmt und damit der auf
den Zementgehalt bezogene Sulfatgehalt berechnet werden konnte.

83
Die Prismen wurden nur bis zum Prüftermin 2,5 Jahre planmäßig entnommen und untersucht,
nicht jedoch nach 3,5 Jahren, weil zu diesem Zeitpunkt noch keinerlei Schadensanzeichen
sichtbar waren und die Lagerungsdauer daher verlängert wurde (s. Kapitel 9.1.2). Anhand der
Prismen wurden zunächst die Biegezug- und dann die Druckfestigkeit bestimmt. Die geprüften
Proben sind sofort in Kunststoffsäcken verpackt worden und aus ihnen wurde anschließend
die Porenlösung ausgepresst. Die Porenlösungen wurden hinsichtlich der Konzentrationen der
Ionen Hydroxid (OH-), Chlorid (Cl-), Sulfat (SO42-), Natrium (Na+), Kalium (K+) und Calcium
(Ca2+) untersucht.
Für die Untersuchungen zur Auffindung einer Prüfmethode zur nasschemischen Bestimmung
der Sulfatbeständigkeit wurden Betonbalken mit 70 x 15 x 15 cm und W/Z-Werten von 0,45,
0,50 bzw. 0,55 hergestellt. Als Zementsorten wurden CEM I 52,5 R, CEM I 42,5 N/C3A-frei
(HS-Zement) und der Mischzement aus 70% HS-Zement/30% Fluamix verwendet. Aus den
Balken sind nach dem Aushärten Bohrkerne (Ø 70 mm) entnommen, von diesen 25 mm dicke
Scheiben angeschnitten und planparallel geschliffen worden. Diese Prüfkörper sind sodann in
gesättigter Gipslösung bei Raumtemperatur gelagert worden. Nach 33, 66, 99 und 152 Tagen
wurde je Rezeptur zwei Scheiben entnommen und der Sulfatgehalt in den Tiefen 0,4-0,6 mm,
1,0-1,2 mm und 1,4-1,6 mm bestimmt. Anhand der Zunahme des Sulfatgehaltes erfolgte die
Beurteilung des Widerstandes der Betone gegen das Eindringen von Sulfat. Zudem wurden
Untersuchungen an Baustellenbetonen durchgeführt, für die Betonbohrkerne aus den
Innenschalen von drei Tunneln zur Verfügung standen.
Neben diesen Laboruntersuchungen wurden noch die in Tunneln vorgefundenen Schäden
(Abbildungen 6, 10, 11 und 12) untersucht. Dazu sind aus den Tunneln Proben des
augenscheinlich vollkommen zerstörten Betons, der an der sichtbaren Innenoberfläche eines
Tunnels anhaftenden Ausblühungen (Abbildung 7), der Gebirgswässer sowie Bohrkerne des
Spritzbetons (Ø 200 mm) entnommen worden. Ein Bohrkern wurde an einer Stelle
entnommen, an der kein Verbund des Spritzbetons zum Untergrund gegeben war. Wie sich
nach der Entnahme zeigte, haftete an der rückwärtigen Oberfläche eine weiße Substanz von
Reaktionsprodukten an (Abbildung 10). An diesem Bohrkern konnten die mit zunehmendem
Abstand von der rückwärtigen Oberfläche gegebenen Veränderungen der
Betonzusammensetzung erfasst werden.
Von ihm sind, wie auch von den aus dem Kanalsystem von Linz entnommenen Bohrkernen,
jeweils ca. 1 cm dicke Scheiben parallel zur Grundfläche abgeschnitten worden. Aus den

84
Abschnitten wurde nach Wassersättigung, dichter Verpackung in Kunststoff-Folien und
nachfolgender zweimonatigen Lagerungszeit bei 20°C die Porenlösung ausgepresst und die
Konzentration der Inhaltsstoffe wie bei den Porenlösungen der Mörtelproben bestimmt (die
lange Lagerungsdauer war notwendig um sicher zu gehen, dass sich das zwischen Feststoffen
und flüssiger Phase einstellende Gleichgewicht erreicht war). Die Feststoffe wurden nicht nur
nasschemisch sondern auch hinsichtlich der enthaltenen Mineralphasen analysiert. Die
zersetzten Betonproben wurden analog untersucht und zudem wurden die aus ihnen
ausgepressten Lösungen wie auch die Bergwasserproben hinsichtlich ausgewählter
hydrochemischer Kennwerte und den Isotopenwerten von Sauerstoff, Wasserstoff,
Kohlenstoff und fallweise Schwefel (δ2H, δ18O, δ13C, δ34S) untersucht.
12.3 Ergebnisse der Untersuchungen
12.3.1 Im Kanalsystem von Linz ausgelagerte Proben
Bei den im Kanalsystem ausgelagerten Proben kam es während der gesamten Lagerungszeit
zu keiner sehr starken Zunahme des Sulfatgehaltes, denn nach 3,5 Jahren lag der SO3-Gehalt
bei allen Proben und Auslagerungsstellen unterhalb von 4% SO3, bezogen auf den
Zementgehalt. Besonders auffällig war, dass der Sulfatgehalt der Proben ab 1,5 Jahren kaum
noch zugenommen hat. Als Ursache hierzu wurde festgestellt, dass innerhalb dieser Proben
nahe der Oberfläche eine Karbonatisierung stattgefunden hat (Abbildung 28; Kapitel 8.2.3.1).
Dies führte zu einer Porenverengung und somit zu einer Abnahme der Geschwindigkeit, mit
der Substanzen von außen in die Proben eindringen konnten. Augenscheinlich konnten
nirgendwo und zu keinem Prüftermin Schädigungsanzeichen festgestellt werden. Die
Festigkeiten haben anfangs zufolge der Nacherhärtung zugenommen und sich dann mit
Ausnahme der im Nachklärbecken gelagerten Proben kaum noch verändert. Dort wurden nach
3,5 Jahren aber entweder erheblich geringere Druck- oder Biegezugfestigkeiten als zuvor
gemessen. Die Druckfestigkeiten haben jedoch nicht mit den Biegezugfestigkeiten korreliert,
so dass hier offenen Fragen bestehen blieben (siehe Kapitel 8.2.2), die nicht einfach mit den
leider erheblichen Messunsicherheiten erklärt werden konnten. Bei den im Nachklärbecken
gelagerten Proben blieben allerdings noch andere Unklarheiten bestehen. So hat hier (nur
hier) die Chloridkonzentration der Porenlösung aus unbekannter Ursache über den gesamten
Versuchszeitraum immer zugenommen, obwohl die Chloridkonzentration des Wassers im
Nachklärbecken wesentlich geringer war. In der Porenlösung, die dort aus dem zu Beginn
entnommenen Betonbohrkern aus dem bis in ca. 1 cm Tiefe reichenden Abschnitt ausgepresst
wurde, war die Chloridkonzentration auch deutlich höher als im Wasser des Nachklärbeckens
(da die Chloridkonzentration nicht mit dem kritischen Sulfatgehalt in Verbindung steht, wurden

85
die Ursachen dieses Phänomens nicht weiter verfolgt). Ansonsten haben sich die
Konzentrationen der in der Porenlösung enthaltenen Ionen erwartungsgemäß verändert. Die
darin zunächst ziemlich konzentriert vorliegenden Ionen von Natrium, Kalium und Hydroxid
sind dem Konzentrationsgradienten folgend aus den Proben hinausdiffundiert, was zu einer
Abnahme der OH--Konzentration der Porenlösung (des pH-Wertes) sowie der Alkaliionen
geführt hat. Dies hat wiederum die Löslichkeit von Calciumhydroxid und Calciumsulfat
verändert, wodurch es mit der Zeit zu einer Zunahme der Ca+2- und SO4-2-Konzentration kam,
wobei besonders die Sulfatkonzentration generell sehr niedrig blieb (Abbildungen 31-33).
12.3.2 Im Labor bei 20°C ausgelagerte Proben (Ettringitbildung)
Die Veränderungen der Druckfestigkeiten entsprachen der Erwartung, haben anfänglich
zufolge der Nacherhärtung zugenommen und haben sich dann bis 2,5 Jahre Lagerungsdauer
nur wenig verändert. Sie waren beim CEM I-Mörtel am höchsten. Der C3A-freie Zement hatte
zwar ebenso wie die Mörtel, die CEM III bzw. das 70/30-Gemisch von HS-Zement mit Fluamix
als Bindemittel enthielten, deutlich niedrigere 28-Tage Festigkeiten, die Nacherhärtung war
aber stark. Die niedrigste Druckfestigkeit wurde zu allen Terminen beim Mörtel mit dem 70/30-
Gemisch von CEM I/C3A-frei mit Fluamix festgestellt. Da die Messunsicherheit aber hoch war,
ist eine nähere Interpretation schwierig und unsicher (siehe Kapitel 9.1.1). Nach 3,5 Jahren
wurden die Druckfestigkeiten des je Rezeptur noch vorhandenen letzten Prismas nicht geprüft
sondern erst nach ca. 4,5 Jahren, weil nach 3,5 Jahren augenscheinlich noch keinerlei
Anzeichen für eine Schädigung erkennbar waren. Da aber der mittlere Sulfatgehalt bei dem
Prisma mit CEM I, W/Z-Wert 0,70, schon sehr hoch war, andeutungsweise schon eine
schwache Zunahme der Länge stattgefunden hat, zudem die Biegezugfestigkeit der bei 6000
mg SO4-2/l gelagerten Proben dieses Mörtels ab 6 Monaten Lagerungsdauer ständig geringer
wurde (Abbildung 36) sowie im Elektronenmikroskop festgestellt wurde, dass innerhalb des
Mörtels auch der bei 3000 mg SO4-2/l gelagerten Proben Risse entstanden waren (Abbildungen
40, 41), war anzunehmen, dass bald auch mit freiem Auge erkennbare erste Dehnungsrisse
entstehen würden und daher eine Verlängerung der Laufzeit des Projektes um 1 Jahr
beantragt. Die verbliebenen Würfelproben sind aber dennoch schon nach 3,5 Jahren
hinsichtlich Gesamtsulfatgehalt, etc. geprüft worden, weil angenommen wurde, dass die an
den Würfeln durchgeführten Prüfungen auch an einer der nach der Prüfung der
Biegezugfestigkeit verbleibenden zwei Prismenhälften gemacht werden können. Dies zog
allerdings nach sich, dass die Porenlösung aus nur einer Hälfte des jeweiligen Prismas
ausgepresst und danach der Gesamtsulfatgehalt der Feststoffe anhand dieses Teils des
jeweiligen Primas bestimmt werden musste. Da die Porenlösung bei den Proben mit W/Z-0,50
nicht immer gelang, konnten nur bei den Mörteln mit W/Z-0,70 alle Prüfungen mit Ausnahme

