



Bibliografische Informationen der Deutschen Bibliothek
Die Deutsche Bibliothek verzeichnet diese Publikation in der Deutschen Nationalbibliografie;
Detaillierte bibliografische Daten sind im Internet über http://dnb.ddb.de abrufbar.
1. Auflage 2006
© 2006 by Verlag: Deutsche Veterinärmedizinische Gesellschaft Service GmbH, Gießen
Printed in Germany
ISBN 3-938026-83-9
Verlag: DVG Service GmbH
Frankfurter Straße 89
35392 Gießen
0641/24466
www.dvg.net

Aus dem
Institut fur Virologie
der Tierarztlichen Hochschule Hannover
Entwicklung einer real-time multiplex multitube
RT-PCR zur Differentialdiagnostikder Klassischen Schweinepest
INAUGURAL-DISSERTATION
Zur Erlangung des Grades
eines Doktors der Veterinarmedizin
(Dr. med. vet.)
durch die Tierarztliche Hochschule Hannover
Vorgelegt von
Andreas Gavrilenko
aus Anapskaja
Hannover 2006

Wissenschaftliche Betreuung: Prof. Dr. V. Moennig
1. Gutachter: Prof. Dr. V. Moennig
2. Gutachter: Prof. Dr. B. Schierwater
Tag der mundlichen Prufung: 23.05.2006

Meiner Familie


Inhaltsverzeichnis
1 Einleitung 1
2 Literaturubersicht 3
2.1 Klassische Schweinepest . . . . . . . . . . . . . . . . . . . . . . . . . . . . 3
2.1.1 Das Virus der Klassischen Schweinepest . . . . . . . . . . . . . . . 4
2.1.1.1 Taxonomie . . . . . . . . . . . . . . . . . . . . . . . . . . 4
2.1.1.2 Morphologie . . . . . . . . . . . . . . . . . . . . . . . . . . 4
2.1.1.3 Genomorganisation und virale Proteine . . . . . . . . . . . 4
2.1.2 Klinik und Pathologie . . . . . . . . . . . . . . . . . . . . . . . . . 8
2.2 Diagnostik der Klassischen Schweinepest . . . . . . . . . . . . . . . . . . . 9
2.2.1 Virus- und Antigennachweis . . . . . . . . . . . . . . . . . . . . . . 9
2.2.2 Antikorpernachweis . . . . . . . . . . . . . . . . . . . . . . . . . . . 10
2.3 Differentialdiagnostik der KSP . . . . . . . . . . . . . . . . . . . . . . . . . 10
2.3.1 Afrikanische Schweinepest . . . . . . . . . . . . . . . . . . . . . . . 12
2.3.1.1 Das Virus der Afrikanischen Schweinepest . . . . . . . . . 12
2.3.1.1.1 Taxonomie, Morphologie und physikalische Ei-
genschaften . . . . . . . . . . . . . . . . . . . . . 12
2.3.1.1.2 Genomorganisation und virale Proteine . . . . . . 12

2.3.1.2 Klinik und Pathologie . . . . . . . . . . . . . . . . . . . . 13
2.3.1.3 Diagnostik . . . . . . . . . . . . . . . . . . . . . . . . . . 14
2.3.2 Aujeszkysche Krankheit . . . . . . . . . . . . . . . . . . . . . . . . 15
2.3.2.1 Suides Herpesvirus Typ 1 . . . . . . . . . . . . . . . . . . 15
2.3.2.1.1 Taxonomie und Morphologie . . . . . . . . . . . . 15
2.3.2.1.2 Genomorganisation und virale Proteine . . . . . . 15
2.3.2.2 Klinik und Pathologie . . . . . . . . . . . . . . . . . . . . 17
2.3.2.3 Diagnostik . . . . . . . . . . . . . . . . . . . . . . . . . . 17
2.3.3 Porzines reproduktives und respiratorisches Syndrom . . . . . . . . 18
2.3.3.1 Das porzine reproduktive und respiratorische Syndrom Virus 18
2.3.3.1.1 Taxonomie und Morphologie . . . . . . . . . . . . 18
2.3.3.1.2 Genomorganisation und virale Proteine . . . . . . 18
2.3.3.2 Klinik und Pathologie . . . . . . . . . . . . . . . . . . . . 19
2.3.3.3 Diagnostik . . . . . . . . . . . . . . . . . . . . . . . . . . 20
2.3.4 Porzine Influenza . . . . . . . . . . . . . . . . . . . . . . . . . . . . 21
2.3.4.1 Das Influenza-A-Virus . . . . . . . . . . . . . . . . . . . . 21
2.3.4.1.1 Taxonomie und Morphologie . . . . . . . . . . . . 21
2.3.4.1.2 Genomorganisation und virale Proteine . . . . . . 21
2.3.4.2 Klinik und Pathologie . . . . . . . . . . . . . . . . . . . . 22
2.3.4.3 Diagnostik . . . . . . . . . . . . . . . . . . . . . . . . . . 23
2.3.5 Porzine Parvovirus - Infektionen . . . . . . . . . . . . . . . . . . . . 23
2.3.5.1 Das Porzine Parvovirus . . . . . . . . . . . . . . . . . . . 23
2.3.5.1.1 Taxonomie und Morphologie . . . . . . . . . . . . 23

2.3.5.1.2 Genomorganisation und virale Proteine . . . . . . 24
2.3.5.2 Klinik und Pathologie . . . . . . . . . . . . . . . . . . . . 25
2.3.5.3 Diagnostik . . . . . . . . . . . . . . . . . . . . . . . . . . 26
2.3.6 Porzine Circovirus-Infektionen . . . . . . . . . . . . . . . . . . . . . 27
2.3.6.1 Das Porzine Circovirus . . . . . . . . . . . . . . . . . . . . 27
2.3.6.1.1 Taxonomie und Morphologie . . . . . . . . . . . . 27
2.3.6.1.2 Genomorganisation und virale Proteine . . . . . . 27
2.3.6.2 Klinik und Pathologie . . . . . . . . . . . . . . . . . . . . 29
2.3.6.3 Diagnostik . . . . . . . . . . . . . . . . . . . . . . . . . . 30
2.4 Polymerasekettenreaktion . . . . . . . . . . . . . . . . . . . . . . . . . . . 31
2.4.1 Prinzip . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 31
2.4.2 Reverse Transkription - Polymerasekettenreaktion . . . . . . . . . . 32
2.4.3 Real-time PCR . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 32
2.4.3.1 Prinzip und Techniken . . . . . . . . . . . . . . . . . . . . 32
2.4.3.2 SYBRTM-Green real-time PCR . . . . . . . . . . . . . . . 33
2.4.3.3 TaqMan real-time PCR . . . . . . . . . . . . . . . . . . . 34
2.4.3.4 Primer-probe energy transfer PriProET . . . . . . . . . . 35
2.4.3.5 Quantitative real-time PCR . . . . . . . . . . . . . . . . . 36
2.4.3.5.1 Absolute Quantifizierung . . . . . . . . . . . . . . 37
2.4.3.5.2 Relative Quantifizierung . . . . . . . . . . . . . . 37
2.4.4 Design und Optimierung einer PCR . . . . . . . . . . . . . . . . . . 38
2.4.4.1 Auswahl der nachzuweisenden Genomregionen . . . . . . . 38
2.4.4.2 Primer- und Sondendesign . . . . . . . . . . . . . . . . . . 38

2.4.4.3 Ermittlung von DNA-Sekundarstrukturen . . . . . . . . . 39
2.4.4.4 Optimierungsschritte . . . . . . . . . . . . . . . . . . . . . 40
2.4.5 Multiplex real-time PCR . . . . . . . . . . . . . . . . . . . . . . . . 41
2.4.6 Interne Kontrollen . . . . . . . . . . . . . . . . . . . . . . . . . . . 43
2.5 Prinzipien der Validierung von Diagnosetests . . . . . . . . . . . . . . . . . 43
2.5.1 Allgemein . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 43
2.5.2 Validierung von PCR . . . . . . . . . . . . . . . . . . . . . . . . . . 44
3 Material und Methoden 47
3.1 Material . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
3.1.1 Zelllinien . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
3.1.1.1 PK(15)A . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
3.1.1.2 EFN-R . . . . . . . . . . . . . . . . . . . . . . . . . . . . 47
3.1.1.3 MARC-145 . . . . . . . . . . . . . . . . . . . . . . . . . . 48
3.1.1.4 Rie 255 . . . . . . . . . . . . . . . . . . . . . . . . . . . . 48
3.1.2 Untersuchungsmaterial . . . . . . . . . . . . . . . . . . . . . . . . . 48
3.1.2.1 Virusisolate und klinische Proben . . . . . . . . . . . . . . 48
3.1.2.2 Plasmide . . . . . . . . . . . . . . . . . . . . . . . . . . . 51
3.1.2.3 Antikorper und weitere Materialien . . . . . . . . . . . . . 52
3.1.2.4 Kits zur Nukleinsaureisolierung . . . . . . . . . . . . . . . 52
3.2 Methoden . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 52
3.2.1 Klassische virologische Methoden . . . . . . . . . . . . . . . . . . . 52
3.2.1.1 Zellkultur . . . . . . . . . . . . . . . . . . . . . . . . . . . 52
3.2.1.1.1 PK(15)A . . . . . . . . . . . . . . . . . . . . . . 52

3.2.1.1.2 EFN-R . . . . . . . . . . . . . . . . . . . . . . . 53
3.2.1.1.3 MARC-145 . . . . . . . . . . . . . . . . . . . . . 53
3.2.1.2 Virustitration . . . . . . . . . . . . . . . . . . . . . . . . . 53
3.2.1.3 Peroxidase-Linked-Assay . . . . . . . . . . . . . . . . . . . 53
3.2.1.4 Virusvermehrung . . . . . . . . . . . . . . . . . . . . . . . 54
3.2.2 Molekularbiologische Methoden . . . . . . . . . . . . . . . . . . . . 55
3.2.2.1 Nukleinsaureisolierung . . . . . . . . . . . . . . . . . . . . 55
3.2.2.1.1 RNA Isolierung aus Zellkultur und Serum . . . . 55
3.2.2.1.2 RNA-Isolierung aus Organmaterial und Blut . . . 55
3.2.2.1.3 DNA-Isolierung aus Zellkultur und Serum . . . . 56
3.2.2.1.4 DNA Isolierung aus Organmaterial und Blut . . . 57
3.2.2.2 Reverse Transkription . . . . . . . . . . . . . . . . . . . . 58
3.2.2.3 Primer- und Sondendesign . . . . . . . . . . . . . . . . . . 58
3.2.2.3.1 Sequenzsuche und Alignment . . . . . . . . . . . 58
3.2.2.3.2 Auswahl von Primern und Sonden . . . . . . . . 59
3.2.2.3.3 Evaluierung von Primern und Sonden . . . . . . . 59
3.2.2.4 Polymerasekettenreaktion . . . . . . . . . . . . . . . . . . 61
3.2.2.4.1 Gel-basierte PCR . . . . . . . . . . . . . . . . . . 61
3.2.2.4.2 SYBRTM-Green real-time PCR . . . . . . . . . . 61
3.2.2.4.3 TaqMan real-time PCR . . . . . . . . . . . . . . 63
3.2.2.4.4 Positive und negative Kontrollen . . . . . . . . . 64
3.2.2.5 Agarose-Gelelektrophorese . . . . . . . . . . . . . . . . . . 64
3.2.2.6 Reinigung der PCR-Fragmente . . . . . . . . . . . . . . . 65

3.2.2.7 TA-Klonierung . . . . . . . . . . . . . . . . . . . . . . . . 66
3.2.2.8 Kolonie-PCR . . . . . . . . . . . . . . . . . . . . . . . . . 67
3.2.2.9 Plasmidpraparation . . . . . . . . . . . . . . . . . . . . . 68
3.2.2.10 Restriktionsenzymverdau . . . . . . . . . . . . . . . . . . 69
3.2.2.11 Dephosphorylierung . . . . . . . . . . . . . . . . . . . . . 69
3.2.2.12 Sequenzierung . . . . . . . . . . . . . . . . . . . . . . . . 70
3.2.2.13 Bestimmung der Kopienzahl der Plasmide . . . . . . . . . 70
4 Ergebnisse 73
4.1 Ergebnisse der Literaturrecherche . . . . . . . . . . . . . . . . . . . . . . . 73
4.2 Entwicklung der multiplex real-time TaqMan PCRs . . . . . . . . . . . . . 78
4.2.1 Ergebnisse der Sequenz-, Primer- und Sondensuche . . . . . . . . . 78
4.2.2 Entwicklung der TaqMan PCRs . . . . . . . . . . . . . . . . . . . . 84
4.2.2.1 Gelbasierte PCRs . . . . . . . . . . . . . . . . . . . . . . . 84
4.2.2.2 Sequenzierung der PCR Produkte . . . . . . . . . . . . . . 84
4.2.2.3 Optimierung . . . . . . . . . . . . . . . . . . . . . . . . . 84
4.2.2.3.1 Primertitration . . . . . . . . . . . . . . . . . . . 84
4.2.2.3.2 Sondentitration . . . . . . . . . . . . . . . . . . . 90
4.2.2.4 Entwicklung interner Kontrollen . . . . . . . . . . . . . . . 90
4.2.2.5 Multiplex real-time PCRs . . . . . . . . . . . . . . . . . . 92
4.3 Validierung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 94
4.3.1 Sensitivitat . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 94
4.3.2 Spezifitat . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 95
4.3.3 Klinische Proben . . . . . . . . . . . . . . . . . . . . . . . . . . . . 98

5 Diskussion 105
5.1 Gesamtkonzept . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 105
5.2 Auswahl der Nachweismethode . . . . . . . . . . . . . . . . . . . . . . . . . 106
5.3 Literaturrecherche und Auswahl der Erreger . . . . . . . . . . . . . . . . . 108
5.3.1 Auswahl der Genomregionen . . . . . . . . . . . . . . . . . . . . . . 109
5.4 Aufteilung des multitube PCR-Ansatzes . . . . . . . . . . . . . . . . . . . . 112
5.5 Nukleinsaureisolierung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 113
5.6 Optimierung der PCRs . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 114
5.6.1 Optimierung der Reaktionsbedingungen . . . . . . . . . . . . . . . 114
5.6.2 Optimierung der Primerkonzentrationen . . . . . . . . . . . . . . . 115
5.6.3 Optimierung der Sondenkonzentrationen . . . . . . . . . . . . . . . 115
5.6.4 Real-time PCR Effizienzen . . . . . . . . . . . . . . . . . . . . . . . 115
5.7 Auswahl der internen Kontrollen . . . . . . . . . . . . . . . . . . . . . . . 116
5.8 Auswahl der Isolate und Organproben . . . . . . . . . . . . . . . . . . . . 117
5.9 Validierung . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 119
5.9.1 Sensitivitat . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 120
5.9.2 Spezifitat . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 122
5.9.3 Klinische Proben . . . . . . . . . . . . . . . . . . . . . . . . . . . . 125
5.10 Schlussfolgerung und Ausblick . . . . . . . . . . . . . . . . . . . . . . . . . 128
6 Zusammenfassung 131
7 Summary 133
Literaturverzeichnis 135

A Anhang 177
A.1 Verwendete Virusisolate, Organ- und Blutproben, Antikorper . . . . . . . . 177
A.2 Enzyme und kommerzielle Kits . . . . . . . . . . . . . . . . . . . . . . . . 178
A.3 Medien, Puffer, Losungen, Antibiotika . . . . . . . . . . . . . . . . . . . . 180
A.3.1 Zellkulturmedien . . . . . . . . . . . . . . . . . . . . . . . . . . . . 180
A.3.2 Puffer und Losungen . . . . . . . . . . . . . . . . . . . . . . . . . . 183
A.4 Sonstige Reagenzien . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 184
A.5 Primer- und Sondensequenzen . . . . . . . . . . . . . . . . . . . . . . . . . 184
A.6 Gerate und Verbrauchsmittel . . . . . . . . . . . . . . . . . . . . . . . . . 184
A.6.1 Gerate . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 184
A.6.2 Verbrauchsmittel . . . . . . . . . . . . . . . . . . . . . . . . . . . . 186
A.7 Software . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 186
A.8 Tabellen und Abbildungen . . . . . . . . . . . . . . . . . . . . . . . . . . . 187
Abbildungsverzeichnis 203
Tabellenverzeichnis 205

Abkurzungsverzeichnis
A. bidest. Aqua bidestillata
AK Aujeszkysche Krankheit
AP Alkalische Phosphatase
AS Aminosaure
ASP Afrikanische Schweinepest
ASPV Virus der Afrikanischen Schweinepest
ATP Adenosintriphosphat
ATV adjusted versen-trypsin
BD Border Disease
BDV Border Disease-Virus
BHV-1 Bovines Herpesvirus Typ 1
bp Basenpaare
BSA Bovines Serumalbumin
BVD Bovine Virusdiarrhoe
C Reaktionszyklus (in der real-time PCR)
cDNA komplementare DNA
Ct Schwellenwert (cycle threshold)
DEPC Diethylpyrocarbonat
DMEM Dulbecco’s modified Eagle medium
DNA Desoxyribonukleinsaure
dNTPs Desoxynukleosid-Triphosphate
dpi Tage post infectionem
dRn (∆Rn) baseline-korrigierte, normalisierte Fluoreszenz
dsDNA doppelstrangige DNA

D-SN diagnostische Sensitivitat
D-SP diagnostische Spezifitat
DTT Dithiothreitol
EAV Virus der infektiosen equinen Arteritis
E. coli Escherichia coli
EDTA Ethylendiamintetraessigsaure
ELISA”enzyme-linked immuno sorbent assay“
EMEM Eagle’s Minimum Essential Medium
EURL EU Referenzlabor (EU Reference Laboratory)
FAT fluorescent antibody test
FKS fetales Kalberserum
G/C Guanin/Cytosin
HA Hamagglutination
HAE hamagglutinierende Einheiten
IK interne Kontrolle
IPTG Isopropyl-thio-galaktosid
KID50 kulturinfektiose Dosis50
kb Kilobasen
KSP Klassische Schweinepest
KSPV Virus der Klassischen Schweinepest
LDV lactate-dehydrogenase elevating-Virus
mAk monoklonaler Antikorper
MES 2(N-Morpholino)ethansulfonsaure
min Minute
M-Protein Membran-Protein
mRNA”messenger RNA“
N/D nicht durchgefuhrt
N-Protein Nukleokapsid-Protein
NTR nichttranslatierte Region
OD optische Dichte
OIE Office International des Epizooties
ORF offener Leserahmen (open reading frame)

PAM porzine alveolare Makrophagen
PBS Phosphat-gepufferte Kochsalzlosung
PBSM PBS ohne Calcium und Magnesium
PCR Polymerasekettenreaktion (polymerase chain reaction)
PCV-1 Porcines Circovirus Typ 1
PCV-2 Porcines Circovirus Typ 2
PDNS porzines Dermatitis und Nephropathie Syndrom
PFU Plaque-bildende Einheiten (plaque forming units)
p.i. post infectionem, nach Infektion
PK porcine kidney
PMWS postweaning multisystemic wasting syndrome
PPV Porzines Parvovirus
PRDC porcine respiratory disease complex
PRRS Porzines reproduktives und respiratorisches Syndrom
PRRSV Porzines reproduktives und respiratorisches Syndrom Virus
PRRSV-EU PRRSV europaische Stamme
PRRSV-NA PRRSV nordamerikanische Stamme
R native Fluoreszenz
R2 Bestimmheitsmaß, Pearson Correlation Coefficient
Rn Fluoreszenz abgeglichen mit einem Referenzfarbstoff
RNA Ribonukleinsaure
RT Reverse Transkriptase, reverse Transkription
sec Sekunde
SHV-1 Suides Herpesvirus Typ 1
SMEDI stillbirth, mummification, embryonic death, infertility
SPF spezifisch pathogenfrei
TCID zellkulturinfektiose Dosis (tissue culture infectious dose)
TBE Tris-Borat-EDTA
TiHo Tierarztliche Hochschule
Tm Schmelztemperatur
TRIS Tris-Hydroxymethylaminomethan
U Einheit (unit)

u. a. unter anderem
VNT Virusneutralisationstest
WWW World Wide Web (Internet)

1. Einleitung
Die Klassische Schweinepest (KSP) wird durch ein behulltes RNA-Virus aus dem Genus
Pestivirus der Familie Flaviviridae verursacht und gehort weltweit zu den wichtigsten vi-
ralen Erkrankungen der Schweine. Die KSP ist eine anzeigepflichtige Seuche. Ausbruche
der KSP haben in den letzten Jahrzehnten enorme wirtschaftliche Verluste in Europa
verursacht.
Das klinische Bild einer KSPV-Infektion ist sehr variabel und haufig unspezifisch. Daher
kommen viele andere Erkrankungen viraler, bakterieller und nicht-infektioser Genese dif-
ferentialdiagnostisch in Frage. Zu den viralen Differentialdiagnosen gehoren Afrikanische
Schweinepest (ASP), porzine Circovirusinfektionen, porzine Parvovirose, porzine Influ-
enza, Infektionen mit dem porzinen respiratorischen und reproduktiven Syndrom Virus
(PRRSV) und Aujeszkysche Krankheit.
Die Vielzahl moglicher Differentialdiagnosen macht die Diagnose aufgrund des klinischen
Bildes nahezu unmoglich. Daher ist sowohl fur die Uberwachung der Schweinepopulation
als auch zur Detektion eines KSP-Ausbruchs eine moderne und effiziente Labordiagno-
stik von herausragender Bedeutung. Zu den schnellsten und empfindlichsten Methoden
des Virusgenomnachweises gehort die Polymerasekettenreaktion (PCR). Die technischen
Entwicklungen der letzten Jahre machten es moglich, verschiedene Erreger in einer so ge-
nannten”multiplex“ PCR nachzuweisen und durch die Nutzung fluoreszierender Sonden
”real-time“ schnell und effizient sichtbar zu machen.
Ziel der vorliegenden Arbeit war es eine moderne und schnelle Nachweismethode differen-
tialdiagnostisch relevanter viraler Erreger auf der Grundlage dieser Technik zu entwickeln.
1