86
der Festigkeiten sowohl nach 3,5 und 4,5 Jahren durchgeführt werden.
Wie aus Abbildung 37 (Kapitel 9.1.2) ersichtlich, ist der Sulfatgehalt der Proben mit W/Z-Wert
0,70 nur bei den in der Lösung mit 6000 mg SO4-2/l kontinuierlich angestiegen, während es bei
den übrigen SO4-2-Konzentrationen zwischen 6 Monaten und 1,5 Jahren zu keinem Anstieg
gekommen ist. Die Untersuchung einer solchen Probe hat ergeben, dass sich an der
Probenoberfläche eine dünne Kalkschicht [Ca(CO)3, Abbildung 38] gebildet hat, in deren
abdichtender Wirkung die Ursache dafür gesehen wird, dass bei den meisten Proben keine
messbare Zunahme des Sulfatgehaltes stattgefunden hat. Im Gegensatz zu den im Kanal
ausgelagerten Proben hat sich die Kalkschicht an der Probenoberfläche ausgebildet, weil das
ausdiffundierende Calciumhydroxid nicht abtransportiert wurde wie im fließenden Wasser und
daher direkt mit dem in der Lagerlösung vorhandenen CO2 zu unlöslichen Calciumkarbonat
reagieren konnte. Zu einer vollständigen Abdichtung ist es aber nicht gekommen, denn die
Konzentration der Alkali- und OH--Ionen der Porenlösung hat auch zwischen 6 Monaten und
1,5 Jahren weiter abgenommen.
Um die Sulfataufnahme wieder in Gang zu bringen und dauerhaft aufrecht zu erhalten, ist die
Kalkschicht der Proben forthin alle 6 Monate durch Eintauchen in verdünnter Ameisensäure
abgeätzt worden. Danach ist es bei allen Proben wieder zu einer Sulfataufnahme gekommen,
wobei die Zunahme des Sulfatgehaltes in allen Lagerlösungen erwartungsgemäß beim CEM I
mit W/Z-Wert 0,70 am stärksten war. Bei dieser Probe betrug der auf den Zementgehalt
bezogene Sulfatgehalt nach 3,5 Jahren Lagerung in der Lösung mit 6000 mg SO4-2/l über 10
% SO3. Interessant war auch das Ergebnis, dass der Anstieg des Sulfatgehaltes bei
Wechsellagerung zwischen 600 und 6000 mg SO4-2/l (Mittelwert 3300 mg SO4
-2/l) gut mit dem
bei konstant 3000 mg SO4-2/l gelagerten Proben übereinstimmte.
Bindemittelrelevante Unterschiede kamen klar zum Ausdruck. So ist der Sulfatgehalt bei den
Prüfmörteln mit 70% HS-Zement/30% Fluamix (im Diagramm als Mix bezeichnet) als
Bindemittel bei einer Sulfatkonzentration der Lagerlösung von 3000 mg SO4-2/l deutlich
weniger stark angestiegen (SO3-Zunahme 1,68 % v. Zement; Abbildung 39) als beim
vergleichbaren Mörtel mit CEM I (SO3-Zunahme 2,78 % v. Zement). Das bedeutet, dass der
im Fluamix enthaltene puzzolanische Anteil entweder eine so dichte Struktur bewirkt hat, dass
das Sulfat wesentlich langsamer eindrang als beim CEM I gebundenen Mörtel und/oder dass
die Sulfatbindung bei diesem Zement anderen, langsamer ablaufenden Mechanismen folgt.
Bei CEM III weisen die Ergebnisse in dieselbe Richtung, können aber derzeit nicht belegt
werden (siehe Kapitel 9.1.2). Aus Abbildung 39 ist auch ersichtlich, dass die Ergebnisse bei

87
Lagerung in gesättigter Gipslösung (Sättigungskonzentration: ~1200 mg SO4-2/l) dieselbe
Tendenz zeigen und dass der Sulfatgehalt entsprechend der geringeren SO4-2-Konzentration
der Lösung deutlich weniger stark angestiegen ist als bei 3000 mg SO4-2/l. Der Einfluss der
Zementsorte und des W/Z-Wertes kam auch aus den Untersuchungen zur Auffindung einer
Prüfmethode zur nasschemischen Bestimmung der Sulfatverteilung in Beton, bei denen die
Veränderungen des Sulfatgehaltes in unterschiedlichen Betontiefen gemessen wurden, klar
zum Ausdruck und bestätigten die an den Mörtelproben erhaltenen Unterschiede. Auch bei
den Betonproben stieg der Sulfatgehalt am stärksten beim CEM I-Beton, gefolgt vom Beton
mit C3A-freiem Zement und am wenigsten stark beim Beton mit 70% HS-Zement/30% Fluamix.
Hinweise auf Treiberscheinungen wurden bei keiner der Betonproben festgestellt.
Das aufgenommene Sulfat lag innerhalb der Proben naturgemäß nicht gleichmäßig verteilt vor
sondern es bestand ein deutlicher Gradient in der SO4-2-Verteilung mit von außen nach innen
abnehmenden Werten (Abbildungen 42, 43), wobei der Verteilungsgradient bei den Proben
mit W/Z-0,70 im Verlauf der Lagerung schwächer wurde, weil ab dem Erreichen der
Probenmitte der Sulfatgehalt dort weiter anstieg, nicht jedoch in den äußeren Probezonen. Die
im Elektronenmikroskop mittels Elektronenstrahlmikroanalyse ermittelte Sulfatverteilung
ergab, dass das Sulfat nicht überall gleich schnell eindrang und nicht kontinuierlich von außen
nach innen abnahm sondern dass lokal überraschend hohe Konzentrationsunterschiede
bestanden (Abbildungen 42, 43). Dies ist nur durch Inhomogenität erklärbar, die in der
Mikrostruktur offenbar von Anfang an vorhanden waren.
Die Veränderungen der Zusammensetzung der Porenlösung folgten dem erwarteten Trend,
der auch bei der Lagerung im Abwasserkanal mit Ausnahme der Lagerstelle 4
(Nachklärbecken) festgestellt wurde. Interessant war der Umstand, dass die
Sulfatkonzentration der Porenlösung sogar bei der Lagerlösungskonzentration von 6000 mg
SO4-2/l immer wesentlich tiefer lag als in der Lagerlösung (Abbildung 45). Das bedeutet, dass
eindiffundiertes Sulfat gebunden wurde und daher die Sulfataufnahmefähigkeit auch zu
Versuchsende noch nicht erschöpft war.
12.3.3 Im Labor bei 5°C ausgelagerte Proben (Thaumasitbildung)
Die bei der Sulfatkorrosion durch Thaumasit entstehenden neuen Sulfatverbindungen
(Thaumasit bzw. Woodfordit, Gips) sind schwer löslich bzw. unlöslich, weshalb die SO4-2- und
Ca+2-Konzentration der Porenlösung unabhängig vom Gesamtsulfatgehalt immer sehr gering
blieb und daher während der Lagerungsdauer immer neues Sulfat eindiffundieren konnte. Das
eingedrungene Sulfat reagierte vermutlich Großteils zunächst mit noch vorhandenem C3A zu

88
Ettringit und in der Folge wurde der Ettringit in Thaumasit als Endstufe der
festigkeitsmindernden chemischen Reaktionen umgewandelt. Die Schadensbildung zeigte
sich zuerst im Auftreten einer weißen Schicht von Reaktionsprodukten an der Oberfläche der
Mörtel, die nur lose anhafteten und mit der Zeit abfielen, sodass sich am Gefäßboden eine
Schicht von Mörtelresten ansammelte.
Weil die Mörtel, die mit CEM I bzw. mit HS-Zement gebunden waren (W/Z-Wert 0,70), nach
3,5 Jahren Lagerung bei 3000 mg SO4-2/l teilweise schon so stark geschädigt waren, dass die
Prüfung der Festigkeiten nicht mehr möglich war bzw. nicht sinnvoll erschien, wurden
Festigkeitsprüfungen nur bis zum Prüftermin 2,5 Jahre durchgeführt (wie sich im Verlauf des
Forschungsprojektes herausgestellt hat, beginnt die Thaumasitschädigung wie ein lösender
Angriff an der Probenoberfläche und setzt sich ins Innere fort, sodass die Untersuchung des
nicht schadhaften Probenteils keine schädigungsrelevanten Erkenntnisse ergibt). Wohl aber
wurden von den Würfelproben die lose anhaftenden Reaktionsprodukte abgebürstet und der
verbliebene Probenteil hinsichtlich des Sulfatgehaltes untersucht. Dabei stellte sich heraus,
dass die Zunahme des Sulfatgehaltes recht gut mit den bei Raumtemperatur bei 3000 mg
SO4-2/l eingetretenen Veränderungen korrelierten (Abbildungen 37 bzw. 46). Auch an den
Proben mit W/Z-Wert 0,45 sind Schäden entstanden, die aber ganz wesentlich geringer
blieben als bei den Proben mit W/Z-Wert 0,70. Auf die, bei der Mörtelherstellung vornehmlich
bei W/Z-Wert 0,70 eingetretenen sichtbaren Sedimentation und des darauf zurückzuführenden
gegenüber der Rezeptur erhöhten W/Z-Wertes im Bereich der abgezogenen Oberfläche ist es
wohl auch zurückzuführen, dass der Angriff immer an der abgezogenen Oberfläche ihren
Ausgang genommen hat und sich von ihr aus in die Tiefe fortsetzte.
Die bei Lagerung bei 3000 mg SO4-2/l entstandenen Schäden (Abbildung 47) waren bei den
kalksteinhaltigen Proben etwa gleich stark wie bei den kalksteinfreien. Interessant war der
Umstand, dass es bei Lagerung in gesättigter Gipslösung zu einer deutlich stärkeren
Schädigung kam als bei 3000 mg SO4-2/l obwohl die Sulfatkonzentration der gesättigten
Gipslösung wesentlich geringer war (max. ~1200 mg SO4-2/l) und daher auch der
Gesamtsulfatgehalt weniger stark angestiegen ist, als bei den bei 3000 SO4-2/l gelagerten
Proben [92, 93]. Offensichtlich hat die Anwesenheit des für die Thaumasitbildung auch
benötigten Calciums im Sulfat-Salz (Gips: CaSO4.2H2O) einen wesentlichen Einfluss auf die
Geschwindigkeit der Schädigung. Die Proben mit Fluamix bzw. CEM III-B als Bindemittel
wurden unvergleichlich weniger angegriffen, wobei an der Probe mit CEM III-B überhaupt
keine Angriff stattgefunden hat. Diese Bindemittel zeichnen sich somit nicht nur durch eine
wesentlich langsamere Sulfataufnahme als auch durch eine hohe Resistenz gegen

89
Thaumasitschädigung aus. Weiter stellte sich heraus, dass auch an der Oberfläche der Proben
die bei 20°C und 6000 mg SO4-2/l ausgelagert waren, stellenweise etwas Thaumasit
entstanden ist. Das zeigt, dass sich Thaumasit auch bei Temperaturen über 15°C bilden kann
[1, 23, 29, 37, 94-100]. Allerdings erfolgt die Bildung dabei so langsam, dass sie praktisch
vernachlässigt werden kann.
Nach 3,5 Jahren wurden die Untersuchungen plangemäß beendet, weil bei den bei 3000 mg
SO4-2/l gelagerten Proben teilweise schon nach 1,5 Jahren deutliche erkennbare Schäden
vorhanden waren, die sich sehr verstärkten (Abbildung 47), sodass kein Grund bestand die
Untersuchungen fortzuführen. Zu Schäden kam es aber nur bei den bei 3000 mg SO4-2/l und
bei in ges. Gipslösung gelagerten Proben, während die Proben, die bei 200 und 600 mg
SO4-2/l gelagert waren, während des gesamten Versuchszeitrums schadensfrei geblieben
sind.
12.3.4 Betonschäden in Tunneln
Anhand der entnommenen Proben zerstörten Betons wurde mittels Röntgendiffraktometrie
nachgewiesen, dass die Betonschädigung bei allen Proben durch die Bildung von Thaumasit
verursacht worden ist. Der durchschnittliche Sulfatgehalt in den aus den Tunneln
entnommenen Gebirgswässern betrug ca. 400 – 500 mg SO4-2/l. Ansonsten enthielt das
Wasser hauptsächlich nur Ionen von Calcium und Hydrogenkarbonat, aber praktisch keine
Alkalien (Natrium, Kalium: <10 mg/l). Die aus den zersetzten Betonproben ausgepressten
Wässer hingegen enthielten sehr viel Natrium, Sulfat und Chlorid. Die Sulfatkonzentration lag
in allen Fällen weit über der Sättigungskonzentration von Gips von ~1.200 mg SO4-2/l und
reichte bis zu etwa 30.000 mg SO4-2/l. Da das Sulfat nur aus dem Gebirgswasser in den Beton
gelangen konnte, musste die Frage geklärt werden, warum und auf welchem Weg das aus
dem Gebirge gelöste Calciumsulfat (Gips; Anhydrit) in leicht lösliches Natriumsulfat
umgewandelt werden konnte [54, 55].
Die festgestellten Konzentrationen der im zersetzten Beton enthaltenen wässrigen Phase
ließen vermuten, dass die Ursache des Konzentrationsanstieges auf einen
Verdunstungsprozess zurück zu führen ist, da verschiedene Inhaltsstoffe höher konzentriert
vorlagen als im Gebirgswasser und weil die Bildung des an verschiedenen Stellen der
Spritzbetonoberfläche vorgefundenen kristallinen Natriumsulfates (Mirabilit), das sehr gut
wasserlöslich ist, ein starkes Indiz für einen Verdunstungsvorgang ist. Der Beweis dafür ergab
sich aus der Konzentrationsbestimmung der stabilen Isotope von Sauerstoff (18O/16O
Verhältnis) und Wasserstoff (2H/1H Verhältnis) in den untersuchten Grund(Gebirgs)wässern

90
und den aus den geschädigten Betonproben ausgepressten Wässern (Abbildung 54). Das ist
möglich, weil es im Zuge eines Verdunstungsprozesses zu einer Anreicherung der schwereren
Isotope 18O und 2H kommt (der Dampfdruck der leichten Wassermoleküle ist höher als der der
schwereren), sodass aus solchen Messungen der jeweils gegebene Verdunstungsgrad
festgestellt werden kann, der in den aus den zerstörten Betonproben ausgepressten Lösungen
sehr unterschiedlich war. Da darin die Konzentration der an der Betonschädigung nicht
beteiligte Inhaltsstoffe (Rubidium, Lithium, etc.) wesentlich höher war, als dem jeweiligen
Verdunstungsgrad entspricht, muss Wasser wiederholt verdampft und durch neu
hinzutretendes Wasser wieder ergänzt worden sein [54].
Die Ergebnisse der nasschemischen Untersuchung der Abschnitte des in Abbildung 10
gezeigten Spritzbeton-Bohrkerns mit 200 mm Durchmesser (Tabelle 11) ergaben in
Übereinstimmung mit den diffraktometrischen Untersuchungen, dass der Betonzuschlag
karbonatischer Natur war (Kalkstein und Dolomit). In der dem Gebirge zugewandten
Randzone (~0-1 cm Tiefe) war der Sufatgehalt weit höher, als bei normgemäßer
Betonzusammensetzung zulässig ist. Mit zunehmender Betontiefe wurden die SO3-Werte
rasch kleiner (Tabelle 11), aber vermutlich war der SO3-Gehalt auch in einem Abstand von ca.
5,5 cm von der äußersten Zone noch überhöht. Der Gehalt an Natrium war in allen Abschnitten
höher als der von Kalium, und die Konzentration beider Ionen ist mit zunehmender Betontiefe
angestiegen. Dies ließ vermuten, dass die leicht löslichen Alkalien durch den Kontakt zum
Gebirgswasser offensichtlich fortlaufend „ausgewaschen“ worden sind. Thaumasitschäden
waren aber nicht nur am Spritzbeton entstanden sondern überall dort, wo sulfathaltiges
Gebirgswasser Kontakt zum Beton hatte und verdunsten konnte. Es sind also in den
Rohstoffen eines jeden dem österreichischen Regelwerk entsprechenden Betons so viel
Alkalien vorhanden, dass die Sulfatkonzentration des mit ihm in Kontakt stehenden Wassers
auf so hohe Werte ansteigen kann, dass das Wasser betonaggressiv wird (Alkalien sind an
der Schadensbildung selbst nicht beteiligt sondern ihre Anwesenheit ist Voraussetzung dafür,
dass die Konzentration des aggressiven Sulfats auf ungewöhnlich hohe Werte ansteigen
kann).
Die Ergebnisse der Analysen der aus den Abschnitten ausgepressten Porenlösungen (Tabelle
12) zeigten, dass die Konzentration der OH--, Na+- und K+-Ionen (der pH-Wert) von der dem
Gebirge zugewandten Randzone (0-1 cm Tiefe) nach innen zugenommen hat. Im Gegensatz
dazu war der Sulfatgehalt der Porenlösung gering und nur im äußersten Abschnitt höher als
in den übrigen Abschnitten. Die Konzentration an Calcium war noch geringer und hat zufolge
des mit steigender OH--Konzentration abnehmenden Löslichkeit des Calciums von außen