2. Literaturubersicht
2.1 Klassische Schweinepest
Die KSP ist eine der wichtigsten Tierseuchen weltweit und ist in die Liste der Erkrankun-
gen eingruppiert, die bei der OIE und der EU anzeigepflichtig sind (Anonym, 2005a).
Auf nationaler Ebene besteht in Deutschland ebenfalls Anzeigepflicht. Das Virus der Klas-
sischen Schweinepest (KSPV) ist weltweit verbreitet, konnte jedoch in einigen Landern
erfolgreich getilgt werden. So sind z. B. Australien, Neuseeland und die USA KSPV frei
(van Oirschot, 1999a; Edwards, 2000). Aktuelle Informationen zum Seuchenstatus
und -geschehen konnen dem OIE Handistatus II 1 entnommen werden. Die erste offizielle
Feststellung der KSP wurde wahrscheinlich in Ohio, USA, im Jahre 1833 gemacht, wie
aus einem Bericht des Bureau of Animal Industry (United States Department of Agri-
culture) aus dem Jahre 1887 hervorgeht (Liess, 1981). Der genaue Ursprung der Seuche
ist unbekannt (van Oirschot, 1999b). Die Erregercharakterisierung als filtrierbares Vi-
rus erfolgte durch de Schweinitz und Dorset (1904). Naturliche Wirte des KSPV
sind ausschließlich Haus- und Wildschweine (Liess, 1981; Laddomada, 2000), es wurde
allerdings auch von subklinischen Infektionen bei anderen Tieren wie Kalbern, Ziegen,
Schafen, Hirschen und Pekaris berichtet (Loan und Storm, 1968).
1http://www.oie.int/hs2/report.asp
3

4 2. Literaturubersicht
2.1.1 Das Virus der Klassischen Schweinepest
2.1.1.1 Taxonomie
Das Virus der Klassischen Schweinepest (KSPV) gehort zum Genus Pestivirus (Horzinek,
1973; Westaway et al., 1985) in der Familie Flaviviridae (Wengler, 1991; Pringle,
1999). Zu diesem Genus gehoren daruber hinaus die eng antigenverwandten Viren der Bo-
vinen Virusdiarrhoe (BVDV) und der Border Disease (BDV) der Schafe (Darbyshire,
1960; Horzinek et al., 1967; Enzmann und Rehberg, 1977; Ridpath und Bolin,
1997). Die Familie Flaviviridae umfasst neben dem Genus Pestivirus die Genera Flavi-
virus und Hepacivirus (Rice et al., 1985). Letzterem gehort ausschließlich das humane
Hepatitis-C-Virus an (Bartenschlager und Lohmann, 2000). Angehorige des Genus
Flavivirus sind u. a. das Gelbfiebervirus und das Fruhsommer-Meningoenzephalitis-Virus
des Menschen, das Louping-Ill-Virus, das Wesselsbron-Disease-Virus der Schafe sowie das
Meningoenzephalitis-Virus der Pute (Wengler et al., 1995).
2.1.1.2 Morphologie
Das Virus der KSP ist ein behulltes RNA-Virus von spharischer Gestalt und misst im
Durchmesser zwischen 40 und 60 nm. Drei strukturelle Glykoproteine erscheinen als un-
regelmaßig gestaltete Projektionen an der Oberflache (s. Abbildung 2.1) (Horzinek et al.,
1967; Moormann und Hulst, 1988; Meyers et al., 1989; Moennig und Plagemann,
1992).
2.1.1.3 Genomorganisation und virale Proteine
Das Genom der Pestiviren, dessen Lange ca. 12,5 Kilobasen betragt, liegt als ein ein-
zelstrangiges, lineares RNA-Molekul in der Plusstrangorientierung vor (Moormann und
Hulst, 1988). Dabei kodiert ein einzelner offener Leserahmen (ORF - open reading frame)
fur ein Polyprotein mit einer Lange von etwa 3900 Aminosauren (Collett et al., 1988;
Meyers et al., 1989; Rumenapf et al., 1993). Das Polyprotein wird co- und posttrans-
lationell in vier Struktur- und sieben Nichtstrukturproteine prozessiert (Elbers et al.,
1996; Meyers und Thiel, 1996). Das Genom wird am 3′- und 5′-Ende von nichttrans-

2.1. Klassische Schweinepest 5
Abbildung 2.1: Schematische Darstellung des Virus der Klassischen Schweinepest.
latierten Regionen (NTR) flankiert (Moormann und Hulst, 1988; Rumenapf et al.,
1989; Moormann et al., 1990; Deng und Brock, 1992) (s. Abbildung 2.2). Die Lange
der 5′NTR betragt bei Pestiviren 372 bis 385 Basen (Becher et al., 1998), die der 3′NTR
etwa 200 Basen. Die beiden NTRs sind unter den Pestiviren hoch konserviert (Meyers
et al., 1989; Moormann et al., 1990; Hofmann et al., 1994; Becher et al., 1995;
Vilcek et al., 1997). So betragt die Identitat im 5′NTR-Bereich zwischen BDV-Isolaten
und KSPV-Isolaten bis zu 80 %, zwischen BDV-Isolaten und BVDV-1-Isolaten bis 65 %
(Vilcek et al., 1997).
Durch Sequenzvergleiche bestimmter Genomabschnitte lasst sich das Virus der klassi-
schen Schweinepest unter Einbeziehung geographischer und epidemiologischer Daten in
Abbildung 2.2: Schematische Darstellung des Pestivirusgenoms. Modifiziert nach
Meyers und Thiel (1996)

6 2. Literaturubersicht
verschiedene genetische Gruppen einteilen (s. Abbildung 2.3) (Lowings et al., 1996;
Greiser-Wilke et al., 1998; Paton et al., 2000b). Basierend auf der Einteilung von
Lowings et al. (1996), wurden drei Gruppen mit jeweils drei bzw. vier Untergruppen
gebildet (Paton et al., 2000b). Die Gruppe 1 mit den Untergruppen 1.1 bis 1.3 beinhal-
tet Isolate aus den 40–60er Jahren, Isolate aus der Ukraine, Kroatien, Mexiko, Thailand
und China aus den 90er Jahren des vergangenen Jahrhunderts, sowie ein Isolat aus dem
Jahr 2004 aus Kolumbien. Die Gruppe 2 mit den Untergruppen 2.1 bis 2.3 beinhaltet
westeuropaische Isolate aus den 1980er und 1990er Jahren, sowie asiatische Isolate aus
den Jahren 1980–2004. Die Gruppe 3 mit den Untergruppen 3.1 bis 3.4 enthalt ein Iso-
lat aus Großbritannien, sowie Isolate aus Thailand, Japan, Korea und Taiwan aus den
1970–1990er Jahre (Lowings et al., 1996; Paton et al., 2000b; Greiser-Wilke et al.,
2005).

2.1.K
lassische
Sch
wein
epest
7
Abbildung 2.3: Genetische Diversitat der KSPV-Isolate. Die Einteilung basiert auf den Sequenzen des E2-gp Gens. Paton et al.
(2000a), vervollstandigt von Greiser-Wilke et al.

8 2. Literaturubersicht
2.1.2 Klinik und Pathologie
Die Erscheinungsformen der KSP sind sehr unterschiedlich, wobei das Alter der Tiere,
die Virusdosis und die Virulenz des Virusstammes sowie Wirtsfaktoren eine Rolle spie-
len (Dahle und Liess, 1995; van Oirschot, 1999b). Es konnen akute, chronische und
pranatale Verlaufsformen unterschieden werden (van Oirschot et al., 1988; Depner
et al., 1997a; Moennig et al., 2003). Junge Tiere zeigen haufig die akute Form der KSP,
die mit hohem Fieber, Anorexie, Konjunktivitis, vergroßerten Lymphknoten, Diarrhoe
und Verstopfung einhergeht. In der Finalphase der Erkrankung kommt es zu petechia-
len und ekchymatosen Blutungen der Haut im Bereich der Ohren, des Schwanzes, am
Bauch und an den Extremitaten (Depner et al., 1997a; Moennig et al., 1993, 2003).
Die Tiere verenden nach acht bis zwanzig Tagen. Die Mortalitat liegt zwischen 20 und
80 %. Die korrespondierenden pathologisch-anatomischen Befunde umfassen stark ver-
großerte Lymphknoten im gesamten Organismus, petechiale und ekchymatose Blutungen
v. a. in den Nieren, der außeren Haut, der Magen-, Blasen- und Gallenblasenwand sowie
Milzrandinfarkte (Trautwein, 1988). Des Weiteren treten durch Sekundarinfektionen
hervorgerufene Veranderungen des Respirationstraktes und eine nichteitrige Enzephalitis
auf (Gruber et al., 1995).
Die chronische Form der KSP endet immer todlich. Die anfanglichen Symptome sind
denen der akuten Form ahnlich. Im weiteren Verlauf werden die Anzeichen immer un-
spezifischer. Die betroffenen Tiere zeigen intermittierendes Fieber, chronische Enteritiden
und Kummern (Moennig et al., 2003). Im pathologisch-anatomischen Bild zeigen chro-
nisch erkrankte Schweine weniger ausgepragte Befunde. Hamorrhagien und Infarkte fehlen
haufig. Zu den typischen Befunden gehoren eine Depletion der lymphatischen Organe so-
wie nekrotische und ulzerative Veranderungen der Darmwand, so genannte”Boutons“
(van Oirschot, 1999b). Außerdem ist eine Hyperplasie der Nebennierenrinde charak-
teristisch (Cheville und Mengeling, 1969; van der Molen und van Oirschot,
1981). Bei tragenden Sauen, die haufig selbst keine Krankheitszeichen zeigen, ruft die
KSPV-Infektion eine Reihe fetaler Veranderungen hervor. Aborte, Totgeburten oder Miß-
bildungen konnen auftreten (Meyer et al., 1980). Persistent infizierte neugeborene Fer-
kel zeigen haufig keine Symptome, scheiden jedoch ihr Leben lang das Virus aus (van
Oirschot und Terpstra, 1977) und sterben letztendlich nach bis zu elf Monaten an

2.2. Diagnostik der Klassischen Schweinepest 9
der so genannten”late-onset“ Form der KSP (Meyer et al., 1981). Das Auftreten und die
Auspragung der klinischen Symptome, vor allem bei der akuten Verlaufsform, sind außerst
variabel, so dass man keines der Krankheitssymptome als pathognomonisch bezeichnen
kann.
2.2 Diagnostik der Klassischen Schweinepest
Die klinische Diagnostik ist v. a. bei alteren Tieren und in Zusammenhang mit moderat
oder schwach virulenten KSPV-Stammen schwierig, daher ist eine rasche und zuverlassige
labordiagnostische Bestatigung außerordentlich wichtig (Paton und Greiser-Wilke,
2003). Fur die labordiagnostische Untersuchung stehen sowohl Methoden zum Virus- und
Antigennachweis als auch serologische Nachweismethoden zur Verfugung. Die Methoden
sind auf Ebene der EU in der Richtlinie 2002/106/EC2 und dem angehangten technischen
Teil weitestgehend verbindlich festgelegt. Auf weltweiter Ebene beinhaltet das Manual of
Diagnostic Tests und Vaccines for Terrestrial Animals (mammals, birds and bees) (im
Weiteren Manual of Diagnostic Tests) des OIE in Kapitel 2.1.13 eine Auflistung und
genaue Erlauterung der Methoden3.
2.2.1 Virus- und Antigennachweis
Der Goldstandard des Virusnachweises ist nach wie vor die Virusisolierung aus Leukozyten
oder Organsuspensionen auf KSPV-permissiven Zellen (Moennig, 2000). Da das Virus
keinen zytopathischen Effekt induziert, erfolgt der Nachweis mit spezifischen Antikorpern
(Moennig, 2000), wobei die Abgrenzung zu anderen Pestiviren mit monoklonalen An-
tikorpern vorgenommen werden kann (Cay et al., 1989; Edwards et al., 1991). Ein
schneller Test ist der Nachweis viralen Antigens in Organschnitten mittels fluoreszieren-
der Antikorper (fluorescent antibody test, FAT) (Solorzano et al., 1966; Turner et al.,
1968; Bouma et al., 2001; Anonym, 2004a; Teifke et al., 2005). Der FAT ist weniger sen-
sitiv und bedarf geschulten und erfahrenen Personals (Moennig, 2000). In ahnlicher Wei-
se kann der Nachweis auch mittels Immunperoxidasefarung erfolgen (Anonym, 2004a).
2http://europa.eu.int/eur-lex/pri/en/oj/dat/2002/l_039/l_03920020209en00710088.pdf3http://www.oie.int/eng/normes/mmanual/A_00036.htm

10 2. Literaturubersicht
Fur Screeningtests großer Probenzahlen wird haufig ein Antigen-ELISA verwendet, der
jedoch im Vergleich zur Virusisolierung weniger sensitiv ist und in der Inkubationszeit, bei
chronisch erkrankten Tieren und bei niedrigen Virustitern zu falsch negativen Ergebnissen
fuhren kann (Kaden et al., 1999). Der Nachweis viraler RNA mittels RT-PCR ist eine
schnelle und sensitive Nachweismoglichkeit, die in den letzten Jahren an Bedeutung ge-
wonnen hat (Paton et al., 2000a; Moennig, 2000; Paton und Greiser-Wilke, 2003).
In den vergangenen Jahren wurden viele verschiedene PCR-Ansatze entwickelt und teil-
weise fur die Diagnostik validiert (Sandvik et al., 1997; de A. Diaz et al., 1998; Thur
und Hofmann, 1998; McGoldrick et al., 1998, 1999; Paton et al., 2000a; Hofmann,
2003; Risatti et al., 2003; Handel et al., 2004; van Rijn et al., 2004; Gaede et al.,
2005; Hoffmann et al., 2005; Risatti et al., 2005; Ophuis et al., 2006).
2.2.2 Antikorpernachweis
Der Virusneutralisationstest (VNT) gilt als sensitivster und spezifischster Test zur Detek-
tion von Antikorpern gegen das KSPV. Da kreuzreagierende Antikorper gegen ruminante
Pestiviren Probleme in der Diagnostik bereiten konnen, werden i. d. R. differentialdiag-
nostische VNTs parallel durchgefuhrt (Moennig, 2000). Eine schnelle, jedoch weniger
sensitive Methode ist der Antikorper-ELISA, der fur”screening“-Tests eingesetzt werden
kann (Moennig, 2000).
2.3 Differentialdiagnostik der KSP
Aufgrund des vielfaltigen und unspezifischen klinischen und pathologischen Bildes kom-
men mehrere Erkrankungen viraler, bakterieller und nichtinfektioser Genese als Differen-
tialdiagnosen der KSP in Betracht und mussen im Verdachtsfall labordiagnostisch ausge-
schlossen werden.
Zu den Differentialdiagnosen gehort die akute Form der ASP (Kleiboeker, 2002), die
anhand der klinischen und pathologischen Erscheinungen nicht von der KSP zu unterschei-
den ist. Außerdem kommen Rotlauf, PRRSV-Infektionen, Cumarin-Vergiftungen, Purpura
haemorrhagica, das”Postweaning Multisystemic Wasting“ Syndrom (PMWS), das por-

2.3. Differentialdiagnostik der KSP 11
zine Dermatitis und Nephropathie Syndrom (PDNS), Infektionen mit Salmonellen oder
Pasteurellen sowie jede fieberhafte Erkrankung des Respirations- oder Gastrointestinal-
traktes, die nicht auf Antibiotika reagiert, differentialdiagnostisch in Frage (Anonym,
2004a; Moennig et al., 2003). So konnen beispielsweise SHV-1-, porzine Parvovirus- sowie
Influenza-A Infektionen ahnliche Symptome wie KSP hervorrufen. Desweiteren konnen
auch Salmonellen, Leptospiren, andere Erreger viraler Meningoenzephalitiden und Stre-
potkokken einen ahnlichen Symptomkomplex auslosen (Anonym, 2004a).
Naturlich vorkommende Infektionen von Schweinen mit ruminanten Pestiviren (BVDV,
BDV) zeigen eine weltweite Verbreitung (Liess und Moennig, 1990). In Deutschland
traten u. a. BDV-Infektionen bei Schweinen aus gemischten Bestanden auf, die zu schwer-
wiegenden Problemen in der serologischen Diagnostik fuhrten (Oguzoglu et al., 2001).
Bei Schweinen stellen Infektionen mit ruminanten Pestiviren generell weniger ein Krank-
heitsproblem, als ein diagnostisches Problem bei der serologischen Abrenzung zu Infek-
tionen mit dem KSPV dar (Paton und Done, 1994). In Einzelfallen konnen Infektionen
mit BVDV und BDV auch bei Schweinen zu Fruchtbarkeitsstorungen (Snowdon und
French, 1968) und klinischen Erkrankungen fuhren, die der chronischen bzw.”late-
onset“ Form der KSP ahneln (Terpstra und Wensvoort, 1988; Paton et al., 1994).
Ein Virusnachweis ist selten moglich (Paton et al., 1994).

12 2. Literaturubersicht
2.3.1 Afrikanische Schweinepest
Die ASP wurde erstmals 1921 in Ost-Afrika beschrieben (Montgomery, 1921). Das Vi-
rus der ASP (ASPV) ist das einzige bisher bekannte DNA Arbovirus (Arthropod-borne)
(Brown et al., 1986; Dixon et al., 1995). Das Virus infiziert Vertreter der Familie Suidae,
zu der unter anderem das Hausschwein, das Wildschwein sowie Warzen- und Buschschwei-
ne gehoren. Zusatzlich kann sich das Virus in seinem Vektor, den Zecken des Genus Orni-
thodoros vermehren (Plowright et al., 1974). Die ASP wurde in den meisten Landern
sudlich der Sahara sowie West- und Nordafrikanischen Staaten beobachtet. In Europa ist
die Krankheit nach Seuchenausbruchen in den 60er bis 80er Jahren des vergangenen Jahr-
hunderts getilgt (mit Ausnahme von Sardinien) (Commission Decision 2005/363/EC)4.
Die ASP gehort ebenfalls zu den Erkrankungen, die sowohl national als auch international
anzeigepflichtig sind (Anonym, 2005a).
2.3.1.1 Das Virus der Afrikanischen Schweinepest
2.3.1.1.1 Taxonomie, Morphologie und physikalische Eigenschaften
Das ASPV ist der einzige Vertreter des Genus Asfivirus in der Familie Asfarviridae
(Dixon et al., 2000). Die Viruspartikel des ASPV sind ca. 200 nm groß, behullt und
haben einen ikosaedrischen Aufbau (s. Abbildung 2.4) (Modrow et al., 2003). Anhand
der Restriktionsenzymanalyse der Virus-DNA konnen funf ASPV-Gruppen unterschieden
werden, wobei nur die afrikanischen Stamme eine ausgepragte Diversitat zeigen (Murphy
et al., 1999).
2.3.1.1.2 Genomorganisation und virale Proteine
Das ASP-Virus enthalt eine doppelstrangige DNA mit einer Lange von ca. 170–190 kBp.
Die DNA besitzt kovalent geschlossene Enden mit inversen terminalen Wiederholungen
und Haarnadelschleifen (Murphy et al., 1999). Das Genom kodiert fur uber 200 Proteine,
die noch unzureichend charakterisiert sind (Kleiboeker et al., 1998; Modrow et al.,
2003). Das Hauptkapsidprotein VP72 des ASPV liegt mit einer Ubereinstimmung der
4http://europa.eu.int/eur-lex/lex/LexUriServ/site/en/oj/2005/l_118/l_11820050505en00370038.pdf

2.3. Differentialdiagnostik der KSP 13
aaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaa
Abbildung 2.4: Schematische Darstellung des ASP-Virus. (Nach Modrow et al. (2003)
Aminosaurensequenzen zwischen ASPV-Stammen aus allen Teilen der Welt von 97,8 % bis
100 % hochkonserviert vor und ist antigenetisch stabil. Es eignet sich daher als molekulare
Basis fur serologische und molekularbiologische Testmethoden (Yu et al., 1996).
2.3.1.2 Klinik und Pathologie
Die Virusubertragung erfolgt durch infizierte Zecken bzw. zwischen Hausschweinen so-
wohl vertikal als auch horizontal (Mebus, 1988; Kleiboeker et al., 1998; Moennig
et al., 2003). Der Krankheitsverlauf kann je nach Virulenz des Erregers von perakut bis
subklinisch variieren, wobei es einen Zusammenhang zwischen klinischer Auspragung der
Symptome und der Adaptation des Virusstammes an das Hausschwein gibt (Moennig
et al., 2003). Die akute Form ist durch hohes Fieber (40,5–42 ◦C), Leukopenie, Thrombo-