91
nach innen abgenommen.
Der Umstand, dass die Alkalikonzentration der Porenlösung umso geringer war, je näher der
jeweilige Betonabschnitt an der dem Gebirge zugewandten Spritzbetonoberfläche lag, geht
hervor, dass die Alkali-Ionen aus dem Beton in das Gebirgswasser diffundierten und dessen
Alkalikonzentration erhöhten. Konnte an solchen Stellen Wasser verdunsten, erhöhte sich die
Konzentration weiter. Im Gegensatz dazu konnte sich die Calciumkonzentration nicht erhöhen,
weil Calcium entweder durch die chemischen Reaktionen mit dem Beton verbraucht wurde
oder mit CO2 (aus der Luft oder Gebirgswasser) zu unlöslichem CaCO3 reagiert hat und dem
Wasser entzogen worden ist. Dadurch wurden die im Gebirgswasser enthaltenen
Calciumionen mit der Zeit durch Natriumionen ersetzt und das Calciumsulfat mehr und mehr
in leicht lösliches Natriumsulfat umgewandelt, sodass das anstehende Wasser betonaggressiv
werden konnte [54, 58]. Die Sulfatkonzentration des im einmal geschädigten Beton
vorhandenen Wassers konnte im Zuge weiterer Verdunstung bis zu etwa 30.000 mg SO4-2/l
zunehmen. Natürlich war damit auch eine starke Zunahme der Betonaggressivität verbunden,
was sich auf die Betonzerstörung sicher stark beschleunigend ausgewirkt hat.
13. Diskussion der Ergebnisse und Schlussfolgerungen
13.1 Linzer Abwasserkanäle
Der Gesamtsulfatgehalt des Konstruktionsbetons der Abwasserkanäle bzw. des
Nachklärbeckens war an allen vier Beobachtungsstellen trotz des Alters der Betone von über
30 Jahren nicht bzw. nur in 0-1,5 cm Tiefe etwas überhöht (<4% SO3, bezogen auf den
Zementgehalt). Auch der mittlere Sulfatgehalt der ausgelagerten, porösen Mörtelproben
(Würfel mit 4 cm Kantenlänge und Prismen mit 4 x 4 x 16 cm; W/Z-Wert 0,70) blieb während
der 3,5 jährigen Laufzeit des Forschungsprojektes überall unter 4% SO3, bezogen auf den
Zementgehalt. Da auch der Sulfatgehalt der Porenlösung der Beton- und Mörtelproben
unauffällig gering war, kann davon ausgegangen werden, dass es in absehbarer Zeit zu keinen
Betonschäden kommen wird. Dies zumal der Sulfatgehalt der Mörtelproben im Verlauf der
Laborlagerung bei 6000 mg SO4-2/l auf Werte von über 10 % des Zementgehaltes angestiegen
ist, ohne dass es zu mit freiem Auge erkennbaren Schäden gekommen ist. Nach Einschätzung
der Autoren besteht im konkreten Fall bei weiterhin gleichbleibenden Bedingungen keine
Gefahr einer Sulfatkorrosion. Auch die von Industriebetrieben zeitweise in das Kanalsystem

92
eingeleiteten sulfathaltigen Abwässer, wodurch es lokal zu einer erhöhten Sulfatkonzentration
kommt, ändert an dieser Einschätzung nichts, weil sich bei den Laboruntersuchungen
herausstellte, dass der Sulfatgehalt der Proben, die abwechselnd bei hohem und niedrigem
Sulfatgehalt der Lagerlösung lagerten, praktisch gleich stark anstieg wie der von
vergleichbaren Proben, die bei konstanter und etwa dem mittleren Sulfatgehalt der
Wechsellösungen entsprechender Konzentration lagerten. Daraus ergibt sich ja, dass es bei
wechselnden Konzentrationen auf den mittleren Sulfatgehalt ankommt und dass daher zur
Beurteilung der Expositionsklasse nicht der höchste Wert heranzuziehen ist. Interessant und
hinsichtlich der Sulfataufnahme des Betons im Abwassersystem sicherlich von Bedeutung war
die bei den Mörtelproben gemachte Beobachtung, dass es unter der Probenoberfläche zu
einer Abdichtung zufolge der Abscheidung von Calciumkarbonat kam, die die Sulfataufnahme
bremste. Da dies sicherlich auch beim Beton der Fall war, ist darin eine vorteilhafte, nicht
erwartete Schutzwirkung zu sehen. Die Stärke der Bremswirkung konnte aber nicht
quantifiziert werden, weil die Bestimmungsunsicherheiten dafür zu groß waren. Dass
allerdings die Gesamtsituation nicht wirklich vollständig überschaut werden kann, und dass
somit Langzeitprognosen hinsichtlich einer allfälligen Beton-Gefährdung offenbar
grundsätzlich nur mit Vorbehalt getroffen werden können, machen abseits der Sulfat-
Problematik liegende Ergebnisse deutlich, wie dass der Chloridgehalt der Porenlösung der im
Nachklärbecken gelagerten Mörtelproben im gesamten Versuchszeitraum entgegen dem
Konzentrationsgradienten anstieg (die Ursachen dafür blieben im Dunkel) und auch in der
Porenlösung des Betons des Nachklärbeckens klar über der Konzentration lag, die im Wasser
des Nachklärbeckens gegeben war (siehe Kapitel 8.2.4).
13.2 Untersuchungen an Laborproben und aus Tunneln
entnommenen Proben
13.2.1 Im Labor bei 20°C ausgelagerte Proben (Ettringitbildung)
Eine klare Antwort erlauben die Ergebnisse auf die Frage, welcher Expositionsklasse der
Beton bei Kontakt zu Wasser mit schwankendem Sulfatgehalt zuzuorden ist. Dass der Anstieg
des Sulfatgehaltes der Mörtelproben bei Wechsellagerung zwischen 600 und 6000 mg SO4-2/l
(Mittelwert 3300 mg SO4-2/l) vergleichbar war mit dem Anstieg, der bei konstant 3000 mg SO4
-
2/l gelagerten Proben eingetreten ist, zeigt, dass dafür die mittlere Konzentration relevant ist.
Hinsichtlich der Frage, ob es einen für alle Fälle der Praxis gültigen kritischen Sulfatgehalt gibt,
bei dessen Überschreitung eine Betonschädigung eintritt, kann mit großer Sicherheit gesagt

93
werden, dass dies nicht der Fall ist. Ettringit ist ja nicht aus chemischen Gründen schädlich
sondern er wächst in vorhandene Poren hinein, wodurch die Druckfestigkeit des Betons
zufolge abnehmender Porosität zunächst zunimmt und erst wenn der vorhandene Porenraum
gefüllt ist, kommt es durch den mit der Ettringitbildung verbundenen Druck bzw. aus anderen
Gründen zur Dehnung des Gefüges (s. Kapitel 2.1.1.4) und in weiterer Folge zur Bildung von
ersten Rissen, die mit der Zeit größer und mehr werden, bis es letztlich zum Betonzerfall
kommen kann. Je höher die Porosität eines Betons ist (je höher der W/B-Wert ist, etc.) umso
mehr Ettringit wird entstehen können und umso höher wird der Gesamtsulfatgehalt ansteigen
können ohne dass eine Schadensbildung eintritt. Daraus darf aber nicht geschlossen werden,
dass hohe Porosität günstig wäre, denn die Eindringgeschwindigkeit nimmt bei niedriger
Porosität so stark ab, dass die Sulfataufnahme so langsam erfolgt, dass es deshalb nicht bzw.
erst viel später zur Schadensbildung kommt. Das wird aus den Versuchsergebnissen deutlich,
die bei Lagerung bei 6000 mg SO4-2/l bis zum Versuchsende (4,5 Jahre) eine Zunahme des
Sulfatgehaltes beim Mörtel mit CEM I und W/Z-Wert 0,45 von ~2 %, bei W/Z-Wert 0,70 aber
eine von über 7%, jeweils bezogen auf den Zementgehalt ergeben haben (der Gesamtgehalt
an Sulfat betrug bei W/Z-0,45 über 5% und bei W/Z-0,70 über 10% SO3). Der Mörtel mit W/Z-
Wert 0,45 hatte keinerlei feststellbare Schädigung, während beim Mörtel mit W/Z-Wert 0,70
die Biegezugfestigkeit deutlich abgenommenen hatte (die Druckfestigkeit blieb unverändert).
Das zeigt, dass eine niedrige Porosität vorteilhaft ist und dass für die Beurteilung, ob in einem
konkreten Fall eine Schädigung eingetreten ist, der Biegezugfestigkeit offenbar höhere
Relevanz zukommt als der Druckfestigkeit. Weiter war die Zementsorte erwartungsgemäß von
deutlichem Einfluss auf die Geschwindigkeit der Sulfataufnahme, wobei sich latent
hydraulische bzw. puzzolanische Zumahlstoffe günstig ausgewirkt haben. Natürlich hängt die
Expansionsgefahr bzw. deren Ausmaß auch vom C3A-Gehalt des Zementes ab. Da Ettringit
nicht die einzige mögliche Sulfatkomponente ist, die mit eindringendem Sulfat entstehen kann,
sondern auch AFt-Phase (hier ist das Aluminium im Ettringit durch Eisen ersetzt) oder Gips
entsteht, wird es bei gegebenem Porenraum nicht nur auf den Gesamtsulfatgehalt ankommen,
wann die Schädigung beginnt. Auch andere Bedingungen, wie den pH-Wert der Porenlösung,
etc., bei denen möglicherweise andere Sulfatverbindungen als Ettringit bevorzugt entstehen
können, sind zu beachten. So ist in einer Arbeit beschrieben, dass die Sulfatbeständigkeit
eines CEM III-Betons nur bei ständiger Unterwasserlagerung besser ist als die eines OPC-
Betons, dass bei wechselnden Lagerungsbedingungen mit feucht/trocken-Wechseln aber der
OPC-Beton weniger angegriffen wurde [101]. Daher erscheint es auch als eher
unwahrscheinlich, dass die erhaltenen Laborergebnisse der Proben mit CEM III und der
Mischung des HS-Zementes mit Fluamix, die ständig zur Gänze in den Sulfatlösungen
eintauchten, auf wechselnde Bedingungen mit feucht/trocken-Phasen übertragen werden

94
können [101-103].
Aus dem Ergebnis, dass an den Mörteln nach einer Lagerungsdauer von 4,5 Jahren trotz eines
teilweise auf über 10% SO3, bezogen auf den Zementgehalt, angestiegenen Sulfatgehaltes
keine Schadensanzeichen sichtbar waren, wohl aber innere Risse vorhanden waren und die
Biegezugfestigkeit abgenommen hatte, ergibt sich, dass der Sulfatgehalt für die Beurteilung,
ob ein Beton sulfatgeschädigt ist, nicht ausreicht sondern zusätzlich die
elektronenmikroskopische Untersuchung der Mikrostruktur hinsichtlich innerer Risse
notwendig ist. Wenn möglich, sollte auch die Biegezugfestigkeit des Betons bestimmt werden,
während die Kenntnis der Druckfestigkeit als weniger wichtig erscheint.
Dass keinem der Projektbeteiligten auch nur ein in Österreich vorgekommener Fall einer
Ettringitschädigung eines Konstruktionsbetons zur Kenntnis gelangt ist, wird zusammen mit
den Ergebnissen der Laboruntersuchungen als deutlicher Hinweis dafür aufgefasst, dass die
Schädigungsgefahr nicht ganz so dramatisch ist, wie aus den in der EN 206-1 [70] bzw. der
ÖN B 4710-1 [71] enthaltenen, die Expositionsklassen kennzeichnenden SO4-2-Grenzwerten,
erscheinen mag, und dass die Sinnhaftigkeit dieser Grenzwerte daher in Frage zu stellen ist.
13.2.2 Im Labor bei 5°C ausgelagerte Proben (Thaumasitbildung)
Die durch Reaktion mit dem Bindemittel des Betons entstehenden neuen Sulfatverbindungen
(Thaumasit bzw. Woodfordit, Gips) sind unlöslich bzw. schwer löslich, weshalb die SO4-2- und
Ca+2-Konzentration der Porenlösung unabhängig vom Gesamtsulfatgehalt unter 1000 mg
SO4-2-/l blieb (Lagerlösungskonzentration bei geschädigten Mörteln: 3000 mg SO4
-2/l) und
daher während der Lagerungsdauer immer neues Sulfat eindiffundieren und der Sulfatgehalt
der Mörtel zufolge chemischer Bindung immer mehr ansteigen konnte. Die Schädigung hat
aber an der Oberfläche begonnen und setzte sich ins Innere fort, wobei das eingedrungene
Sulfat zunächst zu Ettringit bzw. zur AFt-Phase reagierte und in der Folge in Thaumasit als
Endstufe der festigkeitsmindernden chemischen Reaktionen umgewandelt wurde (auch beim
Mörtel mit C3A-freien Zement). Diese Art der Sulfatschädigung war leicht erkennbar und zeigte
sich zuerst im Auftreten einer weißen Schicht von Reaktionsprodukten an der Oberfläche der
Mörtel, die nur lose anhafteten und mit der Zeit abfielen, sodass sich am Gefäßboden eine
Schicht von Mörtelresten ansammelte.
Der Grad der Schäden war bei den kalksteinhaltigen Mörtelproben etwa gleich stark wie bei
den kalksteinfreien Proben (augenscheinliche Beurteilung). Die Anwesenheit einer
karbonatischen Gesteinskörnung ist somit keine notwendige Voraussetzung für eine

95
Thaumasitschädigung. Als ungünstig stellte sich allerdings dolomitische Gesteinskörnung
heraus, die angegriffen wurde (siehe Kapitel 10). Offenbar diffundierte aus der Luft immer
ausreichend CO2 in die Gefäße, obwohl sie mit einem Deckel abgedeckt waren.
Bei Lagerung in gesättigter Gipslösung kam es zu einer deutlich stärkeren Schädigung als bei
3000 mg SO4-2/l obwohl die Sulfatkonzentration der Gipslösung wesentlich geringer war (max.
~1200 mg SO4-2/l) und daher auch der Gesamtsulfatgehalt weniger stark angestiegen ist, als
bei den bei 3000 SO4-2/l gelagerten Proben. Offensichtlich hat die Anwesenheit des für die
Thaumasitbildung auch benötigten Calciums im Sulfat-Salz (Gips: CaSO4) einen wesentlichen
Einfluss auf die Geschwindigkeit der Schädigung. Die Proben mit Fluamix bzw. CEM III-B als
Bindemittel wurden während des Versuchszeitraumes in beiden Lagerlösungen (gesättigte
Gipslösung bzw. 3000 mg SO4-2/l) unvergleichlich weniger (70% HS-Zement/30% Fluamix)
bzw. überhaupt nicht angegriffen (CEM III-B). Diese Bindemittel zeichnen sich somit nicht nur
durch ein dichteres Gefüge aus, in welches Sulfat langsamer eindringt und der Sulfatgehalt
daher langsamer ansteigt als bei einem Mörtel mit anderem Zement aber ansonsten gleicher
Rezeptur, sondern auch durch eine hohe Resistenz gegen Thaumasitschädigung. Der Einfluss
des Bindemittels auf die Widerstandsfähigkeit gegen einen Thaumasitschaden ist aus diesem
Grund als sehr hoch einzustufen. Dies ist allerdings nur bei ständig feuchten Bedingungen als
gesichert anzusehen (s. 13.2.1).
Interessant war, dass der Gesamtsulfatgehalt der bei 3000 mg SO4-2/l und 5°C gelagerten
Proben unterhalb der anhaftenden Schicht von bereits zerstörtem Mörtel etwa gleich stark
anstieg wie bei den bei 20°C gelagerten Proben gleicher Rezeptur. Unterhalb der Oberfläche
der noch nicht sichtbar geschädigten Probenreste, wo kein Berührungskontakt zur
Lagerlösung bestand, konnte sich offensichtlich zufolge zu geringen Sulfatgehaltes der
Porenlösung kein Thaumasit bilden, was erklärt, dass die Thaumasitschädigung von der
Oberfläche der Mörtel ausgehend nach Innen verläuft.
13.2.3 Aus Tunneln mit Betonschäden entnommene Proben
Es wurde nachgewiesen, dass die Betonschädigung an allen untersuchten Stellen durch die
Bildung von Thaumasit verursacht worden ist und dass es nur dort zu Schäden kommen
konnte, wo eine Verdunstung des Gebirgswassers möglich war. Da Wasser durch Spritzbeton
mit normaler Schichtdicke hindurch nicht verdunsten kann, war das nur an Fehlstellen möglich
(undichte Fugen, Risse, etc.). Dass es tatsächlich zur Wasserverdunstung gekommen ist,
wurde u.a. durch (aufwändige) Konzentrationsbestimmungen der stabilen Isotope von
Sauerstoff (18O/16O Verhältnis) und Wasserstoff (2H/1H Verhältnis) in den untersuchten

96
Grund(Gebirgs)wässern und den aus den geschädigten Betonproben ausgepressten Wässern
nachgewiesen (siehe Kapitel 11). Da der mittlere Sulfatgehalt der Gebirgswassers aller
untersuchter Tunnel etwa 400 – 500 mg SO4-2/l betrug, ist anzunehmen, dass dieser
Konzentrationsbereich für alle österreichischen Gebirgswässer gilt. Er ist aber für eine
Schadensbildung zu gering. Dafür ist, wie die Laboruntersuchungen ergaben, eine
Sulfatkonzentration zwischen 600 und ~1200 mg SO4-2/l notwendig. Außerdem hat sich
gezeigt, dass der im Gebirgswasser gelöste Gips im Verlauf der Schädigung in leicht lösliches
Alkalisulfat umgewandelt wurde, was sich auf die Schadensbildung sicherlich stark
beschleunigend ausgewirkt hat, denn in den, aus zersetzten Betonproben ausgepressten
Wässer waren bis zu 30.000 mg SO4-2/l enthalten [54, 55, 58, 76, 78].
Das Schadensbild war an verschiedenen Stellen nicht immer gleich sondern teilweise sehr
unterschiedlich (Kapitel 6.2.2), aber die Thaumasitschädigung hat auch hier immer an der
Betonoberfläche begonnen, sich dann ins Innere fortgesetzt bis sie die sichtbare
Innenoberfläche erreichte oder es ging die Haftung zum Untergrund verloren. Da damit auch
der Kontakt zum Gebirgswasser weitgehend verloren ging, hörte der Angriff vermutlich auf. Da
in intakten Beton eingedrungenes Sulfat fast vollständig gebunden wurde, blieb die
Sulfatkonzentration der Porenlösung niedrig, weshalb sich im Betoninneren kein Thaumasit
bilden konnte. Daher war auch der aus einem Bereich mit hohl liegendem Spritzbeton
entnommene Betonbohrkern unterhalb der dem Gebirge zugewandten Oberfläche, an der
Reaktionsprodukte anhafteten, trotz relativ hohen Sulfatgehaltes augenscheinlich nicht
schadhaft.
14. Empfehlungen für die Praxis
Zunächst werden die aus dem Forschungsvorhaben gewonnenen und für die Praxis
relevanten Erkenntnisse nochmals kurz zusammengefasst.
Ettringitkorrosion
Die Ettringitschädigung von Beton kann bei allen oberhalb des Gefrierpunktes liegenden
Außentemperaturen stattfinden. Dabei kommt es zu einer physikalischen Schädigung durch
Gefügedehnung, wobei zunächst Risse als sichtbares Schadensmerkmal entstehen und es im
Extremfall zu einem völligen Zerfall des Betons kommen kann. Der wirksamste Schutz ist die
Verwendung eines erhöht sulfatbeständigen Zementes (HS-Zement; enthält C3A-armen bzw.
-freien Zementklinker). Wie sich herausgestellt hat, gibt es keinen bestimmten Sulfatgehalt von