14 2. Literaturubersicht
zytopenie, Hyperamie und petechiale Blutungen im Bereich der Ohren, des Schwanzes, an
den Extremitaten sowie am Bauch gekennzeichnet. Gastrointestinale Storungen, Aborte
und respiratorische Symptome treten auf (Mebus, 1988; Anonym, 2005c). Der Tod tritt
nach 4–8 Tagen ein, die Mortalitat erreicht dabei fast 100 %. Bei der Sektion fallen ver-
großerte, hamorrhagische Lymphknoten, Splenomegalie und petechiale Blutungen in den
Nieren, der Harnblase und dem Larynx auf. Die subakute Form ist durch weniger dramati-
sche Symptome charakterisiert. Die Mortalitat betragt 30–70 %. Die chronische Form wird
von Gewichtsverlusten, undulierendem Fieber, respiratorischen Symptomen, Arthritiden
sowie Veranderungen der Haut begleitet. Die Mortalitat ist dabei gering (Mebus, 1988;
Anonym, 2005c).
2.3.1.3 Diagnostik
Da ASP klinisch und pathologisch nicht von der KSP zu unterscheiden ist, kommen fur
beide Erkrankungen gleiche Differentialdiagnosen in Frage (s. Kapitel 2.3). Zur Abgren-
zung und Bestatigung ist daher eine Labordiagnose notwendig. Es existieren Methoden
zum Nachweis des infektiosen Virus, viralen Antigens, viraler DNA und spezifischer An-
tikorper. Die empfohlenen Methoden sind im Manual of Diagnostic Tests und Vaccines
for Terrestrial Animals der OIE in Kapitel 2.1.125 festgehalten. Die zellkulturelle Isolie-
rung kann auf primaren porzinen Monozytenkulturen oder Knochenmarkszellen erfolgen,
wobei die Isolierung durch einen Hamadsorptionstest erweitert wird (Malmquist und
HAY, 1960; Moennig et al., 2003). Eine immer großere Rolle spielt in der Diagnostik
der Nachweis der viralen Nukleinsaure mittels PCR (Steiger et al., 1992; Gonzague
et al., 2002; Aguero et al., 2003; King et al., 2003; Bastos et al., 2003; Aguero et al.,
2004; Basto et al., 2005; Hjertner et al., 2005; Zsak et al., 2005). Ein Antigennach-
weis ist sowohl mittels FAT als auch in Antigen-ELISAs moglich (Moennig et al., 2003;
Pastor et al., 1990). Der serologische Nachweis kann im Antikorper-ELISA (Wardley
et al., 1979) oder mittels eines indirekten Immunfluoreszenztests (Moennig et al., 2003)
erfolgen.
5http://www.oie.int/eng/normes/mmanual/A_00035.htm

2.3. Differentialdiagnostik der KSP 15
2.3.2 Aujeszkysche Krankheit
Die Aujeszkysche Krankheit (AK) wurde erstmals 1902 von dem ungarischen Tierarzt
Aujeszky beschrieben (Aujeszky, 1902). Die Erkrankung kann fast alle Saugetiere be-
treffen, verlauft jedoch bei den verschiedenen Spezies unterschiedlich. Das Schwein gilt
als Primarwirt und Reservoir des Virus (Murphy et al., 1999; Teuffert et al., 2005).
Die AK ist weltweit verbreitet und gehort zu den wirtschaftlich bedeutsamsten Virus-
infektionen. Sie spielt noch immer eine wichtige Rolle in den osteuropaischen Landern,
Deutschland dagegen gilt seit 2003 als AK frei. Der letzte Ausbruch wurde in Deutschland
im Jahre 2000 verzeichnet (Teuffert et al., 2005). Wie KSP und ASP befindet sich die
AK auf der Liste der Erkrankungen, die bei der OIE anzeigepflichtig sind. Dem Handi-
status der OIE6 zufolge (Stand am 13.01.2006) trat die AK im Jahr 2004 in mehreren
Landern der EU auf.
2.3.2.1 Suides Herpesvirus Typ 1
2.3.2.1.1 Taxonomie und Morphologie
Der Erreger der AK, das Suide Herpesvirus Typ 1 (SHV-1), gehort dem Genus Varicellovi-
rus der Subfamilie α-Herpesvirinae in der Familie Herpesviridae an (Klupp et al., 2004b),
zu dem auch das Bovine Herpesvirus Typ 1 (BHV-1) und die Equinen Herpesviren Typ 1
und 4 gehoren. Die Virionengroße des SHV-1 betragt 150–170 nm. Im zentralen Kern, wel-
cher von einem ikosaedrischen Kapsid umgeben wird, befindet sich ein ca. 150 kbp langes,
doppelstrangiges DNA (dsDNA) Molekul. Zwischen dem Kapsid und der glykoprotein-
haltigen Lipidmembran befindet sich das aus mindestens 15 Proteinen zusammengesetzte
Tegument (Klupp et al., 2004b).
2.3.2.1.2 Genomorganisation und virale Proteine
Das lineare dsDNA-Genom des SHV-1 kann in ein langes und ein kurzes Segment ge-
gliedert werden, wobei jedes dieser Segmente eine als”Unique Long“- (UL) bzw.
”Unique
Short“(US)- Region bezeichnete, Sequenzfolge besitzt. Diese Regionen werden, wie die des
6http://www.oie.int/hs2/sit_mald_cont.asp?c_mald=21&c_cont=6&annee=2004

16 2. Literaturubersicht
Abbildung 2.5: Schematische Darstellung eines Herpesvirus. Nach Modrow et al. (2003)
Herpes-Simplex-Virus des Menschen, von invertierten Einheiten wiederholter Sequenzen
flankiert (Ben-Porat et al., 1983; Davison und Wilkie, 1983; Klupp et al., 2004a,b).
Es beinhaltet eine Vielzahl von open reading frames (ORF, offener Leserahmen) und ko-
diert fur eine große, bisher noch nicht genau bestimmte Anzahl an Proteinen (Klupp
et al., 2004b). Fur das SHV-1 wurden bisher 12 Glykoproteine identifiziert, die mit gB
bis gN bezeichnet sind (Modrow et al., 2003). Der hohe C/G-Gehalt erschwert eine
Sequenzierung des SHV-1-Genoms, dennoch liegt eine komplette Sequenz des SHV-1 vor
(Klupp et al., 2004a).

2.3. Differentialdiagnostik der KSP 17
2.3.2.2 Klinik und Pathologie
Das SHV-1 hat ein breites Wirtsspektrum, das Schweine, Rinder, kleine Wiederkauer,
Fleischfresser, Kaninchen und Nager umfasst. Der Krankheitsverlauf ist beim Schwein
altersabhangig. Die Virusubertragung findet aerogen statt. Die Saugferkel erkranken akut
bis perakut. Sie zeigen typische zentralnervose Storungen mit Opisthotonus, Ataxien und
Krampfen. Die Mortalitat ist mit bis zu 100 % sehr hoch. Mit fortschreitendem Alter der
Tiere mildert sich der Krankheitsverlauf. Es kommt jedoch auch hier zu zentralnervosen
Storungen und Fieber. Sekundare Infektionen konnen das Krankheitsbild zusatzlich er-
schweren. Altere Schweine zeigen meistens einen subklinischen Krankheitsverlauf. Es kom-
men Fieber und respiratorische Symptome vor. Bei tragenden Sauen ruft die Infektion
mit SHV-1 Aborte, Mumifikationen oder Resorption der Fruchte hervor (Shope, 1931a;
Fraser und Ramachandran, 1969; McCracken et al., 1973; Schmidt et al., 1987).
In pathologisch-anatomischen Untersuchungen treten haufig leicht gestaute Lymphknoten
mit kleinen Blutungen auf. Petechien in den Nieren werden ebenfalls beobachtet (Frey,
2003). Zu den regelmaßig auftretenden Befunden gehoren auch entzundliche Verande-
rungen des oberen Respirationstraktes und ein Lungenodem. Eine Panenzephalitis ist
vor allem mikroskopisch nachweisbar. Insgesamt auffallig ist, dass die makroskopischen
Veranderungen auch bei jungen Tieren nicht sehr ausgepragt sind (Gustafson, 1970;
Murphy et al., 1999).
2.3.2.3 Diagnostik
Zur Diagnostik der Aujeszkyschen Krankheit stehen sowohl direkte, als auch indirekte
Nachweismethoden zur Verfugung. Auch fur die AK gibt es von der OIE empfohlene Me-
thoden, die im Manual of Diagnostic Tests and Vaccines for Terrestrial Animals, Kapitel
2.2.27, schriftlich niedergelegt sind. Die klassische Methode des Virusnachweises stellt die
Virusisolierung dar. Das SHV-1 lasst sich in einer Vielzahl von Zellkulturen unter Aus-
bildung eines deutlichen zytopathischen Effektes anzuchten (Gustafson, 1970). Das Vi-
rusantigen lasst sich außerdem mittels Immunfluoreszenztechniken (Gustafson, 1970),
Immunperoxidasetechniken oder PCR nachweisen (Echeverria et al., 2000; Krumb-
7http://www.oie.int/eng/normes/mmanual/A_00041.htm

18 2. Literaturubersicht
holz et al., 2003; van Rijn et al., 2004; Cao et al., 2005b; Huang et al., 2005; Yoon
et al., 2005). Als indirekte Methode bieten sich ELISA und Virusneutralisationstests an
(Frey, 2003). Besonders bei der serologischen Unterscheidung zwischen geimpften und
infizierten Tieren wird ein ELISA eingesetzt (van Oirschot et al., 1988; Lange et al.,
2003).
2.3.3 Porzines reproduktives und respiratorisches Syndrom
Das porzine reproduktive und respiratorische Syndrom (PRRS) ist eine der zur Zeit welt-
weit bedeutendsten Schweinekrankheiten. Es verursacht in Zucht- und Mastbetrieben ho-
he wirtschaftliche Schaden (Terpstra et al., 1991; Prieto und Castro, 2005). Der
Erreger des PRRS, das PRRS Virus (PRRSV), wurde erstmals 1990 in den Niederlanden
und kurz danach in den USA entdeckt (Wensvoort et al., 1991; Collins et al., 1992).
2.3.3.1 Das porzine reproduktive und respiratorische Syndrom Virus
2.3.3.1.1 Taxonomie und Morphologie
Das PRRSV wird zusammen mit dem Virus der infektiosen equinen Arteritis (EAV),
dem lactate-dehydrogenase elevating-Virus (LDV) der Mause und dem simian hemorrha-
gic fever -Virus (SHFV) in die Familie Arteriviridae, Ordnung Nidovirales eingruppiert
(Meulenberg et al., 1993b; Cavanagh, 1997). Das PRRSV ist behullt, besitzt einen
Durchmesser von 50-65 nm und enthalt ein 25-35 nm großes Nukleokapsid (Benfield
et al., 1992; Wensvoort et al., 1992; Dea et al., 1995).
2.3.3.1.2 Genomorganisation und virale Proteine
Das PRRSV-Genom besteht aus einem ca. 15,1 kB langen, linearen, einzelstrangigen, po-
lyadenylierten RNA-Molekul. Die RNA kodiert fur acht ORFs. Von den ORFs la und lb
werden Replikations- und Transkriptionsproteine kodiert. Die ORFs zwei bis sieben ko-
dieren fur die Strukturproteine (Conzelmann et al., 1993; Meulenberg et al., 1993a;
Murtaugh et al., 1995). Man unterscheidet zwischen europaischen (PRRSV-EU) und
nordamerikanischen PRRSV-Isolaten (PRRSV-NA). Diese rufen im Allgemeinen ahnliche

2.3. Differentialdiagnostik der KSP 19
Abbildung 2.6: Schematische Darstellung des PRRS-Virus. Nach Modrow et al. (2003)
Symptome hervor, unterscheiden sich jedoch sowohl in antigenetischen und genetischen
Eigenschaften als auch in der Virulenz (Prieto und Castro, 2005). Die Sequenzanalyse
des ORFs funf, der fur das Strukturglykoprotein GP5 kodiert, ergab eine ca. 55 %ige Se-
quenzhomologie zwischen europaischen und amerikanischen Stammen (Murtaugh et al.,
1995). Dies fuhrte zu Unterteilung des PRRSV in zwei verschiedene Genotypen – eu-
ropaische und amerikanische. Die Genomhomologie innerhalb europaischer Stamme liegt
nach unterschiedlichen Angaben zwischen 2–90 % (Murtaugh et al., 1995; Stadejek
et al., 2002), innerhalb amerikanischer bei ca. 90 % (Andreyev et al., 1997; Meng,
2000; Dee et al., 2001; Forsberg et al., 2002).
2.3.3.2 Klinik und Pathologie
Die klinischen Erscheinungen des PRRS sind sehr variabel und abhangig von dem Al-
ter und der Nutzungsrichtung der Tiere. Typischerweise sind es bei Absatzferkeln und
Laufern Fieber, Dyspnoe, Lethargie, Anorexie und Konjunktivitis. In einigen Fallen tre-

20 2. Literaturubersicht
ten Zyanosen an den Ohren und Extremitaten auf. Mastschweine zeigen ebenfalls Fieber
und Pneumonie, mit fortschreitendem Alter der Tiere treten ofter subklinische Infektio-
nen mit dem PRRSV auf (Benfield et al., 1999; Done et al., 1996; Rossow, 1998;
Anonym, 2004a). Bei tragenden Sauen, die milde Allgemeinsymptome zeigen konnen,
kann das PRRSV die Plazenta uberwinden und zu einer intrauterinen Infektion der Ferkel
fuhren. Fruhgeburten, lebensschwache Ferkel und hohe Ferkelverluste sind kennzeichnend
fur diesen Verlauf der Erkrankung (Yoon et al., 1993; Rossow et al., 1994; Mengeling
et al., 1995, 1996a; Wills et al., 1997; Prieto und Castro, 2005).
2.3.3.3 Diagnostik
Der Nachweis des PRRSV mittels Virusisolierung in permissiven Zellkulturen ist zwi-
schen dem ersten und zehnten Tag nach der Infektion (p.i. - post infectionem) moglich,
spater sinkt die Erfolgsquote rapide (Christopher-Hennings et al., 1995, 1998, 2001;
Mengeling et al., 1996b; Prieto et al., 2003). Die Anzucht in Zellkultur ist aller-
dings problematisch, da viele PRRSV-Stamme nicht in permanenten Zelllinien, sondern
nur in porzinen alveolaren Makrophagen (PAM) wachsen (Zhang et al., 1999). Oft ist
der Antikorpernachweis eine Alternative, jedoch kommt es zu einem schnellen Titerabfall
(Lager et al., 1997). Außerdem konnen die Antikorper gegen das Impfvirus das Ergebnis
bei vakzinierten Tieren beeinflussen (Egli et al., 2001). Des Weiteren besteht die Moglich-
keit, die Nukleinsaure des PRRSV nachzuweisen. Es existieren mehrere konventionelle
und real-time PCR-Protokolle, die den Virusnachweis auch bis zum Tag 31 p.i. erlauben.
Einige gestatten zusatzlich eine Differenzierung zwischen europaischen und amerikani-
schen Stammen (Meulenberg et al., 1993a; Mardassi et al., 1994; Christopher-
Hennings et al., 1995, 1998, 2001; Kono et al., 1996; Larochelle und Magar,
1997; Oleksiewicz et al., 1998; Spagnuolo-Weaver et al., 2000; Egli et al., 2001;
Stadejek et al., 2002; van Rijn et al., 2004; Wasilk et al., 2004; Revilla-Fernandez
et al., 2005; Chung et al., 2005; Kleiboeker et al., 2005).

2.3. Differentialdiagnostik der KSP 21
2.3.4 Porzine Influenza
Die porzine Influenza ist eine akute, fieberhafte, respiratorische Erkrankung. Influenza-
viren wurden in Schweinen erstmals 1918 in den USA beobachtet (Easterday und
Hinshaw, 1992). 1930 gelang es Shope (1931b), das Virus zum ersten Mal aus erkrankten
Tieren zu isolieren. Den porzinen Influenzaviren kommt moglicherweise auch eine Rolle
bei der Entstehung neuer humanpathogener Influenzastamme zu (Truyen, 2003).
2.3.4.1 Das Influenza-A-Virus
2.3.4.1.1 Taxonomie und Morphologie
Das Virus der Spezies porzines Influenzavirus (im Weiteren Influenza-A Virus) gehort
zur Familie Orthomyxoviridae, Genus Influenzavirus-A. Influenza-A-Viren sind behullt
und besitzen ein helikalsymmetrisches Nukleokapsid. Die Virionen sind pleomorph, so
variiert ihre Große von ca. 70-90 nm bis hin zu 3000 nm (Modrow et al., 2003).
2.3.4.1.2 Genomorganisation und virale Proteine
Das Genom der Influenza-A-Viren besteht aus acht unabhangigen einzelstrangigen RNA-
Segmenten, die in negativer Polaritat vorliegen. Sie kodieren fur zehn Hauptstrukturpro-
teine. Die systematische Bezeichnung der Influenzaviren bezieht neben dem Typ auch die
Antigenstruktur der wesentlichen Strukturproteine des Virus, des Hamagglutinins (H) und
der Neuraminidase (N), sowie das Jahr und den Ort der Isolierung mit ein. Daraus ergeben
sich verschiedene H- und N-Typen. Beim Schwein wurden bisher hauptsachlich H1N1 und
H3N2 Influenzaviren gefunden (Olsen et al., 1993; Bikour et al., 1994; Truyen, 2003),
wobei letztere vermutlich auf Infektionen mit humanen Influenzaviren zuruckzufuhren
sind (Truyen, 2003). In den letzten Jahren hat sich die epidemiologische Situation in
Europa dramatisch verandert. H1N1, H3N2 und H1N2 Subtypen kozirkulieren in der eu-
ropaischen Schweinepopulation (Reeth et al., 2004). Klassische H1N1 Subtypen wurden
in den letzten Jahren durch avian-like H1N1 Stamme verdrangt (Pensaert et al., 1981;
Brown et al., 1993).
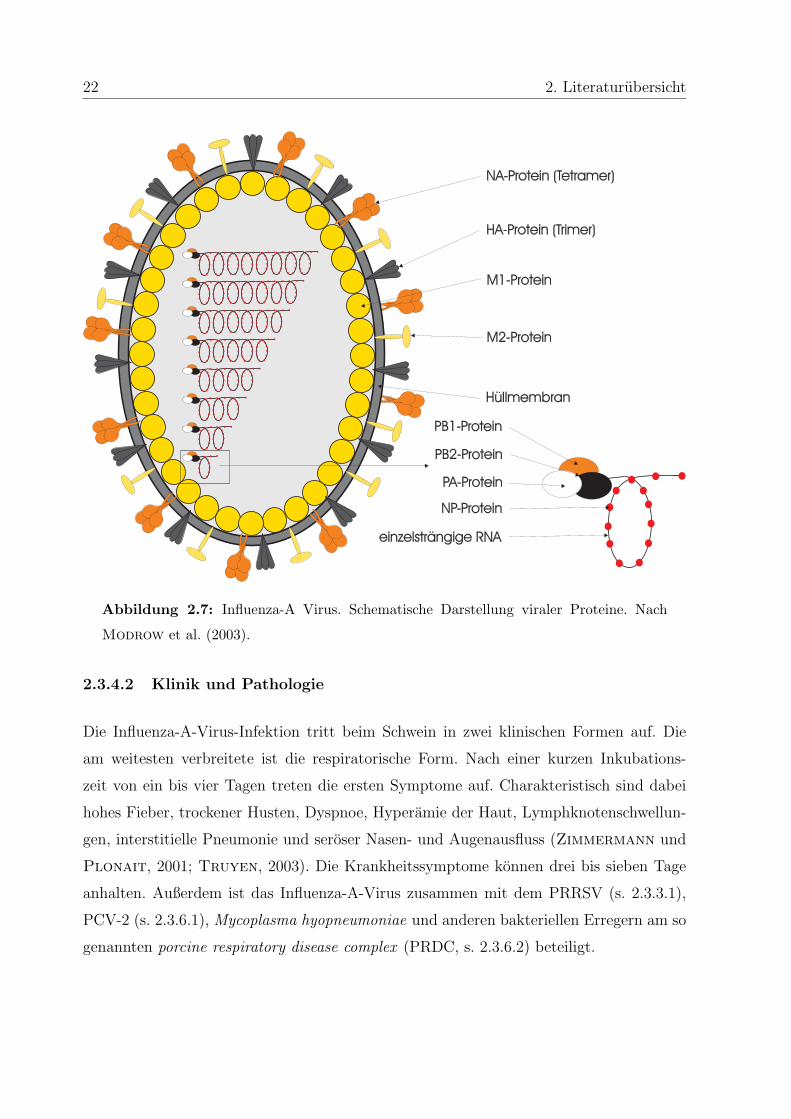
22 2. Literaturubersicht
Abbildung 2.7: Influenza-A Virus. Schematische Darstellung viraler Proteine. Nach
Modrow et al. (2003).
2.3.4.2 Klinik und Pathologie
Die Influenza-A-Virus-Infektion tritt beim Schwein in zwei klinischen Formen auf. Die
am weitesten verbreitete ist die respiratorische Form. Nach einer kurzen Inkubations-
zeit von ein bis vier Tagen treten die ersten Symptome auf. Charakteristisch sind dabei
hohes Fieber, trockener Husten, Dyspnoe, Hyperamie der Haut, Lymphknotenschwellun-
gen, interstitielle Pneumonie und seroser Nasen- und Augenausfluss (Zimmermann und
Plonait, 2001; Truyen, 2003). Die Krankheitssymptome konnen drei bis sieben Tage
anhalten. Außerdem ist das Influenza-A-Virus zusammen mit dem PRRSV (s. 2.3.3.1),
PCV-2 (s. 2.3.6.1), Mycoplasma hyopneumoniae und anderen bakteriellen Erregern am so
genannten porcine respiratory disease complex (PRDC, s. 2.3.6.2) beteiligt.