97
Beton, bei dessen Überschreitung Erhaltungsmaßnahmen nötig werden. Dies ist deshalb so,
weil neben anderen Faktoren die Porosität des Betons von starkem Einfluss ist (je mehr offener
Porenraum vorhanden ist, umso mehr Ettringit kann sich bilden ohne dass es zu einer
Gefügedehnung kommt). Der kritische Sulfatgehalt ist daher von Fall zu Fall unterschiedlich.
Die zur Zuordnung der Expositionsklasse in der EN 206-1 bzw. deren nationaler Umsetzung,
der ÖN B 4710-1 für angreifende Wässer bzw. Böden angegebenen SO4-2-Grenzwerte liegen
offensichtlich sehr weit auf der sicheren Seite. Im Labor sind trotz 4,5 jähriger Auslagerung in
Sulfatlösungen mit bis zu 6000 mg SO4-2/l (obere Grenzkonzentration der Expositionsklasse
XA3) unabhängig von der Zementsorte und Porosität (W/B-Wert) der Prüfkörper keine mit dem
freien Auge sichtbaren Schäden entstanden. Außerdem war keinem der Beteiligten ein Fall
bekannt, wo es in Österreich durch die Einwirkung natürlicher Wässer bzw. Böden zu einer
Sulfatkorrosion von ordnungsgemäß hergestellten Beton gekommen ist. Daher erscheinen die
oberen Grenzkonzentrationen der Expositionsklassen als praxisfern gering.
Thaumasitkorrosion
Die Thaumasitbildung verläuft, wie in Kapitel 2.2 ausgeführt, entweder über die direkte Route
(C-S-H + Calciumsulfat + Karbonat) oder über die AFt-Phase, was im betroffenen Bereich zum
vollständigen Verlust der Festigkeit führt (es entsteht eine breiige Masse). Bei
Umgebungstemperaturen über 15°C ist die Bildungsgeschwindigkeit von Thaumasit allerdings
so gering, dass sie praktisch vernachlässigt werden kann. Unterhalb 15°C kann sowohl
Ettringit- als auch Thaumasitschädigung eintreten, wobei im Fall der Thaumasitbildung die
Schädigung viel schneller sichtbar wird. Obwohl ein C3A-freier Zement gegen einen
Thaumasitangriff keine Schutzwirkung hat, ist auch bei niedrigen Temperaturen die
Verwendung eines sogenannten HS-Zementes notwendig, weil damit der auch unter 15°C
stattfindenden Ettrigitbildung entgegen gewirkt werden muss. Die Schädigung ist im
Gegensatz zur Ettringitkorrosion keine Volumsinstabilität sondern erfolgt wie bei einem
lösenden Angriff von der Oberfläche ausgehend und setzt sich ins Innere fort. Diese Art der
Sulfatkorrosion ist bislang im europäischen Regelwerk nicht verankert. Die in nationalen
Normen gegen einen Sulfatangriff angegebenen betontechnologischen Schutzmaßnahmen
(etwa ÖN B 4710-1) sind daher dafür nicht relevant.
Aus den Untersuchungen aus Labor und Feld (Tunneln) lässt sich ableiten, dass der
Sulfatgehalt der in den untersuchten Tunneln angetroffenen Wässer (Maximalwert: ~550 mg
SO4-2/l) für die Thaumasitbildung nicht ausgereichend hoch war [relevant ist der
Sättigungsindex von Gips (SIGips), der mit zunehmendem pH-Wert abnimmt und im Labor

98
auch unterhalb von 1000 mg SO4-2/l starke Thaumasitschäden verursacht hat (pH-Wert der
Lagerlösung: ~12,5)]. Daher war in den Tunneln eine Konzentrierung des Sulfates durch
Wasserverdunstung nötig. Aus diesem Grund muss bei der Innenauskleidung (Spritzbeton)
und allen übrigen Bereichen, bei denen Gebirgswasser, das an Gips untersättigt ist (SIGips
≤0) und mit Beton in Kontakt kommt (Gänge, etc.) sehr genau darauf geachtet werden, dass
keine undichte Betonfugen oder -risse bzw. bzw. andere Stellen vorhanden sind, durch die
Wasser von der Gebirgsseite nach außen verdunsten kann. Selbstredend muss der Beton so
dicht sein, dass er Wasser nicht kapillar transportieren kann. Andernfalls kann sulfathaltiges
Wasser aufsteigen, an der Verdunstungszone kommt es zur Sulfatanreicherung und in der
Folge zur Betonzersetzung durch Thaumasit. In Fällen mit einem SIGips ≥0 im angreifenden
Wasser müssen spezielle betontechnische Maßnahmen getroffen werden bzw. der Kontakt
solchen Wassers zu Beton unterbunden werden (s. Tabelle 13). Die Untersuchungen haben
weiter gezeigt, dass Gesteinskörner aus Dolomit zersetzt wurden, wobei im zersetzten Korn
neben Brucit [Mg(OH)2] auch Kalzit (CaCO3) nachgewiesen wurde (s. Kapitel 11). Ob der Kalzit
im Zuge des Zersetzungsprozesses neu gebildet wurde, kann nicht eindeutig beantwortet
werden, erscheint aber als sehr wahrscheinlich, weil der Kalzit innerhalb der Umrisse des
zersetzten Dolomitkorns, eingebettet in der Brucit-Masse vorlag (die Zersetzung von Dolomit
hat sehr überrascht, weil dieses Mineral gegen chemische Einwirkungen gewöhnlich
beständiger ist als Kalzit). Daraus ergibt sich die Forderung, dass die Gesteinskörnung bei
Gefahr eines Thaumasitangriffs nicht aus Dolomit bestehen sollte. Nachfolgend sind die für
jeden Angriffstypus notwendigen Gegenmaßnahmen in Tabellenform angeführt.

99
Tabelle 13: Empfehlungen für die Beton-Bestandteile
Schädigung durch Ettringit (3CaO.Al2O3.3CaSO4.32H2O)
Schädigung durch Thaumasit (CaSiO3 CaCO3.CaSO4. 15H2O)
B i n d e m i t e l C3A-freier Klinker
angestrebt: Erschwerung der Sulfateindringung [Verwendung von Zumahl- bzw. Zusatzstoffen; bewirkt eine Reduktion des Porendurchmessers der Bindemittelmatrix und des kapillaren Saugens sowie des Ca(OH)2 - Anteils und damit eine Abnahme der Menge möglicher Gipsbildung].
Zumahlstoffe: puzzolanische bzw. latent hydraulische
günstig; (Hüttensand, Flugasche, Mischungen verschiedener; kein Gesteinsmehl (Typ I))
wie bei Ettringit und zudem arm an Alkalien; keine karbonatischen
Bestandteile kein Gesteinsmehl (Typ I)
G e s t e i n s k ö r n u n g (GK) angestrebt: Die GK soll sich gegenüber Sulfat bei jeder Außentemperatur inert verhalten
alle für Beton geeigneten Gesteinskörnungen können verwendet werden (ÖN EN 12620 [68] bzw. deren österr. Umsetzung ÖN B
3131 [104])
wie bei Ettringit aber keine karbonatischen Gesteinskörnungen; (wenn nicht möglich:
zumindest kein dolomitischer Anteil im Feinbereich)
Z u s a t z s t o f f e angestrebt: Zusatzstoffe sollen das Eindringen von Sulfat erschweren;
Zusatzstoffe vom Typ I sind daher zu vermeiden
Z u s a t z m i t t e l
angestrebt: soll die Eigenschaften des Zementsteines (phys./chem.-Eigenschaften, Porenverteilung, etc.) möglichst nicht verändern (Luftporen zulässig)
alle für Beton geeignete Zusatzmittel können verwendet werden
(ÖN EN 934 I und II [105, 106])
wie bei Ettringit; Zusatzmittel müssen als alkalifrei deklariert sein
Tabelle 14: Betontechnologische Maßnahmen
T u n n e l b e t o n (Frosteinwirkung nur im Portalbereich)
angestrebt: alle Maßnahmen, die das Eindringen von Sulfat erschweren. Möglichst dichtes Betongefüge (geringe Wassereindringtiefe), C3A-freies Bindemittel mit zumindest 25-35%
AHWZ-Anteil, gute Verdichtung und Nachbehandlung, Vermeidung der Ablüftung von Tunnelwasser an der Betonoberfläche
Dichtigkeit des Betons Ettringit: XC4
Thaumasit: max. 20 mm Wassereindringtiefe
Luftgehalt ≥2,5%
L 300 ≥1,0%
(im Portalbereich)

100
15. Literatur
[1] N. Loudon, A review of the experience of thaumasite sulfate attack by the UK Highways Agency, Cement and Concrete Composites, 25 (2003) 1051-1058.
[2] Y. Maltais, E. Samson, J. Marchand, Predicting the durability of Portland cement systems in aggressive environments--Laboratory validation, Cement and Concrete Research, 34 (2004) 1579-1589.
[3] P.K. Mehta, Mechanism of sulfate attack on portland cement concrete — Another look, Cement and Concrete Research, 13 (1983) 401-406.
[4] E. Menéndez, T. Matschei, F.P. Glasser, Sulfate attack on concrete, in: M. Alexander, A. Bertron, N. De Belie (Eds.) Performance of Cement-Based Materials in Aggressive Aqueous Environments, Springer, Dordrecht, 2013, pp. 7-74.
[5] W. Müllauer, R.E. Beddoe, D. Heinz, Sulfate attack expansion mechanisms, Cement and Concrete Research, 52 (2013) 208-215.
[6] A. Neville, The confused world of sulfate attack on concrete, Cement and Concrete Research, 34 (2004) 1275-1296.
[7] M. Santhanam, M.D. Cohen, J. Olek, Sulfate attack research - whither now?, Cement and Concrete Research, 31 (2001) 845-851.
[8] W. Kunther, B. Lothenbach, K.L. Scrivener, On the relevance of volume increase for the length changes of mortar bars in sulfate solutions, Cement and Concrete Research, 46 (2013) 23-29.
[9] W. Kunther, B. Lothenbach, K.L. Scrivener, Deterioration of mortar bars immersed in magnesium containing sulfate solutions, Materials and Structures/Materiaux et Constructions, (2013) 1-9.
[10] B. Lothenbach, B. Bary, P. Le Bescop, T. Schmidt, N. Leterrier, Sulfate ingress in Portland cement, Cement and Concrete Research, 40 (2010) 1211-1225.
[11] S. Lorente, M.P. Yssorche-Cubaynes, J. Auger, Sulfate transfer through concrete: Migration and diffusion results, Cem Concr Compos, 33 (2011) 735-741.
[12] S.J. Barnett, C.D. Adam, A.R.W. Jackson, Solid solutions between ettringite, Ca6Al2(SO4)(3)(OH)(12)center dot 26H(2)O, and thaumasite, Ca3SiSO4CO3(OH)(6)center dot 12H(2)O, J Mater Sci, 35 (2000) 4109-4114.
[13] S.J. Barnett, M.A. Halliwell, N.J. Crammond, C.D. Adam, A.R.W. Jackson, Study of thaumasite and ettringite phases formed in sulfate/blast furnace slag slurries using XRD full pattern fitting, Cement and Concrete Composites, 24 (2002) 339-346.
[14] I. Baur, P. Keller, D. Mavrocordatos, B. Wehrli, C.A. Johnson, Dissolution-precipitation behaviour of ettringite, monosulfate, and calcium silicate hydrate, Cement and Concrete Research, 34 (2004) 341-348.
[15] J. Bensted, Thaumasite - background and nature in deterioration of cements, mortars and concretes, Cem Concr Compos, 21 (1999) 117-121.
[16] J. Bensted, Thaumasite--direct, woodfordite and other possible formation routes, Cement and Concrete Composites, 25 (2003) 873-877.
[17] M.T. Blanco-Varela, J. Aguilera, S. Martinez-Ramirez, Effect of cement C3A content, temperature and storage medium on thaumasite formation in carbonated mortars, Cement and Concrete Research, 36 (2006) 707-715.
[18] P. Brown, R.D. Hooton, Ettringite and thaumasite formation in laboratory concretes prepared using sulfate-resisting cements, Cement and Concrete Composites, 24 (2002) 361-370.
[19] P.W. Brown, R.D. Hooton, B.A. Clark, The co-existence of thaumasite and ettringite in concrete exposed to magnesium sulfate at room temperature and the influence of blast-furnace slag substitution on sulfate resistance, Cement and Concrete Composites, 25 (2003) 939-945.
[20] A.N. Christensen, T.R. Jensen, J.C.J.C. Hanson, Formation of ettringite, Ca6Al2(SO4)3(OH)12[middle dot]26H2O, AFt, and monosulfate, Ca4Al2O6(SO4)[middle dot]14H2O, AFm-14, in hydrothermal hydration of Portland cement and of calcium aluminum oxide--calcium sulfate dihydrate mixtures studied by in situ synchrotron X-ray powder diffraction, Journal of Solid State Chemistry, 177 (2004) 1944-1951.
[21] S. Kohler, D. Heinz, L. Urbonas, Effect of ettringite on thaumasite formation, Cement and Concrete Research, 36 (2006) 697-706.
[22] T. Schmidt, B. Lothenbach, M. Romer, K. Scrivener, D. Rentsch, R. Figi, A thermodynamic and experimental study of the conditions of thaumasite formation, Cement and Concrete Research, 38 (2008) 337-349.
[23] A.M. Ramezanianpour, R.D. Hooton, Thaumasite sulfate attack in Portland and Portland-

101
limestone cement mortars exposed to sulfate solution, Constr Build Mater, 40 (2013) 162-173. [24] C. Bauer, Ursache der Sulfatschäden im Eisenbahntunnel Bosruck, in, 2004. [25] J. Ma, Application of shotcrete linings under sulfate attack environments, Adv Mater Res, 233-
235 (2011) 2061-20672067. [26] F. Bellmann, On the formation of thaumasite CaSiO3-CaSO4-CaCO3-15H2O: part III, Adv Cem
Res, 19 (2007) 139-146. [27] M.T. Blanco-Varela, J. Aguilera, S. Martinez-Ramirez, F. Puertas, A. Palomo, C. Sabbioni, G.
Zappia, C. Riontino, K. Van Balen, E.E. Toumbakari, Thaumasite formation due to atmospheric SO2-hydraulic mortar interaction, Cem Concr Compos, 25 (2003) 983-990.
[28] N.J. Crammond, The thaumasite form of sulfate attack in the UK, Cem Concr Compos, 25 (2003) 809-818.
[29] M.E. Gaze, N.J. Crammond, The formation of thaumasite in a cement : lime : sand mortar exposed to cold magnesium and potassium sulfate solutions, Cem Concr Compos, 22 (2000) 209-222.
[30] M. Romer, Steam locomotive soot and the formation of thaumasite in shotcrete, Cem Concr Compos, 25 (2003) 1173-1176.
[31] A. Skaropoulou, S. Tsivilis, G. Kakali, J.H. Sharp, R.N. Swamy, Long term behavior of Portland limestone cement mortars exposed to magnesium sulfate attack, Cem Concr Compos, 31 (2009) 628-636.
[32] G. Kakali, S. Tsivilis, A. Skaropoulou, J.H. Sharp, R.N. Swamy, Parameters affecting thaumasite formation in limestone cement mortar, Cement and Concrete Composite, 25 (2003) 977–981.
[33] J. Aguilera, S. Martinez-Ramirez, I. Pajares-Colomo, M.T. Blanco-Varela, Formation of thaumasite in carbonated mortars, Cement and Concrete Composites, 25 (2003) 991-996.
[34] S.J. Barnett, D.E. Macphee, N.J. Crammond, Extent of immiscibility in the ettringite-thaumasite system, Cement and Concrete Composites, 25 (2003) 851-855.
[35] F. Bellmann, W. Erfurt, H.M. Ludwig, Field performance of concrete exposed to sulphate and low pH conditions from natural and industrial sources, Cement and Concrete Composites, 34 (2012) 86-93.
[36] P. Brown, R.D. Hooton, B. Clark, Microstructural changes in concretes with sulfate exposure, Cement and Concrete Composites, 26 (2004) 993-999.
[37] N. Crammond, The occurrence of thaumasite in modern construction - a review, Cement and Concrete Composites, 24 (2002) 393-402.
[38] M.A. Eden, The laboratory investigation of concrete affected by TSA in the UK, Cement and Concrete Composites, 25 (2003) 847-850.
[39] D. Heinz, L. Urbonas, About thaumasite formation in Portland-limestone cement pastes and mortars--effect of heat treatment at 95 [degree sign]C and storage at 5 [degree sign]C, Cement and Concrete Composites, 25 (2003) 961-967.
[40] M. Romer, L. Holzer, M. Pfiffner, Swiss tunnel structures: concrete damage by formation of thaumasite, Cement and Concrete Composites, 25 (2003) 1111-1117.
[41] T. Sibbick, D. Fenn, N. Crammond, The occurrence of thaumasite as a product of seawater attack, Cement and Concrete Composites, 25 (2003) 1059-1066.
[42] D.C. Stark, Occurrence of thaumasite in deteriorated concrete, Cement and Concrete Composites, 25 (2003) 1119-1121.
[43] M.D.A. Thomas, C.A. Rogers, R.F. Bleszynski, Occurrences of thaumasite in laboratory and field concrete, Cement and Concrete Composites, 25 (2003) 1045-1050.
[44] B. Ma, X. Gao, E.A. Byars, Q. Zhou, Thaumasite formation in a tunnel of Bapanxia Dam in Western China, Cement and Concrete Research, 36 (2006) 716-722.
[45] M. Santhanam, M.D. Cohen, J. Olek, Effects of gypsum formation on the performance of cement mortars during external sulfate attack, Cement and Concrete Research, 33 (2003) 325-332.
[46] T. Schmidt, B. Lothenbach, M. Romer, J. Neuenschwander, K. Scrivener, Physical and microstructural aspects of sulfate attack on ordinary and limestone blended Portland cements, Cement and Concrete Research, 39 (2009) 1111-1121.
[47] S.M. Torres, C.A. Kirk, C.J. Lynsdale, R.N. Swamy, J.H. Sharp, Thaumasite-ettringite solid solutions in degraded mortars, Cement and Concrete Research, 34 (2004) 1297-1305.
[48] R. Mroz, The effect of sulfate environment and low temperature on the durability of cement mortars with mineral additives, Cem Wapno Beton, 15 (2010) 169-+.
[49] M. Romer, P. Lienemann, Deterioration of shotcrete in the safety gallery of the Gotthard motorway tunnel by salt-containing water, Chimia, 52 (1998) 197-201.
[50] B. Erlin, D.C. Stark, Identification and occurrence of thaumasite in concrete, Highway Res