2.3. Differentialdiagnostik der KSP 23
2.3.4.3 Diagnostik
Die endgultige Diagnose wird durch einen Virusnachweis gefuhrt. Das Virus kann auf
verschiedenen Zelllinien angezuchtet werden, die Virusisolierung erfolgt jedoch meist im
embryonierten Huhnerei. Die Uberprufung und Spezifizierung ist uber den Hamagglutina-
tionshemmtest mit spezifischen Antiseren moglich (Truyen, 2003). In Lungenschnitten
lasst sich das Antigen mittels Immunfluoreszenz-, bzw. Immunperoxidasetechnik nach-
weisen (Zimmermann und Plonait, 2001). Fur die serologische Untersuchung von Se-
rumpaaren steht der Hamagglutinationshemmtest zur Verfugung (Long et al., 2004). In
den vergangenen Jahren wurde eine Vielzahl an PCR-Protokolle zum Nachweis der Nu-
kleinsauren des Influenza-A Virus entwickelt (Fouchier et al., 2000; van Elden et al.,
2001; Choi und Chae, 2002; Matsuzaki, 2003; Richt et al., 2004; Stone et al., 2004;
Watzinger et al., 2004; Chen et al., 2005; Hindiyeh et al., 2005; Krafft et al., 2005;
Landolt et al., 2005; Zhang et al., 2005; Payungporn et al., 2006).
2.3.5 Porzine Parvovirus - Infektionen
Intrauterine Infektionen mit dem porzinen Parvovirus (PPV) gelten heute als wichtigs-
te Ursache fur das so genannte SMEDI-Syndrom (stillbirth, mummification, embryo-
nic death, infertility). Betroffen sind vor allem Bestande mit hohem Jungsauenanteil
(Plonait, 2001). Der Erreger wurde 1967 erstmals in England isoliert und ist weltweit
verbreitet. Die Seropravalenz betragt in deutschen Schweinehaltungen ca. 70 bis 80 %
(Truyen, 2003).
2.3.5.1 Das Porzine Parvovirus
2.3.5.1.1 Taxonomie und Morphologie Das PPV gehort zur Familie Parvoviri-
dae, Subfamilie Parvovirinae. Die Familie enthalt eine weitere Subfamilie - Densovirinae
mit den Genera Brevidensovirus, Densovirus und Iteravirus; die Subfamilie Parvovirinae
umfasst die Genera Dependovirus, Erythrovirus und Parvovirus (Murphy et al., 1995).
Die Parvoviren sind kleine, unbehullte, DNA Viren mit einem Virionendurchmesser von
ca. 20 nm (Mayr et al., 1968; Molitor et al., 1983).

24 2. Literaturubersicht
2.3.5.1.2 Genomorganisation und virale Proteine Das Genom des PPVs besteht
aus einer linearen, einzelstrangigen DNA mit negativer Polaritat und ist ca. 5000 Basen
lang (Molitor et al., 1984; Bergeron et al., 1993). Es besitzt zwei große und einen
kleinen ORF. Alle drei ORFs liegen auf dem komplementaren (positiven) Strang und
uberlappen sich gegenseitig (s. Abbildung 2.8). ORF1 kodiert fur Nichtstrukturprotei-
Abbildung 2.8: Schematische Darstellung des Genoms des Porzinen Parvovirus.
ne und fur die ersten zehn Aminosauren des Strukturproteins VP1. ORF2 kodiert fur
die Strukturproteine VP1 und VP2 (Bergeron et al., 1993). Das Genom der Parvovi-
ren ist von einem Kapsid umgeben, das sich aus den Strukturproteinen VP1 und VP2
zusammensetzt. VP2 ist das Hauptstrukturprotein. Das PPV besitzt außerdem die Nicht-
strukturproteine NS1, NS2 und NS3 (s. Abbildung 2.8), deren Funktionen jedoch noch
nicht genau geklart sind (Berns, 1990; Wilson et al., 1991). Die Aminosauresequenz
des VP2 ist vollstandig in der des VP1 enthalten. Beide Kapsidproteine werden uber die
gleiche”messenger“-RNA (mRNA) transkribiert. Wird die mRNA vom ersten Startcodon
aus translatiert, entsteht das VP1-Kapsidprotein; VP2 wird gebildet, wenn vom zweiten

2.3. Differentialdiagnostik der KSP 25
Startcodon die mRNA translatiert wird. VPl und VP2 sind antigenetisch eng verwandt
(Molitor et al., 1983). Die Proteine werden durch alternatives Spleißen prozessiert, d. h.,
der gleiche Genomabschnitt wird in verschiedenen Leserastern abgelesen. Dabei dienen so-
wohl gespleißte als auch ungespleißte Transkripte als mRNA. Dieser Mechanismus macht
es den Parvoviren moglich, ihre gesamte Erbinformation innerhalb des kleinen Genoms
zu plazieren (Berns, 1990).
2.3.5.2 Klinik und Pathologie
Infektionen mit dem PPV fuhren zu Fruchtbarkeitsstorungen, die durch Umrauschen, Ab-
orte, Totgeburten, Mumifikation, neonatalen Tod und reduzierte neonatale Lebensfahig-
keit gekennzeichnet sind (Cartwright und Huck, 1967; Johnson und Collings,
1969; Morimoto et al., 1972; Narita et al., 1975; Forman et al., 1977). PPV wird
als haufigste Ursache fur Fruchtbarkeitsstorungen beim Schwein angesehen. In der Klinik
gilt es als Hauptursache des”SMEDI“ Syndroms. Die akute Infektion neonataler und adul-
ter Schweine mit dem PPV verlauft in der Regel subklinisch (Mengeling und Cutlip,
1976; Joo et al., 1977). Pathologische Veranderungen nach einer PPV-Infektion sind nur
bei Embryonen und Feten festzustellen.
Der Grad der klinischen Veranderungen hangt von der Virulenz des PPV-Stammes ab.
Avirulente Stamme konnen die Plazentarschranke nicht ubertreten (Mengeling und
Cutlip, 1976). Virulente Stamme sind nur pathogen fur noch nicht immunkompetente
Feten (Mengeling und Cutlip, 1976), nach Erreichen der Immunkompetenz werden
neutralisierende Antikorper gebildet (Bachmann et al., 1975). Hochvirulente Stamme
verursachen Lasionen bzw. fuhren auch nach Erreichen der Immunkompetenz zum Tod
(Kresse et al., 1985). Je nach Immunstatus und Alter der betroffenen Tiere kommt
es zu unterschiedlichen Auspragungen der klinischen Symptome. Die Palette reicht von
Fruchtresorbtion und Umrauschen, uber Aborte mumifizierter Fruchte bis hin zur Ge-
burt lebensschwacher Ferkel (Rodeffer et al., 1975; Cropper et al., 1976; Wrathall
und Mengeling, 1979; Mengeling et al., 1980; Kuiper, 1985). Der haufigste Krank-
heitsbefund ist die Mumifikation (Mengeling, 1975; Mengeling und Cutlip, 1976;
Donaldson-Wood et al., 1977; Cutler et al., 1983). Das PPV wird mit dem PMWS
(s. 2.3.6.2) in Zusammenhang gebracht, dabei zeigte sich, dass eine Koinfektion von neo-

26 2. Literaturubersicht
natalen Ferkeln mit PPV und PCV-2 (s. Kapitel 2.3.6.1) das Krankheitsbild von PMWS
verstarkt. PPV bewirkt dabei eine Immundysfunktion, die den Viruseintritt von PCV-2
in Zellen erleichtert (Ellis et al., 1999; Allan et al., 1999; Kennedy et al., 2000).
2.3.5.3 Diagnostik
Fur die PPV-Diagnostik stehen sowohl direkte als auch indirekte Nachweismethoden zur
Verfugung. Die Methode der Wahl fur den Nachweis von PPV ist die PCR, da mit ihr
auch der Nachweis viraler Nukleinsauren in mumifizierten Feten moglich ist, wenn kein in-
fektioses Virus mehr nachweisbar ist (Truyen, 2003). Es sind bisher mehrere gel-basierte
und real-time PCR-Protokolle publiziert worden (Molitor et al., 1991; Gradil et al.,
1994; Bergeron et al., 1996; Belak et al., 1998; Arnauld et al., 1998; Soares et al.,
1999; Choi und Chae, 2000; Ellis et al., 2000; Kim und Chae, 2001; Lyoo et al.,
2001; Kim und Chae, 2003; Kim et al., 2003b; Prikhod’ko et al., 2003; Huang et al.,
2004; Kim und Chae, 2004; Cao et al., 2005b). Alternativ besteht die Moglichkeit das
PPV in der Zellkultur zu isolieren. Mengeling (1975, 1978) stellte jedoch fest, dass die
Infektiositat progressiv nach den Tod des Fetus abnimmt und die Isolierung in Zellkul-
tur daher haufig erfolglos verlauft. Wegen der hohen Tenazitat ist bei diesen Verfahren
auch das Kontaminationsrisiko sehr hoch (Cartwright et al., 1969). Generell konnen
mit zunehmender Reife der Feten Antikorper auftreten, die den Virusnachweis storen
(Truyen, 2003). Relativ unspezifisch und weniger sensitiv ist der Hamagglutinationstest
bzw. der spezifische Hamagglutinationshemmtest (Jenkins, 1992; Mengeling et al.,
1993; Truyen, 2003). Die Immunfluoreszenztechnik erlaubt den Nachweis von PPV in
Geweben (Joo und Johnson, 1977; Mengeling, 1978), wobei sich v. a. Lungen eignen
(Truyen, 2003). Oraveerakul et al. (1990) haben ein”slot-blot hybridization“ - Pro-
tokoll mit nichtradioaktiven Sonden zum Antigennachweis in Gewebe und in Zellkultur
entwickelt. Zum indirekten Erregernachweis stehen kommerzielle ELISA-Kits zum semi-
quantitativen Nachweis von PPV-Antikorpern zur Verfugung. Da viele Schweine durch
Impfung, subklinische Infektionen oder maternale Antikorper seropositiv sind, ist eine
einmalige serologische Untersuchung nicht aussagekraftig. Daher wird die Untersuchung
von Serumpaaren durchgefuhrt (Truyen, 2003).

2.3. Differentialdiagnostik der KSP 27
2.3.6 Porzine Circovirus-Infektionen
Das porzine Circovirus (PCV) wurde 1974 von Tischer et al. (1974) als Kontaminan-
te der stabilen porzinen Nierenzelllinie (PK-15) entdeckt. Es existieren zwei Typen des
PCVs. Wahrend das PCV Typ 1 als apathogen beschrieben wird, ist das PCV Typ 2
fur klinisch manifeste Infektionen verantwortlich. Das porzine Circovirus Typ 2 wurde
erstmals 1991 in Kanada als Ausloser klinischer Erkrankungen beschrieben. Seither fand
man ahnliche Bilder nahezu weltweit. Der Erreger ist beteiligt am PMWS, PDNS und
PRDC (Haas, 2003).
2.3.6.1 Das Porzine Circovirus
2.3.6.1.1 Taxonomie und Morphologie
Die porzinen Circovirus Typen 1 und 2 (PCV-1 bzw. PCV-2) gehoren zusammen mit den
Circoviren der Taube (Pigeon circovirus), der Gans (Goose circovirus), des Kanarienvogels
(Canary circovirus), der Ente (Duck circovirus) und dem Psittacine Beak and Feather
Disease Virus dem Genus Circovirus an (Mankertz et al., 2000; Todd et al., 2001a,b;
Ritchie et al., 1989). Das Chicken Anaemia Virus (Todd et al., 1990) ist der alleinige
Vertreter des Genus Gyrovirus (Pringle, 1999). Die beiden Genera sind in der 1993
etablierten Familie Circoviridae untergebracht (Studdert, 1993; Lukert et al., 1995).
Das PCV ist ein unbehulltes Virus mit einem Durchmesser der Viruspartikel von lediglich
17 nm (Tischer et al., 1982). Es ist damit das kleinste bekannte Virus. Das Nukleokapsid
hat ein Molekulargewicht von 36 kDa und besitzt eine ikosaedrische Symmetrie (Tischer
et al., 1982).
2.3.6.1.2 Genomorganisation und virale Proteine
Die Nukleinsaure der porzinen Circoviren liegt in Form einer zirkular geschlossenen, ein-
zelstrangigen DNA mit positiver Strangorientierung vor (Buhk et al., 1985; Tischer
et al., 1987; Meehan et al., 1997). Die Genomlange betragt beim PCV-1 1759 Basen
und beim PCV-2 1768 Basen (Hamel et al., 1998; Morozov et al., 1998). Beide Typen
sind phylogenetisch eng miteinander verwandt, die Nukleotidsequenzhomologie zwischen
ihnen betragt nach unterschiedlichen Aussagen ca. 68 % bis 80 % (Morozov et al., 1998;

28 2. Literaturubersicht
Hamel et al., 1998; Meehan et al., 1998). Die Nukleotidsequenzhomologien von Feld-
isolaten des PCV-2 liegen bei 96 % (Morozov et al., 1998). Porzine Circoviren besitzen
insgesamt elf ORFs innerhalb ihres Genoms (Hamel et al., 1998).
Im Bereich des ORF1 betragt die Homologie zwischen PCV-1 und PCV-2 85 % (Morozov
et al., 1998; Meehan et al., 1998). In den ORFs 5, 6, 9, 10 und 11 besitzen die bei-
den PCV Typen keinerlei Homologie. In den anderen ORFs ist sie unterschiedlich hoch
ausgepragt (Hamel et al., 1998). Der ORF2 besitzt zwischen PCV-1 und PCV-2 eine
hohere Sequenzvariabilitat. Sie liegt in disem Teil des Genoms bei ca. 66 % (Morozov
et al., 1998; Hamel et al., 1998). Wie Ilyina und Koonin (1992) zeigten, kodiert der
ORF1 fur ein replikationsassoziiertes Protein, die ORF2 kodiert fur Hauptkapsidproteine
(Nawagitgul et al., 2000; Meehan et al., 2001).
bbbbbbbbbbbbbbbbbbbbbbbbbb
bbbbbbbbbbbbbbbbbbbbbbbbbb
bbbbbbbbbbbbbbbbbbbbbbbbbb
bbbbbbbbbbbbbbbbbbbbbbbbbb
bbbbbbbbbbbbbbbbbbbbbbbbbb
bbbbbbbbbbbbbbbbbbbbbbbbbb
bbbbbbbbbbbbbbbbbbbbbbbbbb
dddddddddddddddddddddddddd
dddddddddddddddddddddddddd
dddddddddddddddddddddddddd
dddddddddddddddddddddddddd
dddddddddddddddddddddddddd
dddddddddddddddddddddddddd
dddddddddddddddddddddddddd
aaaaaaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaaaaaa
cccccccccccccccccccccccccc
cccccccccccccccccccccccccc
cccccccccccccccccccccccccc
cccccccccccccccccccccccccc
cccccccccccccccccccccccccc
cccccccccccccccccccccccccc
cccccccccccccccccccccccccc
bbbbbbbbbbbbbbbbbbbbbb
bbbbbbbbbbbbbbbbbbbbbb
bbbbbbbbbbbbbbbbbbbbbb
bbbbbbbbbbbbbbbbbbbbbb
bbbbbbbbbbbbbbbbbbbbbb
bbbbbbbbbbbbbbbbbbbbbb
bbbbbbbbbbbbbbbbbbbbbb
dddddddddddddddddddddd
dddddddddddddddddddddd
dddddddddddddddddddddd
dddddddddddddddddddddd
dddddddddddddddddddddd
dddddddddddddddddddddd
dddddddddddddddddddddd
aaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaa
cccccccccccccccccccccc
cccccccccccccccccccccc
cccccccccccccccccccccc
cccccccccccccccccccccc
cccccccccccccccccccccc
cccccccccccccccccccccc
cccccccccccccccccccccc
bbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbb
ddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddd
aaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaa
ccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccc
bbbbbbbbbbbbbbbbbbbbbbbbbb
bbbbbbbbbbbbbbbbbbbbbbbbbb
bbbbbbbbbbbbbbbbbbbbbbbbbb
bbbbbbbbbbbbbbbbbbbbbbbbbb
bbbbbbbbbbbbbbbbbbbbbbbbbb
bbbbbbbbbbbbbbbbbbbbbbbbbb
bbbbbbbbbbbbbbbbbbbbbbbbbb
dddddddddddddddddddddddddd
dddddddddddddddddddddddddd
dddddddddddddddddddddddddd
dddddddddddddddddddddddddd
dddddddddddddddddddddddddd
dddddddddddddddddddddddddd
dddddddddddddddddddddddddd
aaaaaaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaaaaaa
cccccccccccccccccccccccccc
cccccccccccccccccccccccccc
cccccccccccccccccccccccccc
cccccccccccccccccccccccccc
cccccccccccccccccccccccccc
cccccccccccccccccccccccccc
cccccccccccccccccccccccccc
bbbbbbbbbbbbbbbbbbbbbb
bbbbbbbbbbbbbbbbbbbbbb
bbbbbbbbbbbbbbbbbbbbbb
bbbbbbbbbbbbbbbbbbbbbb
bbbbbbbbbbbbbbbbbbbbbb
bbbbbbbbbbbbbbbbbbbbbb
bbbbbbbbbbbbbbbbbbbbbb
dddddddddddddddddddddd
dddddddddddddddddddddd
dddddddddddddddddddddd
dddddddddddddddddddddd
dddddddddddddddddddddd
dddddddddddddddddddddd
dddddddddddddddddddddd
aaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaa
cccccccccccccccccccccc
cccccccccccccccccccccc
cccccccccccccccccccccc
cccccccccccccccccccccc
cccccccccccccccccccccc
cccccccccccccccccccccc
cccccccccccccccccccccc
bbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbb
ddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddd
aaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaa
ccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccc
bbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbb
ddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddd
aaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaa
ccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccc
bbbbbbbbbbbbbbbbbbbbbb
bbbbbbbbbbbbbbbbbbbbbb
bbbbbbbbbbbbbbbbbbbbbb
bbbbbbbbbbbbbbbbbbbbbb
bbbbbbbbbbbbbbbbbbbbbb
dddddddddddddddddddddd
dddddddddddddddddddddd
dddddddddddddddddddddd
dddddddddddddddddddddd
dddddddddddddddddddddd
aaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaa
aaaaaaaaaaaaaaaaaaaaaa
cccccccccccccccccccccc
cccccccccccccccccccccc
cccccccccccccccccccccc
cccccccccccccccccccccc
ccccccccccccccccccccccbbbbbbbbbbbbbb
bbbbbbbbbbbbbb
bbbbbbbbbbbbbb
bbbbbbbbbbbbbb
bbbbbbbbbbbbbb
bbbbbbbbbbbbbb
bbbbbbbbbbbbbb
bbbbbbbbbbbbbb
dddddddddddddd
dddddddddddddd
dddddddddddddd
dddddddddddddd
dddddddddddddd
dddddddddddddd
dddddddddddddd
dddddddddddddd
aaaaaaaaaaaaaa
aaaaaaaaaaaaaa
aaaaaaaaaaaaaa
aaaaaaaaaaaaaa
aaaaaaaaaaaaaa
aaaaaaaaaaaaaa
aaaaaaaaaaaaaa
aaaaaaaaaaaaaa
cccccccccccccc
cccccccccccccc
cccccccccccccc
cccccccccccccc
cccccccccccccc
cccccccccccccc
cccccccccccccc
cccccccccccccc
bbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbbb
dddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddddd
aaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaaa
cccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccccc
Abbildung 2.9: Schematische Darstellung eines PCV-2. Nach Modrow et al. (2003).

2.3. Differentialdiagnostik der KSP 29
2.3.6.2 Klinik und Pathologie
Trotz der weltweiten Verbreitung des PCV-1 (Allan et al., 1998), wird das Virus als
apathogen angesehen (Tischer et al., 1986; Allan et al., 1995; Krakowka et al., 2000).
Weder in PCV-1-positiven Betrieben, noch bei experimentell infizierten Tieren konnten
Krankheitssymptome festgestellt werden (Allan et al., 1995). Die pathogene Rolle des
PCV-2 beim Schwein wird dagegen oft diskutiert (Allan et al., 1998; Meehan et al.,
1998; Kim und Chae, 2001, 2002; Pallares et al., 2002; Chae, 2004). Zur Ausbildung
eines Krankheitskomplexes sind aber weitere Koinfektionen oder Kofaktoren erforderlich
(Allan et al., 2000; Choi und Chae, 2000; Krakowka et al., 2001; Kim und Chae,
2003).
Postweaning multisystemic wasting syndrome (PMWS)
Das PCV-2 wurde erstmals von Ellis et al. (1998) mit dem PMWS in Zusammenhang
gebracht. Obgleich nachgewiesen wurde, dass das PCV-2 die Hauptursache des PMWS
ist, sind fur eine Ausbildung des Krankheitskomplexes weitere Kofaktoren erforderlich
(Ellis et al., 2004). Betroffen sind sechs bis 15 Wochen alte Schweine, typischerweise
sind es jedoch junge Tiere nach dem Absetzen (post-weaner) (Harding et al., 1998;
Harding, 2004). Die Diagnose PMWS kann nur gestellt werden, wenn die drei folgenden
Kriterien erfullt werden: typische klinische Symptome, Vorhandensein charakteristischer
mikroskopischer Lasionen und Anwesenheit des PCV-2 in den Lasionen (Segales et al.,
2004; Chae, 2004). Die sechs Hauptsymptome des PMWS sind Kummern, Dyspnoe,
vergroßerte Lymphknoten, Diarrhoe, Blasse und Ikterus (Harding, 2004). Bei der Sektion
fallen die nicht kollabierten, braun-marmorierten Lungen und vergroßerte Lymphknoten
auf (Rosell et al., 1999). Die Leber kann sowohl vergroßert, als auch verkleinert sein
und an den Nieren sind multiple helle Punkte verschiedener Große feststellbar (Rosell
et al., 2000; Segales et al., 2004).
Porzines Dermatitis und Nephropathie Syndrom (PDNS)
PDNS wurde erstmals von Smith et al. (1993) in Großbritannien beschrieben. Mittler-
weile kommt die Krankheit weltweit vor (Segales et al., 2004). Das Leitsymptom ist die

30 2. Literaturubersicht
Ausbildung von runden oder unregelmaßig geformten, rot bis violettgefarbten Hautarea-
len. Meistens sind sie an den Extremitaten, am Unterbauch, an den Flanken oder auch
an den Ohren lokalisiert (Harding, 2004). In schweren Fallen kommt es zu Abmagerung,
Anorexie und Fieber. Selten treten Todesfalle auf. Es kommt zu einer nekrotisierenden
Vaskulitis im Bereich der Haut und in den Nieren. Letztere sind dabei vergroßert, blass
und weisen petechiale Blutungen auf (Harding, 2004).
Porcine respiratory disease complex (PRDC)
Der PRDC ist durch Kummern, Husten und Pneumonie charakterisiert (Thacker et al.,
2000). Betroffen sind meistens Schweine im Alter von 16 bis 20 Wochen. Neben PCV-2
sind auch das PRRSV (s. Kapitel 2.3.3), das porzine Influenza Virus (s. Kapitel 2.3.4.1)
und Mycoplasma hyopneumoniae am Geschehen beteiligt (Thacker et al., 2000). Die
klinischen Erscheinungen sind von der Anzahl der beteiligten Erreger abhangig. In den
meisten Fallen ist es schwierig, PRDC von PMWS oder PRRS zu unterscheiden. Die
pathologischen Veranderungen in den Lungen sind mit denen von PRRS, oder bakteriellen
Infektionen (wie Salmonellose) identisch (Harms et al., 2001).
2.3.6.3 Diagnostik
Zum Nachweis des PCV-2 stehen sowohl direkte, als auch indirekte Nachweismethoden zur
Verfugung. Immunhistochemie und in situ Hybridisierung geben neben dem Erregernach-
weis auch eine Auskunft uber das pathologisch-histologische Geschehen im betroffenen
Gewebe (Choi und Chae, 1999, 2000; Kim und Chae, 2001; Kim et al., 2003a; Kim und
Chae, 2004). Ellis et al. (1998) und Choi et al. (2000) gelang der Nachweis von PCV-2
in verschiedenen Organen mittels Immunhistochemie unter Verwendung von mono- und
polyklonalen Antikorpern. In situ Hybridisierung erlaubt sowohl den Nachweis, als auch
Differenzierung zwischen PCV-1 und PCV-2 (Kim und Chae, 2001, 2002). Die PCR
bleibt im Vergleich zu in situ Hybridisierung die sensitivere Methode (Calsamiglia
et al., 2002). Es sind mehrere gel-basierte PCR-Protokolle publiziert (Ouardani et al.,
1999; Kim et al., 2001; Kim und Chae, 2004).