102
Record, 113 (1965) 108–113. [51] G.C. Long, Y.J. Xie, D.H. Deng, X.K. Li, Deterioration of concrete in railway tunnel suffering
from sulfate attack, J Cent South Univ Technol, 18 (2011) 881-888. [52] S. Tsivilis, K. Sotiriadis, A. Skaropoulou, Thaumasite form of sulfate attack (TSA) in limestone
cement pastes, Journal of the European Ceramic Society, 27 (2007) 1711-1714. [53] N. Thaulow, S. Sahu, Mechanism of concrete deterioration due to salt crystallization, Materials
Characterization, 53 (2004) 123-127. [54] F. Mittermayr, A. Baldermann, C. Kurta, T. Rinder, D. Klammer, A. Leis, J. Tritthart, M. Dietzel,
Evaporation — a key mechanism for the thaumasite form of sulfate attack, Cement and Concrete Research, 49 (2013) 55-64.
[55] F. Mittermayr, C. Bauer, D. Klammer, M.E. Boettcher, A. Leis, P. Escher, M. Dietzel, Concrete under sulphate attack: an isotope study on sulphur sources, Isotopes in Environmental and Health Studies, 48 (2012) 105-117.
[56] F. Mittermayr, D. Klammer, M. Dietzel, C. Bauer, M. Böttcher, M. Koch, S.J. Köhler, A. Mayer, Thaumasitbildung in Tunnelbauten – Hydrogeochemie und stabile Isotope, Gruppe Geotechnik Graz, (2008) 115-132.
[57] F. Mittermayr, D. Klammer, D. Höllen, S. Köhler, M. Böttcher, A. Leis, M. Dietzel, Deterioration of Concrete: Application of Stable Isotopes, in: M.A.T.M. Broekmans (Ed.) Proceedings of the 10th International Congress for Applied Mineralogy (ICAM), Springer Berlin Heidelberg, 2012, pp. 435-443.
[58] F. Mittermayr, T. Rinder, D. Klammer, A. Leis, M. Dietzel, A Carbon Isotope Study of Thaumasite and Calcite Sinter Formation in Underground Construction, in: H. Justnes, S. Jacobsen (Eds.) International Congress on Durability of Concrete, NTNU, Trondheim, 2012, pp. No. C6-1: 1-14.
[59] ÖNORM EN 196-1: Prüfverfahren für Zement – Teil 1: Bestimmung der Festigkeit, 2005. [60] ÖNORM EN 450-1, in: Teil 1: Definition, Anforderungen und Konformitätskriterien, 2012. [61] ÖNORM B 3309-1: Aufbereitete hydraulisch wirksame Zusatzstoffe für die Betonherstellung
(AHWZ) – Teil 1: Kombinationsprodukte (GC/GC-HS), 2010. [62] J. Stark, B. Wicht, Schadensmachanismus des Sulfatangriffs auf Beton, in: F.A. Finger-Institut-
f.-Baustoffkunde-d.-Bauhaus-Universität-Weimar (Ed.) Dauerhaftigkeit Von Beton: Der Baustoff Als Werkstoff, Birkhäuser, Basel, 2001, pp. 119–147.
[63] H. Taylor, Cement Chemistry, 2nd ed., Thomas Telford Publishing, 1997. [64] W. Lukas, Betonzerstörung durch SO3-Angriff unter Bildung von Thaumasit und Woodfordit,
Cement and Concrete Research, 5 (1975) 503-517. [65] S.J. Barnett, C.D. Adam, A.R.W. Jackson, An XRPD profile fitting investigation of the solid
solution between ettringite, Ca6Al2(SO4)3(OH)12[middle dot]26H2O, and carbonate ettringite, Ca6Al2(CO3)3(OH)12[middle dot]26H2O, Cement and Concrete Research, 31 (2001) 13-17.
[66] S.J. Barnett, D.E. Macphee, E.E. Lachowski, N.J. Crammond, XRD, EDX and IR analysis of solid solutions between thaumasite and ettringite, Cement and Concrete Research, 32 (2002) 719-730.
[67] ÖNORM EN 197-1, in: Zement – Teil 1: Zusammensetzung, Anforderungen und Konformitätskriterien von Normalzement, 2011.
[68] ÖNORM EN 12620: Gesteinskörnungen für Beton, 2013. [69] ÖNORM EN 1008: Zugabewasser von Beton, 2002. [70] ÖNORM EN 206-1: Beton – Teil 1: Festlegung, Eigenschaften, Herstellung und Konformität,
2005. [71] ÖNORM B 4710-1: Beton – Teil 1: „Festlegung, Herstellung, Verwendung und
Konformitätsnachweis, 2007. [72] ÖNORM B 3305: Betonangreifende Wässer Böden und Gase – Beurteilung und chemische
Analyse, 1972. [73] ÖNORM B 5017: Hochleistungsbeton im Siedlungswasserbau (HL-SW-Beton) – Herstellung,
Verwendung und Gütenachweis, 2000. [74] ÖNORM B 4200-10: Beton Herstellung und Überuradiung, 1983. [75] F. Mittermayr, D. Klammer, S. Köhler, M. Böttcher, A. Leis, M. Dietzel, Sulfatangriff: Die Bildung
von Thaumasit und die Auflösung von dolomitischen Zuschlagstoffen, in: 17 IBAUSIL, F.A. Finger Institut für Baustoffkunde, Weimar, 2009.
[76] F. Mittermayr, PhD thesis: Why thaumasite is forming in concrete structures, in: IAG, TU Graz, Graz, 2012, pp. 120.
[77] W. Lukas, H. Huber, Sulfatbeständiger Spritzbeton für Straßentunnelauskleidungen, in: Schriftenreihe Straßenforschung des Bundesministerium für Bauten und Technik, Heft 192,

103
1982, pp. 117. [78] J. Tritthart, D. Klammer, F. Mittermayr, A. Brunnsteiner, A Casestudy of Thaumasite Formation
in an Austrian Tunnel, in: 13th International Congress on the Chemistry of Cement, Madrid 3-8 July 2011, 2011, pp. 1-6.
[79] J. Tritthart, Chloride binding in cement II. The influence of the hydroxide concentration in the pore solution of hardened cement paste on chloride binding, Cement and Concrete Research, 19 (1989) 683-691.
[80] J. Tritthart, Chloride binding in cement I. Investigations to determine the composition of porewater in hardened cement, Cement and Concrete Research, 19 (1989) 586-594.
[81] J. Tritthart, Choridinduzierte Betonstahlkorrosion, in: Schriftenreihe Straßenforschung des BMfwA, Heft 346, 1988, pp. 117.
[82] J. Tritthart, Changes in pore water composition and in total chloride content at different levels of cement paste plates under different storage conditions, Cement and Concrete Research, 22 (1992) 129-138.
[83] J. Tritthart, Korrosionsrelevante Mechanismen und Beurteilungskriterien von chloridverseuchtem Stahlbeton (Corrosion relevant mechanisms and criteria for the assessment of corrosion risk of Cl-contaminated concrete), in: Schriftenreihe Straßenforschung des BMfwA, Heft 414, 1993, pp. 131.
[84] F. Mittermayr, D. Klammer, D. Höllen, S. Köhler, M. Böttcher, A. Leis, M. Dietzel, Deterioration of concrete – application of stable isotopes, in: 10th International Congress for Applied Mineralogy, Trondheim, 2011.
[85] M. Dietzel, F. Mittermayr, D. Klammer, D. Höllen, S. Köhler, A. Leis, What do Stable Isotopes tell us about Deterioration of Concrete, in: 13th International Congress on the Chemistry of Cement, Madrid, 2011, pp. 1-6.
[86] F. Mittermayr, D. Klammer, S. Köhler, A. Leis, D. Höllen, M. Dietzel, Dissolution of Dolomite in alkaline cementitious media, in: Á. Palomo (Ed.) 13th International Congress on the Chemistry of Cement, CSIC, Madrid, 2011, pp. 1-6.
[87] H. Haynes, R. O'Neill, M. Neff, P.K. Mehta, Salt weathering distress on concrete exposed to sodium sulfate environment, ACI Mater J, 105 (2008) 35-43.
[88] G.W. Scherer, Stress from crystallization of salt, Cement and Concrete Research, 34 (2004) 1613-1624.
[89] I.K. Iden, P. Hagelia, C, O and S isotopic signatures in concrete which have suffered thaumasite formation and limited thaumasite form of sulfate attack, Cement and Concrete Composites, 25 (2003) 839-846.
[90] F. Goetz-Neunhoeffer, J. Neubauer, P. Schwesig, Mineralogical characteristics of Ettringites synthesized from solutions and suspensions, Cement and Concrete Research, 36 (2006) 65-70.
[91] W.J. French, Presidential Address 2003: Why concrete cracks - geological factors in concrete failure, Proc Geol Assoc, 116 (2005) 89-105.
[92] D.W. Hobbs, Thaumasite sulfate attack in field and laboratory concretes: implications for specifications, Cem Concr Compos, 25 (2003) 1195-1202.
[93] D.W. Hobbs, M.G. Taylor, Nature of the thaumasite sulfate attack mechanism in field concrete, Cement and Concrete Research, 30 (2000) 529-533.
[94] J. Aguilera, M.T.B. Varela, T. Vázquez, Procedure of synthesis of thaumasite, Cement and Concrete Research, 31 (2001) 1163-1168.
[95] F.P. Glasser, J. Marchand, E. Samson, Durability of concrete — Degradation phenomena involving detrimental chemical reactions, Cement and Concrete Research, 38 (2008) 226-246.
[96] H. Justnes, Thaumasite formed by sulfate attack on mortar with limestone filler, Cement and Concrete Composites, 25 (2003) 955-959.
[97] D.E. Macphee, S.J. Barnett, Solution properties of solids in the ettringite--thaumasite solid solution series, Cement and Concrete Research, 34 (2004) 1591-1598.
[98] S. Sahu, S. Badger, N. Thaulow, Mechanism of thaumasite formation in concrete slabs on grade in Southern California, Cement and Concrete Composites, 25 (2003) 889-897.
[99] S.M. Torres, J.H. Sharp, R.N. Swamy, C.J. Lynsdale, S.A. Huntley, Long term durability of Portland-limestone cement mortars exposed to magnesium sulfate attack, Cement and Concrete Composites, 25 (2003) 947-954.
[100] S. Sahu, S. Badger, N. Thaulow, Evidence of thaumasite formation in Southern California concrete, Cement and Concrete Composites, 24 (2002) 379-384.
[101] E. Gruyaert, P. Van den Heede, M. Maes, N. De Belie, Investigation of the influence of blast-

104
furnace slag on the resistance of concrete against organic acid or sulphate attack by means of accelerated degradation tests, Cement and Concrete Research, 42 (2012) 173-185.
[102] E. Gruyaert, P. Van den Heede, N. De Belie, Carbonation of slag concrete: Effect of the cement replacement level and curing on the carbonation coefficient – Effect of carbonation on the pore structure, Cement and Concrete Composites, 35 (2013) 39-48.
[103] E. Gruyaert, M. Maes, N. De Belie, Performance of BFS concrete: k-Value concept versus equivalent performance concept, Constr Build Mater, 47 (2013) 441-455.
[104] ÖNORM B 3131: Gesteinskörnungen für Beton, 2010, pp. 16. [105] ÖNORM EN 934-1: Zusatzmittel für Beton, Mörtel und Einpressmörtel Teil 1: Gemeinsame
Anforderungen, 2008. [106] ÖNORM EN 934-2: Zusatzmittel für Beton, Mörtel und Einpressmörtel Teil 2: Betonzusatzmittel
- Definitionen, Anforderungen, Konformität, Kennzeichnung und Beschriftung, 2006.