2.4. Polymerasekettenreaktion 31
2.4 Polymerasekettenreaktion
2.4.1 Prinzip
Die Polymerasekettenreaktion (PCR - Polymerase chain reaction), die zum ersten Mal
von Mullis und Faloona (1987) vorgestellt wurde, stellt eine in vitro Technik dar.
Dabei wird ein von zwei spezifischen Oligonukleotiden, den Primern, eingerahmtes DNA-
Fragment in einer enzymatischen, zyklischen Reaktion mehrfach vervielfaltigt. Im ers-
ten Schritt, der Denaturierung bei 92-95 ◦C, entsteht aus einer doppelstrangigen DNA
(dsDNA) einzelstrangige Matrizen-DNA (ssDNA). Danach wird der Reaktionsansatz auf
eine bestimmte Temperatur herunter gekuhlt, die von der Schmelztemperatur der jewei-
ligen Primer abhangig ist. Dabei kommt es zur Anlagerung (Annealing) der Primer an
die komplementaren Sequenzen der Matrizen-DNA. Im nachsten Schritt werden die Pri-
mer von einer DNA-abhangigen DNA-Polymerase in Anwesenheit freier Desoxynukleosid-
Triphosphate (dNTPs) und Magnesium-Ionen verlangert (Elongation). Die im ersten Zy-
klus entstandenen Reaktionsprodukte (Amplifikate) dienen in nachfolgenden Zyklen selbst
als Matrize. Die Polymerase verlangert den DNA-Strang solange sie sich nicht von ihm
lost, oder die Reaktion durch eine Temperaturerhohung unterbrochen wird. Damit wird
ein neuer Zyklus eingeleitet. Ublicherweise wird die Reaktion 20-40 mal wiederholt. In der
Theorie verdoppelt sich die Anzahl neu entstandener DNA-Kopien von Zyklus zu Zyklus.
Anders als in der Theorie, liegt in der Praxis der durchschnittliche Multiplikationsfaktor
in der Gesamtreaktion bei ca. 1,6 bis 1,7 je Zyklus (Kainz, 2000). Das spielt in der quan-
titativen PCR (s. 2.4.3.5), bei der die genaue Zahl der Ausgangskopien bestimmt werden
kann, eine entscheidende Rolle.
Die entstandenen DNA-Fragmente lassen sich anschließend durch Auftrennung anhand
der Fragmentgroße in einem Agarose-Gel (3.2.2.5) und anschließender Farbung mit einem
fluoreszierenden Farbstoff, wie z. B. Ethidiumbromid (Kemp et al., 1989) unter UV-
Licht bei 254 nm sichtbar machen (Sambrook et al., 1989). Durch einen parallel aufge-
tragenen Großenmarker lasst sich die Große des DNA-Amplifikats bestimmen (Mullis
und Faloona, 1987). Eine weitere Verifizierung der amplifizierten DNA laßt sich durch
Restriktionsenzymschnittmusteranalyse (Sambrook et al., 1989), Bestimmung der Nu-
kleotidabfolge durch Sequenzierung (Innis et al., 1988) oder Hybridisierung mit einer

32 2. Literaturubersicht
spezifischen Gensonde (Saiki et al., 1985; Sambrook et al., 1989) durchfuhren.
2.4.2 Reverse Transkription - Polymerasekettenreaktion
Einen großen Beitrag zur Erfolgsgeschichte der PCR leistete die Entdeckung der reversen
Transkriptase (RT) oder RNA-abhangigen DNA-Polymerase (Baltimore, 1970; Temin
und Mizutani, 1970). Die reverse Transkriptase ermoglichte in der Kombination mit
der PCR eine neue Untersuchungstechnik, die Reverse Transkription - Polymeraseket-
tenreaktion (RT-PCR). Durch die reverse Transkription lasst sich jede beliebige RNA
als PCR-Vorlage nutzen. Dabei wird die RNA zunachst in eine komplementare DNA
(cDNA) transkribiert, die anschließend in der PCR vervielfaltigt wird. Dadurch konnen
auch RNA-Viren mittels PCR nachgewiesen werden (Gao und Moore, 1996).
2.4.3 Real-time PCR
2.4.3.1 Prinzip und Techniken
Die real-time PCR (auch qPCR - quantitative PCR genannt) stellt eine Weiterentwick-
lung der konventionellen PCR dar. Der wichtigste Unterschied besteht in der Moglichkeit,
die amplifizierte DNA noch wahrend der Reaktion (in Echtzeit - real-time) zu detektie-
ren. Dabei gibt es zwei unterschiedliche Techniken. Bei der einen werden interkalierende
Farbstoffe, die sich in die doppelstrangige DNA einlagern (Higuchi et al., 1992, 1993)
eingesetzt, und bei der anderen sequenzspezifische Oligonukleotide (Sonden), die mit Fluo-
reszenzfarbstoffen markiert sind (Saiki et al., 1985; Sambrook et al., 1989). Mittlerweile
gibt es eine Vielzahl an fluoreszenzfarbstoffmarkierten Sonden auf dem Markt. Eine Uber-
sicht bieten Bente (2003) und Bustin und Nolan (2004a). In der Abbildung 2.10 sind
die Grundbegriffe der real-time PCR erklart. Weitere Informationen sind u.a. bei Bustin
(2000, 2002); Bustin und Nolan (2004c), sowie Bente (2003) zu finden.

2.4. Polymerasekettenreaktion 33
Abbildung 2.10: Schematische Darstellung einer Amplifikationsgrafik: Die gemessene
Fluoreszenz (Ordinate) wird gegen die Zyklenzahl (Abszisse) aufgetragen. In den ersten
Zyklen andern sich die Fluoreszenzwerte nur unwesentlich, hier wird die Basislinie (Ba-
seline) definiert. Aus der Standardabweichung der Fluoreszenz zwischen Zyklus drei und
15 multipliziert mit dem Faktor zehn, wird ein Fluoreszenzwert errechnet, der zur Grund-
fluoreszenz der Proben addiert wird. So ergibt sich der Schwellenwert (Threshold). Der
Schnittpunkt zwischen Amplifikationsgraph und Schwellenwert wird als cycle-threshold
(Ct) bezeichnet. Er markiert einen Anstieg der Fluoreszenz uber den Schwellenwert. R+n ,
R−n - normalisierte Fluoreszenz einer positiven, bzw. negativen Probe. (∆Rn) - baseline-
korrigierte, normalisierte Fluoreszenz. Modifiziert nach Bente (2003).
2.4.3.2 SYBRTM-Green real-time PCR
SYBRTM-Green gehort neben Ethidiumbromid zu den so genannten interkalierenden Farb-
stoffen (Higuchi et al., 1992, 1993). Die Besonderheit dieser Farbstoffe liegt in der Fahig-
keit, unter UV-Licht, interkaliert in doppelstrangiger DNA, sichtbares Licht zu emittieren.
So besitzt SYBRTM-Green eine Anregungswellenlange von 497 nm und eine Emissions-
wellenlange von 520 nm (Higuchi et al., 1992, 1993). Der Vorteil dieser Farbstoffe ist,
dass sie universell einsetzbar sind (Bustin, 2000), andererseits binden sie an jede doppel-
strangige DNA, so dass auch unspezifische PCR-Produkte ein positives Signal emittieren

34 2. Literaturubersicht
(Vandesompele et al., 2002). Aus diesem Grunde wird am Ende einer SYBRTM-Green -
PCR eine Schmelzpunktanalyse durchgefuhrt (Ririe et al., 1997; Vandesompele et al.,
2002). Dabei wird der Reaktionsansatz in 1 ◦C Schritten von ca. 55 ◦C auf ca. 96 ◦C er-
hitzt und kontinuierlich die Fluoreszenz gemessen. Der Punkt, an dem doppelstrangige
DNA denaturiert, ist durch einen Abfall der Fluoreszenz des interkalierenden Farbstoffes
gekennzeichnet. So kann fur jedes PCR-Produkt ein spezifischer Schmelzpunkt bestimmt
werden (Ririe et al., 1997).
2.4.3.3 TaqMan real-time PCR
Die so genannte TaqMan real-time PCR wurde erstmals von Holland et al. (1991) be-
Abbildung 2.11: Schematische Darstellung eines TaqMan Assays

2.4. Polymerasekettenreaktion 35
schrieben. Das Prinzip basiert auf der 5′–3′ Exonukleaseaktivitat der Taq-Polymerase.
Dies bedeutet, dass die Polymerase die vor ihr liegende, hybridisierte Sonde wahrend
der DNA-Strangverlangerung hydrolysiert. Nur bestimmte Polymerasen, unter anderem
die Taq-Polymerase, besitzen diese Fahigkeit (Kreuzer et al., 2000). Die Sonde ist ein
Oligonukleotid, das an seinem 5′-Ende ein Farbstoffmolekul und am 3′-Ende einen so ge-
nannten Quencher tragt (s. Kapitel 2.4.4.2). So lange sich die beiden Molekule auf der
Sonde, in unmittelbarer Nahe, befinden, kommt es zu keinem Anstieg der Fluoreszenz. So-
bald aber das Oligonukleotid von der Taq-Polymerase hydrolysiert wird, werden Farbstoff
und Quencher voneinander getrennt. Folglich kommt es zu einem irreversiblen Anstieg der
Fluoreszenz, der von dem real-time PCR-Gerat gemessen wird (Heid et al., 1996).
TaqMan-PCR ist unter den real-time PCR Methoden am weitesten verbreitet (Terry
et al., 2002). Sie ist ausreichend sensitiv um einzelne Mutationen in einer Sequenz zu
detektieren (Livak, 1999). Desweiteren erlaubt die Auswahl an verfugbaren Fluoreszenz-
farbstoffen TaqMan-Sonden fur multiplex PCRs zu verwenden (s. Kapitel 2.4.5).
2.4.3.4 Primer-probe energy transfer PriProET
Eine Weiterentwicklung der real-time PCR stellt das so genannte primer-probe energy
transfer System (PriProET ) dar (Rasmussen et al., 2003). Dieses System kombiniert die
Moglichkeit, das gebildete PCR-Produkt sondenbasiert in Echtzeit zu detektieren mit
der Option, die Hybridisierung der Sonde mittels Schmelzkurvenanalyse zu bestatigen.
Wie ublich werden zunachst zwei Primer fur die Amplifikation des ausgewahlten Ge-
nomfragments eingesetzt, dazu kommt eine produktspezifische Sonde. Wahrend einer der
beiden Primer mit einem Donor -Farbstoff markiert wird, ist die Sonde an einen Repor-
ter -Farbstoff gekoppelt. Die Primer werden in jedem PCR-Zyklus verlangert, so dass
sich die Sonde an den gebildeten DNA-Strang anlagern kann. Dabei findet die Ener-
gieubertragung vom Donor -Farbstoff auf den Reporter -Farbstoff statt. Dadurch, dass die
Fluoreszenz nur in Anwesenheit des spezifischen Produkts erfolgen kann, wird eine direk-
te PCR-Quantifizierung anhand der vom Reporter-Farbstoff abgestrahlten Lichtmenge
ermoglicht (Rasmussen et al., 2003, 2005).

36 2. Literaturubersicht
Abbildung 2.12: Beim primer-probe energy transfer System (PriProET ) werden zusatz-
lich zum Forward -Primer ein mit Fluoreszenzfarbstoff markierter Reverse-Primer und ei-
ne ebenfalls markierte Sonde verwendet. Der Donor -Farbstoff (FAM) ist am 5′-Ende des
Reverse-Primers, der Reporter -Farbstoff (Cy5) am 3′-Ende der Sonde platziert. Wahrend
der Amplifizierung wird der Primer-Strang verlangert, die FAM-markierte Sonde bindet
daran und es findet eine Energieubertragung von FAM zu Cy5 statt. Damit wird eine
Quantifizierung des Produktes moglich, da das von Cy5 ausgestrahlte Licht direkt pro-
portional zur gebildeten Produktmenge ist. Die Verwendung einer Polymerase ohne 5′-
3′-Exonukleaseaktivitat verhindert die Sondenhydrolyse. Aus 5′–3′-Exonukleaseaktivitat
verhindert die Sondenhydrolyse. Aus Rasmussen et al. (2003).
2.4.3.5 Quantitative real-time PCR
Die quantitative real-time PCR wird bei Genexpressionsstudien, bei der Bestimmung der
Viruslast in der Labordiagnostik, oder im Rahmen von Pathogenesestudien eingesetzt

2.4. Polymerasekettenreaktion 37
(Schang und Osorio, 1993; Heid et al., 1996; Bustin, 2000; van Elden et al., 2001;
Bensaude et al., 2004; Ward et al., 2004; Chung et al., 2005). Dabei wird der Ct-
Wert (s. Abbildung 2.10) als Bezugsgroße genutzt, da er sich umgekehrt proportional
zum Logarithmus der Ausgangsmenge an Nukleinsaure verhalt (Higuchi et al., 1993).
Die Quantifizierung kann relativ oder absolut erfolgen.
2.4.3.5.1 Absolute Quantifizierung
Die absolute Quantifizierung erlaubt eine relativ prazise Bestimmung der Viruslast, der
Kopienzahl oder der gesamten Nukleinsaurekonzentration. Als Ergebnis bekommt man
einen absoluten Zahlenwert mit einer Einheit (z. B. Kopienzahl, µg DNA). Die absolute
Quantifizierung kann sowohl mit als auch ohne interne Kontrolle erfolgen. Die Quantifizie-
rung ohne interne Kontrolle erfolgt anhand einer externen Standardkurve mit bekannten
Konzentrationen (Bustin, 2000; Pfaffl, 2004). Als interne Kontrolle kann eine exogene
DNA (oder RNA) verwendet werden, mit der jede Probe versetzt wird. Sie wird zusam-
men mit dem Zielgen amplifiziert und erlaubt so, falsch negative Ergebnisse zu erkennen
(Anonym, 2005b).
2.4.3.5.2 Relative Quantifizierung
Bei der relativen Quantifizierung erfolgt eine Quantifizierung der zu detektierenden Nu-
kleinsaure relativ zu einem Housekeeping-Gen. Das Hauptcharakteristikum eines Hou-
sekeeping-Gens ist, dass es moglichst in allen Geweben gleich hoch exprimiert wird. So
werden mit Hilfe externer Standards die absoluten Mengen von Ziel- und Referenzgen aus
dem gleichem Probenmaterial kalkuliert. In weiteren Tests werden die Zielgen Konzentra-
tionen als relative Menge gegenuber dem Referenzgen bestimmt (Bustin, 2000; Pfaffl,
2004; Anonym, 2005b).

38 2. Literaturubersicht
2.4.4 Design und Optimierung einer PCR
2.4.4.1 Auswahl der nachzuweisenden Genomregionen
Zum Nachweis von Viren mittels PCR werden spezifische Genomabschnitte ausgewahlt.
Dabei sollten die Primerbindungsstellen moglichst hoch konserviert sein, d.h. die Genom-
sequenzen durfen keine, bzw. nur wenige Unterschiede zwischen Vertretern einer Erreger-
spezies aufweisen. Bei den viralen Erregern stellen nicht kodierende Genomregionen oder
auch Gene fur Nichtstrukturproteine – also Abschnitte, die geringerem Evolutionsdruck
unterliegen, haufig geeignete Bereiche dar (Belak und Thoren, 2001).
2.4.4.2 Primer- und Sondendesign
Die optimale Produktlange fur die real-time RT-PCR liegt zwischen 80 und 100 bp (Bustin,
2000). Es ist auch moglich eine TaqMan-PCR mit Produktlangen bis 400 bp durchzufuhren
(Bustin und McKay, 1999), doch kurzere Fragmente werden effektiver amplifiziert und
sind toleranter in Bezug auf die Reaktionsbedingungen. Dies ist darauf zuruckzufuhren,
dass die kurzeren Fragmente leichter denaturieren, und so leichter von Primern und Son-
den gebunden werden. Die Amplifikationsrate der Taq-Polymerase betragt zwischen 30-
70 Basen/sec (Jeffreys et al., 1988), und so sind 15 sec ausreichend fur den Amplifika-
tionsschritt (Bustin, 2000).
Beim Primerdesign mussen vor allem folgende Kriterien berucksichtigt werden: die Schmelz-
temperatur (Tm), die Amplifikatgroße und die Lokalisation der TaqMan-Sonde. Die Schmelz-
temperatur ist definiert als diejenige Temperatur, bei der die Helixstruktur (zwischen
Primer und Zielstrang) zur Halfte verloren gegangen ist. Sie ist abhangig von der Basen-
zusammensetzung und wird nach Baldino et al. (1989) wie folgt berechnet:
Tm = 81, 5 + 16, 6(log10[J+]) + 0, 41( %G + C) − (675/n)
J = Konzentration monovalenter Kationen in mol/l
(% G + C) = Anteil der Basen Cytosin und Guanin in Prozent
n = Anzahl der Nukleotide

2.4. Polymerasekettenreaktion 39
Die optimale Primerlange betragt ca. 18-25 Basen (bis 30 b), der G/C-Gehalt sollte zwi-
schen 20 % und 70 % liegen und die Schmelztemperatur eines Primerpaares nicht mehr
als ein bis zwei ◦C von der angestrebten Temperatur (60 ◦C bei TaqMan-PCRs) abwei-
chen (Bustin, 2000). Ublicherweise werden die Primer in Konzentrationen von 50 nM bis
300 nM eingesetzt. Zu hohe Primerkonzentrationen begunstigen die Bildung von Primer-
Dimern und unspezifischen Produkten, zu niedrige Konzentrationen resultieren in herab-
gesetzter Sensitivitat der PCR.
Das Sondendesign unterscheidet sich nicht wesentlich von dem der Primer. Wie Bustin
(2000) beschreibt, sollte die Sonde ca. 30 Basen lang sein und eine um ca. 10 ◦C hohere
Schmelztemperatur als die Primer besitzen.
Die Auswahl der Reporterfarbstoffe ist in erster Linie von den detektierbaren Lichtwel-
lenlangen des real-time PCR-Gerates abhangig. Im Falle einer multiplex real-time PCR
mussen die Emissions- und Anregungsspektren verschiedener Farbstoffe weit genug von-
einander getrennt sein (s. Abbildung 2.13), damit keine Interferenzen bei der Fluores-
zenzmessung entstehen (Bustin und Nolan, 2004a; Bente, 2003). Die verschiedenen
Reporterfarbstoffe benotigen ensprechende Quencher. In der real-time multiplex PCR ha-
ben sich nichtfluoreszierende Quencher8 bewahrt. Die so genannten Black Hole Quencher
(BHQTM-1, BHQTM-2 und BHQTM-3; Biosearch Technologies, Novato, USA) besitzen kei-
ne Eigenfluoreszenz und geben das aufgenommmene Licht als Warme ab (Parkhurst
und Parkhurst, 1995a,b). Die Aufnahmespektren betragen bei BHQTM-1 480-580 nm,
BHQTM-2 550-650 nm und bei BHQTM-3 620-730 nm.
Informationen uber weitere Parameter und Richtlinien, die beim Primer- und Sonden-
design zu beachten sind, sind bei Mulhard (2002); Bustin und Nolan (2004d) und
Bente (2003) zu finden.
2.4.4.3 Ermittlung von DNA-Sekundarstrukturen
Fur eine effiziente Amplifikation der Ziel-DNA spielen Sekundarstrukturen, die die DNA
bei der Anlagerungstemperatur (Annealing) ausbildet, eine wichtige Rolle. Wichtig ist da-
bei, dass das 3′-Ende der Primer an einem moglichst linear vorliegenden Teil der Matrize
8Eine Ubersicht von Quenchern ist bei Bustin und Nolan (2004a) oder im WWW zu finden.

40 2. Literaturubersicht
450 500 750700650600550
Cy2 JOE TAMRA Cy5
FAM HEX ROX Cy7
TET Cy3 Texas Red
Abbildung 2.13: Emissonsspektren verschiedener Fluoreszenzfarbstoffe
bindet, um eine problemlose Anlagerung der Polymerase zu gewahrleisten (Peters et al.,
2004). Zuker (2003) entwickelte, basierend auf den Erkenntnissen von SantaLucia
(1998) zu DNA-Sekundarstrukturen unter verschiedenen thermodynamischen Bedingun-
gen ein Programm (mFold), welches die Uberprufung der moglichen Sekundarstrukturen
der Matrizen-DNA bei einer gegebenen Temperatur und Reaktionsbedingungen gestattet.
Dieses Programm ist frei zuganglich unter http://www.bioinfo.rpi.edu/applications/mfold.
2.4.4.4 Optimierungsschritte
Die Optimierungsschritte spielen bei der Entwicklung einer PCR, vor allem einer multi-
plex PCR, eine wichtige Rolle (Nolan, 2004). Der Einsatz von optimierten Primer- und
Sondenkonzentrationen erlaubt eine Reduktion der Konzentration im Ansatz, was wie-
derum die Sensitivitat und Spezifitat erhoht. Nicht zuletzt spielt der finanzielle Faktor
eine wichtige Rolle (Brisson et al., 2004). Die Optimierungsschritte erfordern den Einsatz
geeigneter Ziel-DNA, meistens sind es gereinigte PCR-Produkte oder linearisierte Plasmi-
de mit klonierten Zielsequenzen (s. 3.2.2.7). Nicht linearisierte zirkulare Plasmide stellen
kein geeignetes Ziel dar (Nolan, 2004). Nachdem die Identitat des Amplifikats gesichert
ist (s. 2.4.1), werden die optimalen Primerkonzentrationen mit Hilfe von SYBRTM-Green
real-time PCRs ermittelt (s. 3.2.2.4.2). Dies hat zum Ziel die Primerkonzentrationen zu

2.4. Polymerasekettenreaktion 41
ermitteln, bei denen die Template-DNA (bzw. cDNA) am effektivsten amplifiziert wird
(Nolan, 2004). Anschließend werden die optimalen Konzentrationen fur die Sonde ermit-
telt; je niedriger der Ct-Wert und je hoher der ∆Rn-Wert, umso besser funktioniert die
Sonde (Peters et al., 2004; Nolan, 2004). Doch die Fluoreszenz- und Ct-Werte alleine
geben keine Auskunft uber die Effizienz und die Kinetik der PCR. Die Erstellung einer
Standardkurve kann weitere Informationen uber Effizienz, Sensitivitat und Genauigkeit
der PCR geben (Higuchi et al., 1993; Bustin, 2000). Zur Erstellung der Standardkur-
ve werden die ermittelten Ct-Werte der Standardverdunnung gegen ihre logarithmische
Konzentration (z. B. Kopienzahl) aufgetragen. Die Neigung der Standardkurve korelliert
mit der Effizienz der PCR und wird nach folgender Formel berechnet:
Effizienz = [10(−1/slope)] − 1
slope = Neigung der Standardkurve
Wenn die Effizienz der PCR bei 100 % liegt, betragt die Neigung der Standardkurve -
3,32 (Bustin, 2000; Bustin und Nolan, 2004b). Das Bestimmheitsmaß - R2 (Pearson
Correlation Coefficient), ausgedruckt als eine Zahl zwischen null und eins, gibt Auskunft,
wie nah einzelne Komponenten in einem mehrfach Ansatz dem Mittelwert sind. Anhand
dieser Kriterien werden die einzelnen PCRs beurteilt und ggf. optimiert.
2.4.5 Multiplex real-time PCR
Die multiplex PCR stellt eine material- und zeitsparende Technik zur simultanen Am-
plifikation mehrerer PCR-Ziele in einem Reaktionsansatz dar. Dies spielt v. a. in dia-
gnostischen Tests und wissenschaftlichen Analysen eine Rolle, da die haufig begrenzte
Probenmenge ein limitierender Faktor ist (Bustin, 2000). Die Umsetzung erfolgt durch
den Einsatz eines oder mehrerer Primerpaare mit mehreren, unterschiedlich markierten
Sonden (Edwards und Gibbs, 1994; Mackay et al., 2002). Fur TaqMan-basierte Sys-
teme konnte gezeigt werden, dass bis zu drei bzw. vier PCR-Ziele erfolgreich miteinander
kombiniert werden konnen, ohne die Sensitivitat im Vergleich zu Einzelreaktionen we-
sentlich zu beeintrachtigen (Moody et al., 2000; Grace et al., 2003; van Rijn et al.,
2004). Entscheidend fur eine erfolgreiche Amplifikation ist ein sorgfaltiges Primerdesign.

42 2. Literaturubersicht
Die gewahlten Primer sollten eine maximale Spezifitat besitzen und eine moglichst maxi-
male Amplifikationseffizienz gewahrleisten (Brisson et al., 2004; Peters et al., 2004).
Dabei wird empfohlen, die Primer und Sonden fur das spezielle System neu zu entwerfen
und direkt aufeinander abzustimmen. Es konnte gezeigt werden, dass so entwickelte Pro-
tokolle leistungsfahiger sind (Brisson et al., 2004). Neben den Eigenschaften der Primer
sind auch deren optimale Konzentrationen von Bedeutung (Bej et al., 1990). Die Opti-
mierung der Reaktionskomponenten (dNTPs, MgCl2, Polymerase etc.) erfolgt analog zu
konventionellen PCRs. Besonderes Augenmerk gilt bei der Optimierung einer multiplex
PCR der Minimierung der moglichen Primer- und Sondeninteraktionen (Elnifro et al.,
2000). Real-time multiplex PCR-Protokolle sind in der humanmedizinischen Diagnostik
inzwischen verbreitet (Meng et al., 2001; Candotti et al., 2004; Templeton et al.,
2004). In den vergangenen Jahren wurden auch in der Veterinarmedizin derartige Proto-
kolle entwickelt (van Rijn et al., 2004; Huang et al., 2004; Cao et al., 2005a; Chung
et al., 2005; Belak, 2005; Kleiboeker et al., 2005).
Zu den multiplex real-time PCR-Protokollen, die fur die veterinarmedizinische Diagnostik
konzipiert und publiziert wurden, gehoren unter anderem Testprotokolle, die im Rahmen
eines EU Projektes (Projekt-Nummer QLK2-CT-2000-00486)9 entwickelt wurden, dessen
Ziel die Entwicklung, Standardisierung und Harmonisierung neuer, molekulardiagnosti-
scher Methoden zum Nachweis acht relevanter, porziner Erregern war. Das in so genannte
Workpackages (WP) untergliederte Projekt wurde von sechs europaischen Laboratorien
durchgefuhrt.
Das Konzept schloss das SHV-1, ASPV, KSPV, PRRSV, das Virus der Maul- und Klauen-
seuche (MKS-Virus), PPV, das Virus der Vesikularen Schweinekrankheit (SVDV - swine
vesicular disease virus) sowie Vesikulare Stomatitis Virus (VSV - vesicular stomatitis vi-
rus) ein. Die nachzuweisenden Erreger wurden in diesem Projekt so genannten Clustern
zugeordnet, die die Erreger nach Symptomenkomplexen einteilten. Der respiratorisch-
reproduktive Cluster beinhaltete Molecular Beacon-basierte multiplex-PCRs fur den Nach-
weis von KSPV/ASPV und PRRSV/SHV-1/PPV. Weiterhin wurden so genannte PriPro-
ET (Primer-Probe Energy Transfer system) fur den Nachweis von VSV, SVDV, MKS-
Virus, ASPV, KSPV und PPV etabliert. Mit diesem System wurden Nachweismetho-
den fur Erreger des”hamorrhagischen“ Clusters (ASPV/KSPV) und des
”vesikularen“
9http://www.multiplex-eu.org

2.5. Prinzipien der Validierung von Diagnosetests 43
(ehemalige OIE Liste-A Krankheiten) Clusters (SVDV/VSV/MKS-Virus) in multiplex-
Ansatzen entwickelt.
Neben der Etablierung der einzelnen Protokolle befasste sich das Projekt zudem mit der
Etablierung und Optimierung der Methoden zur Nukleinsaurereinigung und -extraktion
sowie mit der Entwicklung geeigneter Kontrollen.
2.4.6 Interne Kontrollen
Fur die Kontrolle der Nukleinsaureextraktion und Amplifikation empfiehlt es sich, in-
terne Kontrollen einzufuhren (Belak, 2005). Dabei konnen z. B. Housekeeping genes
verwendet werden. Unter Housekeeping genes versteht man eine Reihe konstitutiv expri-
mierter, proteinkodierender Gene, welche von jedem Zelltyp und in jedem Zellstadium
exprimiert werden. Typischerweise sind es Gene des Grundstoffwechsels (Thellin et al.,
1999). Die von diesen Genen kodierten mRNAs werden u. a. auch in Genexpressionsstu-
dien als interne Standards eingesetzt. Haufig verwendete Housekeeping genes sind β-Aktin
und Glycerinaldehyd-3-Phosphat-Dehydrogenase (GAPDH), 18S rRNA und β-2 Mikro-
globulin sowie Ubiquitin C (Schmittgen und Zakrajsek, 2000; Vandesompele et al.,
2002). Fur die Verwendung in quantitativen Studien sollte die Auswahl der internen Re-
ferenz individuell nach Prufung verschiedener Housekeeping genes erfolgen, da kein op-
timaler, allgemein gultiger Standard existiert (Bustin, 2002). Bei der Verwendung der
Housekeeping genes als interne Referenz in qualitativen Ansatzen, erfolgt der Einsatz
v. a. aufgrund der kontinuierlichen Expression und Nachweisbarkeit (Thellin et al.,
1999; Vandesompele et al., 2002).
2.5 Prinzipien der Validierung von Diagnosetests
2.5.1 Allgemein
Das Internationale Tierseuchenamt - OIE (Office International des Epizooties) in Paris
gibt (sinngemaß) folgende Definition eines validierten Tests: Die Validierung von Diagno-
setests ist eine Evaluierung eines diagnostischen Tests zur Bestimmung seiner Fahigkeit

44 2. Literaturubersicht
fur einen speziellen Einsatz. Ein validierter Test liefert konsequent Ergebnisse, die es er-
lauben, Tiere als positiv oder negativ einzustufen, und als Schlussfolgerung eine prazise
Aussage uber deren Infektionsstatus gibt. Das von OIE empfohlene Validierungsschema
(Anonym, 2004b) umfasst die folgenden funf Stufen und ist in Abbildung 2.14 dargestellt.
1. Feststellung der Durchfuhrbarkeit und Einsatzfahigkeit der Methode fur die Frage-
stellung
2. Wahl, Optimierung und Standardisierung der Reagenzien, Techniken und Methoden
3. Bestimmung der Testleistung
4. Fortlaufende Beobachtung der Testleistung
5. Fortfuhrung und Ausweitung der Validierung
2.5.2 Validierung von PCR
Der Validierung von PCR-Tests ist ein Kapitel im Manual of Diagnostic Tests and Vacci-
nes for Terrestrial Animals10 gewidmet (Anonym, 2004c), welches eine Spezifikation des
oben genannten Funf-Stufen-Konzepts darstellt.
10 http://www.oie.int/eng/normes/mmanual/A_00014.htm
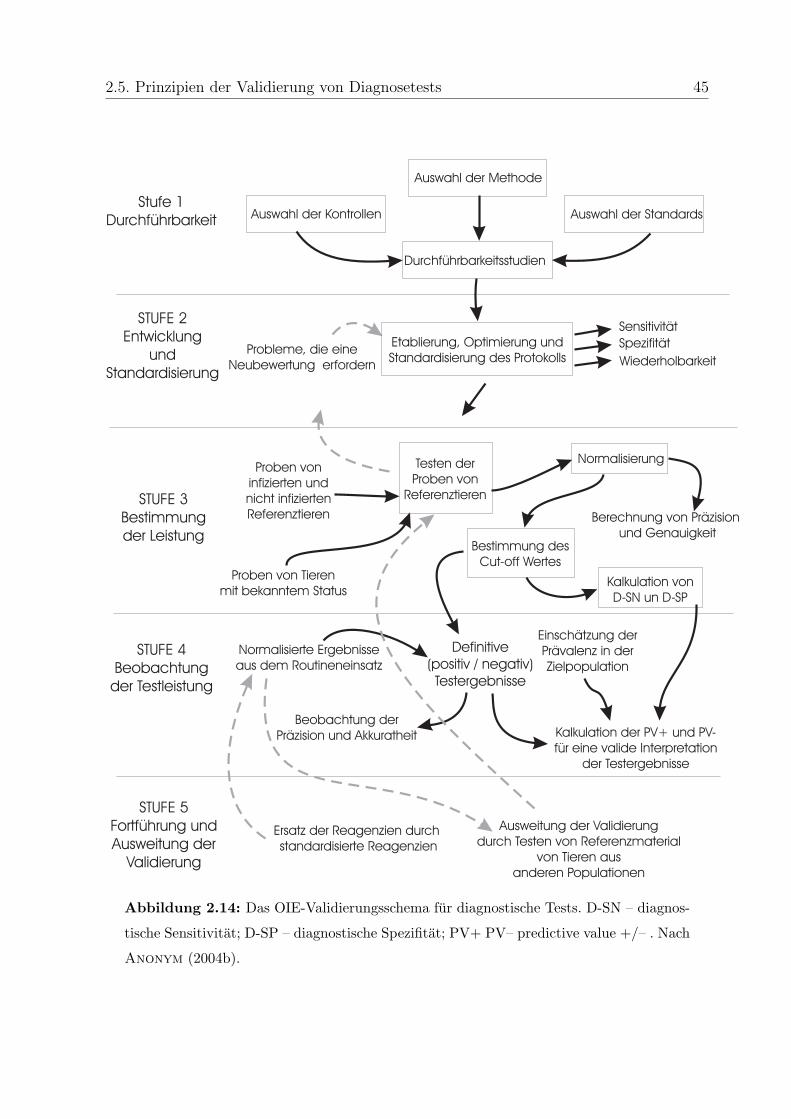
2.5. Prinzipien der Validierung von Diagnosetests 45
Abbildung 2.14: Das OIE-Validierungsschema fur diagnostische Tests. D-SN – diagnos-
tische Sensitivitat; D-SP – diagnostische Spezifitat; PV+ PV– predictive value +/– . Nach
Anonym (2004b).


3. Material und Methoden
3.1 Material
3.1.1 Zelllinien
3.1.1.1 PK(15)A
Die permanente epitheloide Zelllinie PK(15)A (porcine kidney (15) Amsterdam) wurde
1974 durch Einzelzellklonierung hergestellt1. Sie ist frei von BVDV, Mykoplasmen, Bak-
terien und Pilzen und wird im europaischen Referenzlabor (EURL) fur KSP als Referenz-
zelllinie fur die KSP-Diagnostik und KSPV-Vermehrung eingesetzt. Im Rahmen dieser
Arbeit diente sie ebenfalls der Vermehrung von KSPV.
3.1.1.2 EFN-R
Die EFN-R-Zellen stellen eine permanente epitheloide Zelllinie aus fetalen Schweinenie-
ren dar. Die Zelllinie ist frei von Mykoplasmen, Bakterien, Pilzen und einer Reihe boviner
und porziner Viren. Sie ist empfanglich fur verschiedene porzine Viren. Im Rahmen die-
ser Arbeit wurde sie zur Vermehrung von porzinem Influenza und porzinem Parvovirus
verwendet.
1Liess, personliche Mitteilung
47

48 3. Material und Methoden
3.1.1.3 MARC-145
Fur die Anzucht von PRRSV wurden die Zelllinie MARC-145 verwendet. Es handelt sich
hierbei um fetale Nierenzellen des Rhesusaffen, genauer um einen Klon der epitheloiden
Zelllinie MA-104. Die Zelllinie ist frei von Mykoplasmen, Bakterien, Pilzen, acht bovinen
und sieben porzinen Viren.
3.1.1.4 Rie 255
Die Anzucht der PPV-Isolate erfolgte auf der permanenten porzinen Hodenzelllinie STE,
die aus der Zellbank des Friedrich-Loeffler-Institutes (Bundesforschungsinstitut fur Tier-
gesundheit) stammte und dort unter der Katalognummer RIE 255 gefuhrt wird. Diese
polymorph-epitheloide Zelllinie ist permissiv fur PPV, SHV-1, verschiedene Enteroviren,
das Virus der transmissiblen Gastroenteritis und KSPV. Der Linienpass bestatigt die
Freiheit von Mykoplasmen und sieben porzinen und acht bovinen Viren.
3.1.2 Untersuchungsmaterial
3.1.2.1 Virusisolate und klinische Proben
Fur die Etablierung und Validierung der im Rahmen dieser Arbeit entwickelten PCRs wur-
den verschiedene Virusisolate, positives und negatives Probenmaterial sowie bestimmte
Plasmide verwendet. Die Isolate des Suiden Herpesvirus 1 (s. Tabelle 3.1) wurden freund-
licherweise vom Nationalen Referenzlabor fur Aujeszkysche Krankheit, Friedrich-Loeffler-
Institut, Wusterhausen zur Verfugung gestellt. Vier verschiedene Isolate des porzinen
Parvovirus wurden von Prof. U. Truyen, Universitat Leipzig, zur Verfugung gestellt. Im
Einzelnen handelt es sich um PPV–Stamm NADL-2 und drei Stamme mit der Bezeich-
nung”PPV-Nr.2“,
”PPV-Nr.3“ und
”PPV-Nr.4“.
Ein weiteres PPV- sowie ein PRRSV-Isolat, sowie funf Schweineseren in denen PCV-2
nachgewiesen wurde, wurden freundlicherweise von Dr. S. Kenklies, Landesamt fur Ver-
braucherschutz Sachsen-Anhalt zur Verfugung gestellt.
Drei Influenza-A Virusisolate mit der Bezeichnung”H3N2-Schwein“,
”H1N1-Schwein“ und
”KP“ stammten aus der Sammlung des Institutes fur Virologie der Stiftung Tierarztliche

3.1. Material 49
Hochschule Hannover.
Insgesamt zwolf porzine Influenzavirusisolate (s. Tabelle 3.2) wurden freundlicherweise
von der”IDT / Impfstoffwerk Dessau-Tornau GmbH“ zur Verfugung gestellt. Es han-
delte sich um sechs H3N2, vier H1N1 und zwei H1N2-Stamme, die uber embryonier-
te Huhnereier isoliert, und auf MDBK-Zellen kultiviert wurden. Die zellkulturinfektiose
Dosis50 (TCID50) lag bei allen Stammen uber 105,0 TCID50/ml. Die im Rahmen die-
Tabelle 3.1: SHV-1 Isolate.
Isolat-Nr. Orig. Nr. Herkunft
I 1 SA 229 / 92 Cottbus
I 18 AUZ 269 Frankfurt an der Oder
I 37 Vi 1804 / 95 Oldenburg / Stade
I 57 V 50 Stendal
I 311 Vi 736/37 Hannover
I 321 Vi 433 Hannover
I 351 Vi 936 Hannover
I 386 V 7231/95 Arnsberg
I 387 Vi 816 Berlin
I 397 VO / 12 / 14 Berlin
I 410 3386/95 Kassel
I 423 1325/96 Kassel
I 425 1329/96 Kassel
I 427 I1 / 51 Nurnberg
I 450 SP 9871/90 Osterreich
I 517 Frankreich
I 527 Frankreich
I 537 99,6 Frankreich
I 549 V 46302/98 CSR
I 550 1869 Land Brandenburg
I 611 PA 1520 / 2002 Koblenz
I 612 YSW 185 / 03 Land Brandenburg

50 3. Material und Methoden
ser Arbeit fur die Validierung und Etablierung der PCRs eingesetzten klinischen Proben
und Virusisolate verschiedener Pestiviren stammten aus der Sammlung des EURL fur
KSP, Hannover. Die in (s. Tabelle 3.3) dargestellte Probenauswahl wurde im Rahmen der
Spezifitatsprufung eingesetzt. Ein PPV-Isolat sowie PCV-2 positives Untersuchungsmate-
rial wurden freundlicherweise vom Staatlichen Veterinaruntersuchungsamt Arnsberg zur
Verfugung gestellt.
PCV-2 negatives Untersuchungsmaterial aus PCV-2 spezifisch pathogenfreien (SPF) Schwei-
nen wurde freundlicherweise von Frau Dr. B. Thur, IVI, Mittelhausern, Schweiz uberlassen
und als Negativkontrolle zur Etablierung und Validierung der PCR eingesetzt.
Uber 40 klinische Proben stellte freundlicherweise Frau Dr. E. große Beilage, Außenstelle
fur Epidemiologie, Bakum, Tierarztliche Hochschule Hannover zur Verfugung. Es handel-
te sich dabei um Proben aus der Routinediagnostik.
Zwei PRRSV Impfstamme wurden ebenfalls verwendet. Der Impfstoff Porcilis R© PRRS
Tabelle 3.2: Influenza-A – Isolate. Die Subtypen sind jeweils hinter der Stammbezeich-
nung in Klammern aufgefuhrt. Der Hamagglutinationstiter (HA-Titer) wurde von der IDT
GmbH bestimmt.
Lfd. Nr. Stamm Passage HA-Titer/
Influenza-A virus/swine Originalbestimmung
1 Berlin/5780/00 (H3N2) 2 1:512
2 Lohne/1/98 (H3N2) 2 1:256
3 Bakum/3543/98 (H1N1) 2 1:256
4 Belzig/54/01 (H3N2) 2 1:512
5 Lohne/1/97 (H3N2) 2 1:512
6 Belzig/2/01 (H1N1) 3 1:256
7 Bakum/1362/98 (H3N2) 2 1:256
8 Bakum/909/93 (H3N2) 2 1:64/128
9 Bakum/1832/00 (H1N2) 3 1:512/1024
10 Bakum/1833/00 (H1N2) 3 1:16/32
11 Bakum/5/95 (H1N1) 12 1:512
12 Potsdam/1/81 (H1N1) 3 1:1024

3.1. Material 51
Tabelle 3.3: Pestiviren-Isolate. VS = Virussuspension, OM = Organmaterial.
Datenbank Nr. Urspr. Name Jahr Land Wirt Genotyp Material
CSF0104 Diepholz 1/Han 94 1994 DE HS 2.3 VS
CSF0184 V 487/93 1993 DE WS 2.3 VS
CSF0277 V 1240/97 1997 DE HS 2.1 Serum
CSF0309 Kanagawa 1974 JP - Outgroup VS
CSF0573 Parma98 1998 IT HS 2.2 VS
CSF0634 VI 3837/38 1999 DE HS 2.3 OM
CSF0650 Guatemala HC GT HS 1.3 Serum
CSF0695 759/Ru 1999 RU HS 1.1 OM
CSF0902 Alfort 187 1968 FR 1.1 Serum
CSF0905 Brescia IT 1.2 OM
BVDV NADL BVDV 1a VS
BVDV 7443 BVDV 1 VS
5521-DE BVDV 2 VS
BDV Moredun BDV VS
BDV Frijters BDV VS
BDV S137-4 BDV VS
D15/99 Arnsberg Pestivirus VS
basiert auf europaischem Stamm DV des PRRSV (Fa. Intervet, Unterschleißheim), der
Ingelvac PRRS MLV auf amerikanischen Stammen ATCC VR2332 und ATCC VR2385
(Fa. Boehringer Ingelheim, Ingelheim).
3.1.2.2 Plasmide
Die Plasmide”Dixon 2“,
”Dixon 3“,
”Dixon 5“ und
”Dixon 6“ wurden freundlicherweise
von Dr. Donald P. King, Veterinary Laboratories Agency, Weybridge, UK, zur Verfugung
gestellt. Es handelte sich dabei um pGEM R©-T-easy Plasmide mit klonierter Sequenz des
VP72 Proteins des Virus der Afrikanischen Schweinepest mit einer Lange von 537 bp.

52 3. Material und Methoden
3.1.2.3 Antikorper und weitere Materialien
Die im Rahmen dieser Arbeit eingesetzten diagnostischen Antikorper und weitere Mate-
rialien sind im Anhang A.1 aufgefuhrt.
3.1.2.4 Kits zur Nukleinsaureisolierung
Kits zur Nukleinsaureisolierung und entsprechende Protokolle sind in Kapitel 3.2.2.1 auf-
gefuhrt.
3.2 Methoden
3.2.1 Klassische virologische Methoden
3.2.1.1 Zellkultur
Alle verwendeten Zelllinien wurden bei 37 ◦C und 5 % CO2-Atmosphare in 25 cm2 – Zell-
kulturflaschen (s. Anhang A.6.2) kultiviert und in Intervallen von drei bis vier Tagen
passagiert (falls nicht anders angegeben). Die Methoden und Medien waren fur die jewei-
lige Zelllinie spezifisch.
3.2.1.1.1 PK(15)A
Die Kultivierung der Zellen erfolgte in 75 cm2 Zellkulturflaschen (Greiner) unter Verwen-
dung von Eagle’s Minimum Essential Medium (EMEM) Kulturmedium mit 5 % fetalem
Kalberserum (FKS). Es wurden ca. 1,6 × 106 Zellen in acht ml Medium pro 75 cm2 ein-
gesat. Der Zellrasen war nach drei bis vier Tagen konfluent. Zur Zellpassage wurde das
Medium entfernt und der Zellrasen einmal mit adjusted trypsin-versen (ATV) gespult.
Anschließend wurden zwei ml ATV auf die Zellen gegeben und 15 bei 37 ◦C inkubiert.
Sobald sich der Zellrasen vom Untergrund gelost hatte, wurden die Zellen in acht ml
Medium aufgenommen, resuspendiert und zwei ml der Zellsuspension mit 25 ml Medium
weiterpassagiert.

3.2. Methoden 53
3.2.1.1.2 EFN-R
Die Kultivierung der EFN-R Zellen entsprach der der PK(15)A Zellen. Als Erhaltungs-
medium wurde EMEM-Kulturmedium mit Hanks-Salzen und 5 % FKS verwendet.
3.2.1.1.3 MARC-145
Die Kultivierung der MARC-145 Zellen entsprach in weiten Teilen der der PK(15)A Zellen.
Als Erhaltungsmedium wurde EDulb mit 10 % FKS Antibiotikazusatz verwendet.
3.2.1.2 Virustitration
Zur Titerbestimmung einer Virussuspension wurde eine Endpunktverdunnung eingesetzt.
Die zu titrierende Virussuspension wurde im entsprechenden Kulturmedium in log10 Stu-
fen von 10−1– 10−8 verdunnt. Vier Kavitaten einer Mikrotiterplatte wurden mit je 100 µl
der entsprechenden Verdunnungsstufe beschickt. Anschließend wurden jeder Kavitat 50µl
einer auf 300000 Zellen/ml Medium (unter Zusatz von 10 % FKS) eingestellten Zellsuspen-
sion zugesetzt. Die Kultur wurde 72 h in einer feuchten Kammer bei 37 ◦C und 5 % CO2 im
Brutschrank inkubiert. Der Virusnachweis erfolgte mit den unter 3.2.2.4 und 3.2.1.3 be-
schriebenen Methoden. Die Ermittlung des Titers erfolgte nach der Formel von Kaerber
(1931) in 50 % kulturinfektiosen Dosen (KID50) KID50.
logKID50 = L1,0 − Lint(S − 0, 5)
L1,0 Logarithmus der hochsten Verdunnungsstufe mit der Reaktionsrate 1,0
Lint = Logarithmus des Verdunnungsintervalls
S Summe der Reaktionsraten, begonnen bei der letzten Reaktionsrate, die 1,0 betragt
3.2.1.3 Peroxidase-Linked-Assay
24–72 Stunden nach der Infektion (abhangig von eingesetztem Virus) wurde der Zell-
kulturuberstand abgenommen und die Platten nach einmaligem Waschen mit 1/3 PBS

54 3. Material und Methoden
fur drei Stunden bei 80 ◦C in einem Heissluftsterilisator fixiert. Der Zellrasen der hitze-
fixierten Platte wurde nach dem Abkuhlen auf Raumtemperatur mit PBS-Tween (0,1 %
Tween, s. Anhang A.3.1) benetzt und jede Kavitat mit 50 µl der Konjugat-Losung befullt
(die Konjugatverdunnung war virusabhangig). Anschließend erfolgte eine einstundige In-
kubationzeit bei 37 ◦C. Danach wurde das Konjugat wieder abgenommen, die Platte
dreimal mit PBS-Tween und einmal mit A. bidest. gewaschen. Die Kavitaten wurden
nun mit 50 µl frisch angesetzter 3-Amino-9-Ethylcarbazol (AEC)-Substrat-Losung (AEC-
Dimethylformamid-Stammlosung, Natriumazetatpuffer pH 6,8 und 3 %iges H2O2, s. An-
hang A.3.1) beschickt. Nach etwa 10–15 min Inkubation war das Zytoplasma der infi-
zierten Zellen aufgrund einer peroxidasevermittelten Reaktion rot gefarbt. Anschließend
wurde das Substrat entfernt und die Kavitaten zweimal mit A. bidest. gewaschen und
aufgefullt. Die Auswertung der Platten erfolgte lichtmikroskopisch. Eine Kavitat galt als
positiv, wenn das Zytoplasma mindestens einer Zelle gefarbt war.
3.2.1.4 Virusvermehrung
Fur Virustitrationen, weitere Tests zur Validierung und Etablierung der PCRs und Be-
vorratung wurden die in dieser Arbeit eingesetzten Viren, soweit es moglich war, in der
entsprechenden Zellkultur (s. Kapitel 3.1.1) vermehrt. Dazu wurden die Zellen in 25 cm2
oder 75 cm2 Zellkulturflaschen ausgesat und 24 Stunden spater im semikonfluenten Zu-
stand mit einem ml Virussuspension inokuliert. Nach einer Adsorbtionszeit von 1 h bei
37 ◦C wurden sieben ml Erhaltungsmedium zugegeben und die Zellkultur fur 72 h bei 37 ◦C
und 5 % CO2 inkubiert. Darauf folgend wurde die infizierte Zellkultur einem Gefrier-Tau-
Zyklus (1 h bei −80 ◦C) unterzogen um intrazellular vorliegende Virionen freizusetzen. Das
Gemisch wurde anschließend 15 min bei 1750× g und 4 ◦C zentrifugiert. Der Uberstand
wurde vorsichtig dekantiert, portioniert und bei −80 ◦C gelagert.

3.2. Methoden 55
3.2.2 Molekularbiologische Methoden
3.2.2.1 Nukleinsaureisolierung
Fur die Nukleinsaureisolierung aus den unterschiedlichen Untersuchungsmaterialien wur-
den verschiedene kommerziell erhaltliche Systeme nach Herstellerangaben eingesetzt.
3.2.2.1.1 RNA Isolierung aus Zellkultur und Serum
Fur die RNA Extraktion aus dem Zellkulturuberstand wurde der High Pure Viral RNA
Kit der Fa. Roche, Mannheim (s. Anhang A.2) verwendet. Der Kit funktioniert nach fol-
gendem Prinzip. Nach der Lyse des eingesetzten Materials mit einem Detergenz wird die
virale RNA freigesetzt. Danach bindet die virale RNA in Anwesenheit chaotroper Sal-
ze selektiv an Glasfasern im Mikrozentrifugen-Rohrchen. Die RNA bleibt wahrend der
folgenden Waschschritten an die Membran gebunden. Schließlich wird die RNA mittels
Elutionspuffer von der Membran gelost.
Im Einzelnen wurden folgende Schritte durchgefuhrt. Die Arbeitslosung wurde herge-
stellt, indem aliquotierte poly(A) carrier RNA mit 2,5 ml Bindepuffer versetzt wurde. An-
schließend wurden 200 µl Zellkulturuberstand mit 400 µl Arbeitslosung gemischt, 10 min
bei Raumtemperatur (RT) inkubiert und danach auf das Mikrozentrifugen-Rohrchen mit
Glasfasern gegeben. Im Folgenden wurde 15 sec bei 13000× g zentrifugiert, 500 µl Inhi-
bitor Removal Puffer auf die Saule gegeben und erneut eine min bei 8000× g zentrifu-
giert. Danach wurde die Saule zwei Mal mit 450 µl Waschpuffer gewaschen und eine min
bei 8000× g zentrifugiert; schließlich noch Mal 10 sec bei 13000× g zentrifugiert. Der
Durchlauf und das Auffanggefaß wurden verworfen, die Saule in ein 1,5 ml Reaktionsgefaß
uberfuhrt. Die RNA wurde mit 50 µl Elutionspuffer eluiert und das Gefaß eine min bei
13000× g zentrifugiert. Die RNA wurde sofort in ein Eisbad gestellt und in der reversen
Transkription eingesetzt oder bei −80 ◦C eingefroren.
3.2.2.1.2 RNA-Isolierung aus Organmaterial und Blut
Die RNA-Isolierung aus Organ- und zellhaltigem Material erfolgte mit Hilfe des RNeasy R©
Lipid Tissue Kits.
Der Kit basiert auf der Phenol-Chloroform Extraktion mit anschließender Gesamt-RNA-

56 3. Material und Methoden
Isolierung uber Silica-Gel-Membran. Dabei lysiert das QIAzol -Reagenz (ein Phenol-Guanidin-
Isothiocyanat-Gemisch) das Probenmaterial. Nach Zugabe von Chloroform und darauf
folgender Zentrifugation trennt sich die Losung in wassrige und organische Phasen. Die
die RNA befindet sich in der oberen wassrigen Phase, die DNA in der mittleren und
Proteine in der unteren, organischen, oder in der mittleren Phase. Aus der oberen Phase
wird anschließend die RNA uber die Silica-Gel-Saule isoliert, und dabei durch mehrfaches
Waschen von Verunreinigungen befreit. Die Isolierung erfolgte geringgradig modifiziert
nach dem Protokoll des Herstellers, das folgende Schritte vorsah:
Ein etwa erbsengroßes Gewebestuck wurde steril entnommen, zerkleinert und in ein 1,5 ml
Reaktionsgefaß uberfuhrt. Unter Zugabe von 750 µl QIAzol-Reagenz wurde das Gewe-
bestuck mit einem Mikropistill homogenisiert. Nach 5 min Inkubation bei Raumtempe-
ratur wurden 200 µl Chloroform zugegeben und 15 sec geschuttelt. Es folgten drei min
Inkubation bei Raumtemperatur und anschließend 15 min Zentrifugation bei 12000× g
und 4 ◦C. Durch die Zentrifugation trennte sich die Probe in drei Phasen, eine obere
farblose, wassrige, eine mittlere weiße und eine untere rote, organische Phase. Die RNA
befand sich in der oberen Phase.
Die obere Phase (max. 600 µl) wurde in ein 1,5 ml Reaktionsgefaß uberfuhrt, ein Volu-
menaquivalent (600 µl) 70 %iges Ethanol zugegeben und beides vermischt. Darauf folgend
wurden bis zu 700 µl des Gemisches auf die RNeasy Saule gegeben und 15 sec bei 8000× g
zentrifugiert. Der Durchlauf wurde verworfen. Der Schritt wurde nochmal ein Mal wie-
derholt. Danach wurden 700 µl Puffer RW1 auf die RNeasy-Saule gegeben und 15 sec
bei 8000× g zentrifugiert. Es schloss sich ein zweimaliger Waschschritt mit 500 µl RPE-
Puffer und eine Zentrifugation fur 15 sec bei 8000× g an. Der Durchlauf wurde jeweils
verworfen. Danach wurde die Saule auf ein neues 1,5 ml Reaktionsgefaß gesetzt und 30 µl
RNase-freies Wasser auf die Membran der Saule geben, fur eine min bei Raumtemperatur
inkubiert und anschließend eine min bei 8000× g zentrifugiert. Die eluierte RNA wurde
sofort in ein Eisbad gestellt und entweder direkt in der reversen Transkription eingesetzt
oder bei −80 ◦C eingefroren.
3.2.2.1.3 DNA-Isolierung aus Zellkultur und Serum
Die DNA-Isolierung aus Zellkulturlysat und Serum erfolgte mit Hilfe des QIAamp R© DNA
Mini Kits (A.2). Die Extraktion erfolgte nach Protokoll des Herstellers, das folgende

3.2. Methoden 57
Schritte vorsah:
Die Probe wurde zunachst auf Raumtemperatur gebracht. Danach wurden und 200 µl
der Probe in ein 1,5 ml Reaktionsgefaß mit 20 µl Proteinase K verbracht. Anschließend
wurden 200 µl Puffer AL dazugegeben, gemischt und bei 57 ◦C fur 10 min inkubiert. Die
Probe wurde kurz anzentrifugiert, bevor 200 µl 96 %iges Ethanol dazugegeben und mit der
Probe vermischt wurden. Das Gemisch wurde anschließend auf die QIAamp Saule in einem
Reaktionsgefaß gegeben und bei 6000× g fur eine min zentrifugiert (der Durchlauf und
das Reaktionsgefaß wurden nach jedem folgenden Waschschritt verworfen und die Saule
in ein neues Reaktionsgefaß gesetzt). Danach wurden 500 µl des Puffers AW1 auf die
Saule gegeben und bei 6000× g fur eine min zentrifugiert. Anschließend folgte 500 µl des
Puffers AW2, der wiederum auf die Saule gegeben und bei 13000× g fur 3 min zentrifugiert
wurde. Die Saule wurde auf ein 1,5 ml Reaktionsgefaß gesetzt, 200 µl des Puffers AE
daraufgegeben und fur eine min inkubiert. Anschließend folgte Zentrifugation bei 6000× g
fur eine min. Die eluierte DNA wurde direkt in der PCR eingesetzt oder bei −20 ◦C
eingefroren.
3.2.2.1.4 DNA Isolierung aus Organmaterial und Blut
Fur die DNA-Isolierung wurde der DNeasy R© Tissue Kit verwendet. Dafur wurden ca.
25 µg Gewebe mit einer Skalpellklinge in kleine Stucke geschnitten, in ein 1,5 ml Reak-
tionsgefaß verbracht und mit 180 µl Puffer ATL und 20 µl Proteinase K gemischt. Die
Probe wurde nun bei 55 ◦C bis zu kompletten Lyse des Materials (1-3 h) inkubiert. Nach
kurzem Durchmischen wurden 200 µl Puffer AL zu der Probe dazugegeben, grundlich ge-
mischt und bei 70 ◦C fur 10 min inkubiert; anschließend wurden 200 µl 96 %iges Ethanol
dazupipettiert und nochmals grundlich gemischt. Das Gemisch wurde auf eine DNeasy
mini Saule gegeben und bei bei 6000× g fur eine min zentrifugiert. Der Durchlauf und
das Reaktionsgefaß wurden nach jedem Waschschritt verworfen und die Saule in ein neues
Reaktionsgefaß gesetzt. Danach wurden 500 µl des Puffers AW1 auf die Saule gegeben und
bei 6000× g fur eine min zentrifugiert. Anschließend wurden 500 µl des Puffers AW2 auf
die Saule gegeben und bei 13000× g fur 3 min zentrifugiert. Die Saule wurde in ein neues
1,5 ml Reaktionsgefaß gesetzt, 200 µl des Puffers AE daraufgegeben und fur eine min ste-
hen gelassen. Anschließend wurde sie bei 6000× g fur eine min zentrifugiert. Die eluierte
DNA wurde direkt in der PCR eingesetzt oder bei −20 ◦C eingefroren.

58 3. Material und Methoden
3.2.2.2 Reverse Transkription
Die reverse Transkription erfolgte nach einem laboreigenen Protokoll, welches von K. Fie-
big2 fur den Nachweis von PRRSV in der PCR optimiert wurde. Dafur wurden in einem
0,5 ml Reaktionsgefaß 26 µl des Mastermixes I (bestehend aus 8 µl 5×RT-Puffer, 8 µl
dNTP Mixes 2,5 mM je Nukleotid und 10 µl DEPC Wasser) und 7 µl RNA zusammen-
pippettiert und anschließend bei 80 ◦C fur 5 min inkubiert. Die Mischung wurde danach
sofort wieder in ein Eisbad gestellt. Nach dem Abkuhlen wurde der Mastermix II (beste-
hend aus 3,5 µl 0,1 M Dithiothreitol (DTT), 2 µl 1:15 Hexameren, 0,5 µl 25 mM MgCl2,
0,5 µl RNase Inhibitor und 1,5 µl Reverse Transkriptase (s. Anhang A.2)) dazupipettiert,
der Ansatz zuruck in den Cycler gestellt und das Programm fortgestzt. Es folgten 5 min
bei 25 ◦C, 1 h bei 55 ◦C, 5 min bei 99 ◦C und schließlich eine Abkuhlung auf 4 ◦C. Das
gesamte Programm ist in der Tabelle 3.4 dargestellt. Die auf diese Weise synthetisierte
cDNA wurde direkt in der PCR eingesetzt oder bei −20 ◦C eingefroren.
Tabelle 3.4: Reverse Transkription, thermisches Profil
Temperatur Dauer Anzahl der Zyklen
80 ◦C ∞ 1
80 ◦C 5 min 1
4 ◦C ∞ 1
25 ◦C 5 min 1
55 ◦C 1 h 1
99 ◦C 5 min 1
4 ◦C ∞ 1
3.2.2.3 Primer- und Sondendesign
3.2.2.3.1 Sequenzsuche und Alignment
Bei der Entwicklung der in dieser Arbeit beschriebenen PCRs3 wurden alle frei verfugba-
2K. Fiebig, personliche Mitteilung3Außer PRRSV. Nach Kleiboeker et al. (2005)

3.2. Methoden 59
ren Sequenzen in der Genomdatenbank GenBank 4 gesucht, heruntergeladen und lokal
gespeichert. Anschließend wurden die Sequenzabschnitte mit hoher Konservierung in den
relevanten Genomregionen anhand des mit Hilfe des Programms ClustalW (Thompson
et al. (1994) s. Anhang A.7) erstellten Alignments ermittelt. Das ClustalW wurde aus
dem Bioedit R© Programm (s. Anhang A.7) aufgerufen und bedient, das fur die Arbeit mit
Sequenzen benutzt wurde.
3.2.2.3.2 Auswahl von Primern und Sonden
Die Auswahl von Primern und Sonden erfolgte anhand der Kriterien, die unter 2.4.4.2
beschrieben sind. Bei der computergestutzten Primer- und Sondensuche wurde das Pro-
gramm Beacon Designer 2.13 (s. Anhang A.7) verwendet. Das Programm ermoglichte eine
gezielte Suche nach Primern und TaqMan-Sonden. Es konnten mehrere Sequenzen gleich-
zeitig bearbeitet werden, so dass es fur die Entwicklung von multiplex TaqMan-PCRs
geeignet war. Als Suchkriterien wurden in Tabelle 3.5 dargestellten Werte verwendet.
Da fur Pestiviren kein Primerpaar und TaqMan-Sonde mit Hilfe des Programms gefunden
wurde, wurde die Suche manuell unter Berucksichtigung der Richtlinien (s. Kapitel 2.4.4.2)
durchgefuhrt. Die Ergebnisse der Primer- und Sondensuche sind im Kapitel 4.2.1 zusam-
mengefasst.
3.2.2.3.3 Evaluierung von Primern und Sonden
Ermittlung von unspezifischen Bindungen
Fur die Ermittlung von unspezifischen Bindungen von Primerpaaren, die zu Amplifikation
von unspezifischen Produkten fuhren konnten, wurden das so genannte Basic Local Align-
ment Search Tool (BLAST)5 (Altschul und Koonin, 1998) eingesetzt. Dabei werden
eingegebene Oligonukleotid-Sequenzen mit allen in der BLAST-Datenbank verfugbaren
Sequenzen verglichen, gefundene Homologien ubermittelt und grafisch dargestellt.
4http://www.ncbi.nlm.nih.gov/GenBank/5Im Internet unter http://www.ncbi.nlm.nih.gov/blast verfugbar

60 3. Material und Methoden
Tabelle 3.5: Einstellungen und Parameter der Primer- und Sondensuche im Beacon De-
signer 2.13
Parameter Wert
Primer Length Range 19-30 bp
TaqMan Length Range 20-34 bp
Primer Target Tm 57 ◦C ± 1 ◦C
TaqMan Target Tm +10 ◦C ± 1 ◦C
Amplicon Length 75-120 bp
Hairpin max. ∆G -6 kcal/mol
Self Dimer max ∆G -9 kcal/mol
Run/Repeat Maximum Length 3 bp
Multiplexing ∆G -9 kcal/mol
Maximum bases between Primer and Taqman 9
Ermittlung von sekundaren Strukturen
Die Ermitllung der DNA-Sekundarstrukturen erfolgte mittels mFold-Programms (s. Ka-
pitel 2.4.4.3). Die Voreinstellungen des Programms wurden wie in Tabelle 3.6 gewahlt:
Tabelle 3.6: Einstellungen und Parameter fur das mFold Programm zur Ermittlung von
DNA-Sekundarstrukturen
Parameter Wert
DNA sequence linear
Folding temperature 60 ◦C
Ionic conditions [Na+] 1,0 M; [Mg++] 0 M
Correction type Oligomer
Percent suboptimality number 5
Upper bound on the number of computed foldings 50

3.2. Methoden 61
3.2.2.4 Polymerasekettenreaktion
3.2.2.4.1 Gel-basierte PCR
Bei der Entwicklung der real-time PCRs wurden verschiedene Tests und Optimierungs-
schritte vorgenommen (s. Kapitel 2.4.4.4). Zur Identifizierung der Produkte und zur Her-
stellung von PCR-Produkten fur die Klonierung wurde die konventionelle, gel-basierte
PCR angewandt. Benutzt wurde dabei der Abgene HotStart Mastermix (s. Anhang A.2).
In einem 0,5 ml PCR-Reaktionsgefaß wurden 45 µl des Mastermixes mit einer Hotstart-
Polymerase, drei µl Template und jeweils ein µl Forward- und Reverse-Primer (Konzentra-
tion variierte je nach Ziel) gemischt, kurz anzentrifugiert und in den PCR-Thermocycler
(s. Anhang A.6.1) verbracht. Anschließend wurde in Tabelle 3.7 dargestelltes Programm
gestartet.
Tabelle 3.7: Abgene HotStart PCR. Thermisches Profil
Temperatur Dauer Anzahl der Zyklen
95 ◦C 10 min 1
95 ◦C 1 min ⌉
56 ◦C 1 min 40
72 ◦C 30 sec ⌋
4 ◦C ∞
3.2.2.4.2 SYBRTM-Green real-time PCR
Fur jede PCR wurden die optimalen Primerkonzentrationen mit Hilfe der SYBRTM-Green
real-time PCR ermittelt. Dabei wurde eine Primermatrix verwendet, die die Primerkon-
zentrationen in den Stufen 50, 100, 300, 600 und 900 nM in allen moglichen Kombina-
tionen enthielt. Als Template wurden 104 Plasmidkopien mit entsprechenden klonierten
DNA-Sequenzen eingesetzt. Die Primermatrix ist in Abbildung 3.1 dargestellt.
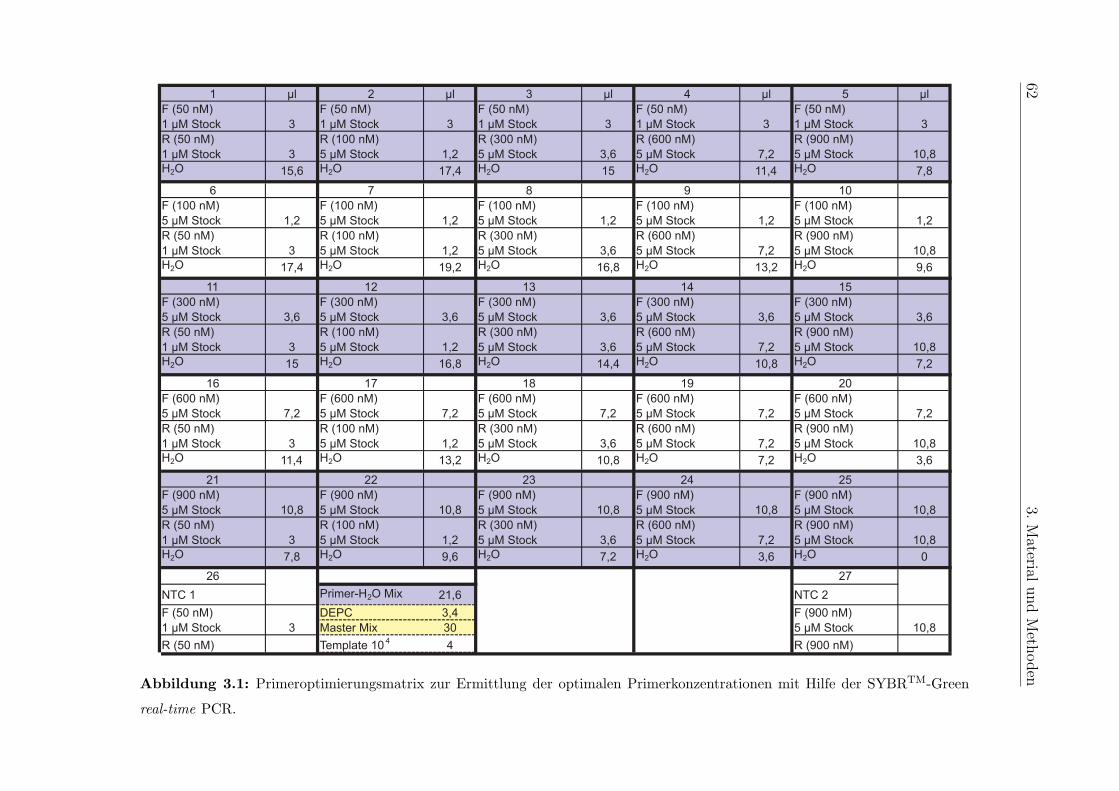
623.
Material
und
Meth
oden
Abbildung 3.1: Primeroptimierungsmatrix zur Ermittlung der optimalen Primerkonzentrationen mit Hilfe der SYBRTM-Green
real-time PCR.

3.2. Methoden 63
Fur die Durchfuhrung wurde der BrilliantTMSYBRTM-Green QPCR Master Mix verwen-
det (s. Anhang A.2). Fur jede Primerkombination wurden jeweils 26,1 µl Primer-Wasser
Mixes, 3,4 µl DEPC Wasser, 30 µl Mastermix und vier µl Template in je 0,5 ml Reakti-
onsgefaßen zusammen gemischt und jeweils 25 µl in zwei 0,2 ml real-time PCR Reakti-
onsgefaße uberfuhrt, in das real-time PCR-Gerat (Mx 4000, s. Anhang A.6.1) verbracht
und in der Tabelle 3.8 dargestelltes Programm gestartet.
Wahrend des Programms erfolgte eine Schmelzpunktanalyse. Das PCR-Produkt wurde
von 55 ◦C auf 95 ◦C in 1 ◦C Schritten erhitzt und dabei die Fluoreszenz gemessen.
3.2.2.4.3 TaqMan real-time PCR
Die TaqMan real-time PCR wurde in Einfach- oder Multiplex-Ansatzen durchgefuhrt. Als
Mastermix wurde der QuantiTect R© Multiplex PCR Kit (s. Anhang A.2), der alle notigen
Komponenten enthielt, verwendet. Es wurden noch 20×Primer-Sonden Mixe, Template
DNA (bzw. cDNA) und DNase/RNase-freies Wasser zu einem 25 µl Ansatz zusammenge-
mischt (s. dazu Tabelle 3.9), kurz anzentrifugiert und in 0,2 ml Reaktionsgefaße uberfuhrt.
Die Gefaße wurden in das real-time PCR-Gerat (Mx 4000, s. Anhang A.6.1) verbracht
und in der Tabelle 4.2 dargestelltes Programm gestartet. Die Fluoreszenz wurde in der
Annealing-Phase 3×fach gemessen.
Tabelle 3.8: SYBRTM-Green PCR. Thermisches Profil
Temperatur Dauer Anzahl der Zyklen
95 ◦C 10 min 1
95 ◦C 1 min ⌉
55 ◦C 1 min 40
72 ◦C 30 sec ⌋
95 ◦C 1 min
55 ◦C ↑ Schmelzpunktanalyse
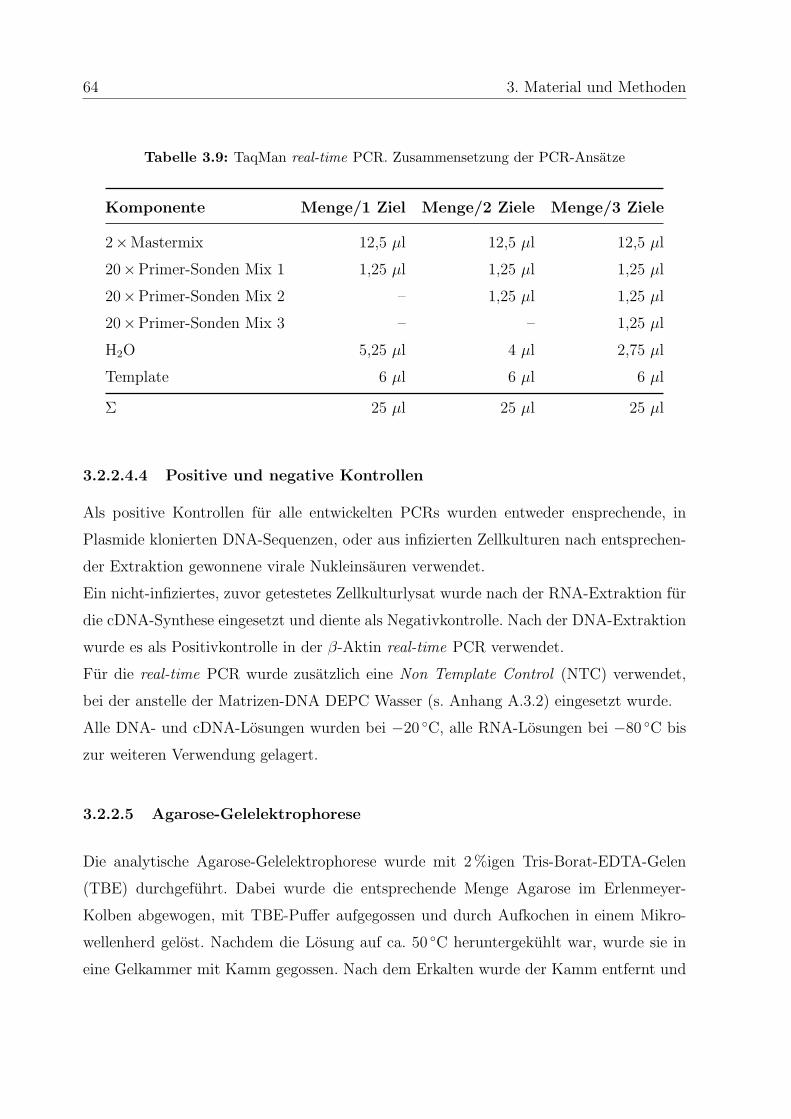
64 3. Material und Methoden
Tabelle 3.9: TaqMan real-time PCR. Zusammensetzung der PCR-Ansatze
Komponente Menge/1 Ziel Menge/2 Ziele Menge/3 Ziele
2×Mastermix 12,5 µl 12,5 µl 12,5 µl
20×Primer-Sonden Mix 1 1,25 µl 1,25 µl 1,25 µl
20×Primer-Sonden Mix 2 – 1,25 µl 1,25 µl
20×Primer-Sonden Mix 3 – – 1,25 µl
H2O 5,25 µl 4 µl 2,75 µl
Template 6 µl 6 µl 6 µl
Σ 25 µl 25 µl 25 µl
3.2.2.4.4 Positive und negative Kontrollen
Als positive Kontrollen fur alle entwickelten PCRs wurden entweder ensprechende, in
Plasmide klonierten DNA-Sequenzen, oder aus infizierten Zellkulturen nach entsprechen-
der Extraktion gewonnene virale Nukleinsauren verwendet.
Ein nicht-infiziertes, zuvor getestetes Zellkulturlysat wurde nach der RNA-Extraktion fur
die cDNA-Synthese eingesetzt und diente als Negativkontrolle. Nach der DNA-Extraktion
wurde es als Positivkontrolle in der β-Aktin real-time PCR verwendet.
Fur die real-time PCR wurde zusatzlich eine Non Template Control (NTC) verwendet,
bei der anstelle der Matrizen-DNA DEPC Wasser (s. Anhang A.3.2) eingesetzt wurde.
Alle DNA- und cDNA-Losungen wurden bei −20 ◦C, alle RNA-Losungen bei −80 ◦C bis
zur weiteren Verwendung gelagert.
3.2.2.5 Agarose-Gelelektrophorese
Die analytische Agarose-Gelelektrophorese wurde mit 2 %igen Tris-Borat-EDTA-Gelen
(TBE) durchgefuhrt. Dabei wurde die entsprechende Menge Agarose im Erlenmeyer-
Kolben abgewogen, mit TBE-Puffer aufgegossen und durch Aufkochen in einem Mikro-
wellenherd gelost. Nachdem die Losung auf ca. 50 ◦C heruntergekuhlt war, wurde sie in
eine Gelkammer mit Kamm gegossen. Nach dem Erkalten wurde der Kamm entfernt und

3.2. Methoden 65
das feste Gel samt Gelkammer in eine mit TBE-Puffer gefullte Kammer verbracht. An-
schließend wurden sieben µl der Proben mit drei µl eines Ladepuffers (s. Anhang A.3.2)
versetzt und in die Taschen gefullt; zusatzlich wurden drei µl eines Langenstandards
(s. Anhang A.4) ebenfalls mit 3 µl des Ladepuffers versetzt und aufgetragen. Es wurde
eine Spannung von 120 V fur ca. 90 min angelegt. Das Gel wurde anschließend in ei-
ner 0,2 %igen Ethidiumbromid-Losung ca. 10 min gefarbt und im Wasserbad ca. 20 min
gewassert. Durch UV-Licht eines Transilluminators (s. Anhang A.6.1) wurden die an-
gefarbten Banden bei 254 nm Wellenlange sichtbar gemacht, mit einem Bilderfassungs-
system (s. Anhang A.6.1) erfasst und zur Archivierung ausgedruckt.
3.2.2.6 Reinigung der PCR-Fragmente
Fur die TA-Klonierung wurden die eingesetzten PCR-Fragmente nach Herstellerempfeh-
lung von der Polymerase, Salzen, Primern und nicht verbrauchten Nukleotiden gereinigt.
Dabei wurden kommerziell erhaltliche Kits eingesetzt. Bei der Reinigung wird die DNA an
eine anionische Silica-Gel-Membran unter definierten niedrigen Salz- und pH-Bedingungen
gebunden und nach Waschschritten und Elution durch einen Puffer wieder gelost. Fur die
Reinigung kleiner Fragmente (ab 17 bp Lange) wurde der QIAquick Nucleotide Removal
Kit R© und fur PCR-Fragmente ab 100 bp Große der QIAquick PCR Purification Kit R©
verwendet. Dabei wurde der vollstandige PCR-Ansatz mit 10 Volumenequivalente Bin-
depuffer (PN-Puffer) versetzt, mit der Pipette gemischt und auf die Saule uberfuhrt.
Anschließend wurde das Auffanggefaß mit der Saule eine min bei 8000× g zentrifugiert.
Der Durchlauf wurde verworfen und 750 µl PE-Puffer erneut auf die Saule gegeben. Es
erfolgte ebenfalls eine Zentrifugation fur eine min bei 8000× g. Der Durchlauf wurde er-
neut verworfen und die Saule bei 13000× g fur eine min trocken zentrifugiert. Auf die
Saule wurden nun 55 µl Elutionspuffer pipettiert, die Saule in ein 1,5 ml Reaktionsgefaß
uberfuhrt und bei 10000× g fur eine min zentrifugiert.
Die eluierte DNA wurde entweder direkt in der TA-Klonierung eingesetzt oder bei −20 ◦C
gelagert.

66 3. Material und Methoden
3.2.2.7 TA-Klonierung
Bei der Klonierung der PCR-Produkte wurden der p-GEM R©-T-easy und der TOPO R© TA
Cloning Kit verwendet (s. Anhang A.2). Als Vektoren dienten im Falle des p-GEM R©-T-
easy-Kits das Standardplasmid p-GEM R©-T-easy, und beim TOPO R© TA Cloning Kit das
pCR R©2.1 Standardplasmid. Die Protokolle waren prinzipiell gleich aufgebaut. Im Einzel-
nen wurden beim p-GEM R©-T-easy-Kit folgende Schritte durchgefuhrt:
Als Erstes wurde der Ligationansatz vorbereitet. Dafur wurden funf µl 2× Rapid Ligation
Buffer, ein µl p-GEM R©-T-easy Vector (50ng), drei µl PCR Produkt, ein µl T4 DNA Ligase
in einem 0,5 ml Reaktionsgefaß zusammenpipettiert und eine Stunde bei Raumtemperatur
inkubiert. Anschließend wurden zwei µl des Ligationsansatzes in ein 1,5 ml Reaktionsgefaß
verbracht und 50 µl der JM109 High Efficiency Competent Cells auf Eis zugegeben. Das
Gefaß wurde kurz geschwenkt und 20 min auf Eis inkubiert. Danach folgte die Hitze-
schocktransformation in einem Wasserbad fur 45–50 sec bei 42 ◦C. Die Gefaße wurden
dann sofort wieder fur 2 min aufs Eis verbracht, danach 950 µl des S.O.C. Mediums
zugesetzt und 1,5 h bei 37 ◦C und 200 rpm in einem Brutschrank inkubiert. Anschlie-
ßend wurden 50 und 100 µl der Transformationskultur auf vorbereiteten IPTG/X-Gal
LB-Agarplatten ausplattiert. Die Platten wurden anschließend uber Nacht (16–24 h) bei
37 ◦C inkubiert. Die Selektion der Bakterienkolonien erfolgte am nachsten Tag anhand der
blau-weiß Farbung der Kolonien. Die Bakterien, die den Vektor mit der inserierten DNA
aufgenommen hatten, waren weiß gefarbt. Sie wurden in der anschließenden Kolonie-PCR
(s. 3.2.2.8) uberpruft und fur spatere Mini-Praparation (s. 3.2.2.9) in 5 ml LB-Medium
uber Nacht bei 37 ◦C und 200 rpm in einem Brutschrank weiter vermehrt.
Die Klonierung mit dem TOPO R© TA Cloning Kit (s. A.2) beinhaltete folgende Schritte:
Fur die Ligation wurden in einem 1,5 ml Reaktionsgefaß drei µl des PCR-Produkts mit
0,9 µl des pCR R©2.1-TOPO R© Vektors, ein µl Salzlosung und 1,1 µl sterilem Wassers ge-
mischt, funf min bei Raumtemperatur inkubiert und anschließend sofort auf Eis gestellt.
Zwei µl des Reaktionsansatzes wurden zu den chemokompetenten Bakterien (TOP10 aus
dem Kit) pipettiert und fur 30 min auf Eis inkubiert. Danach wurden die Bakterien fur
30 sec bei 42 ◦C hitzeschocktransformiert und sofort wieder auf Eis gestellt. Nach Zugabe
von 250 µl des S.O.C.-Mediums und einer Inkubation in einem Brutschrank bei 37 ◦C und
200 rpm fur 1 Stunde wurden die Bakterien auf Selektionsagarplatten in den Verdunnun-

3.2. Methoden 67
gen 1:50 und 1:100 ausplattiert und bei 37 ◦C inkubiert. Den Selektionsagarplatten wurden
50 µg/ml Ampicillin als Selektionsantibiotikum und 40 µg/µl X-Gal als Indikator zugege-
ben. Die Selektion der Bakterienkolonien erfolgte am nachsten Tag anhand der blau-weiß
Farbung der Kolonien analog zu dem p-GEMR©-T-easy-Kit. Die Bakterien, die den Vek-
tor mit der inserierten DNA aufgenommen hatten, waren weiß gefarbt. Sie wurden in
der anschließenden Kolonie-PCR (s. 3.2.2.8) uberpruft und fur spatere Mini-Praparation
(s. 3.2.2.9) in 5 ml LB-Medium uber Nacht bei 37 ◦C und 200 rpm in einem Brutschrank
weiter vermehrt.
3.2.2.8 Kolonie-PCR
Die so genannte Kolonie-PCR wurde eingesetzt, um die Bakterienkolonien, die den Vektor
mit der Produktsequenz bei der TA-Klonierung (s. Kapitel 3.2.2.7) aufgenommen hatten,
zu identifizieren. Fur die PCR-Reaktion wurde in der Tabelle 3.10 dargesteller Ansatz
auf Eis vorbereitet. Zu jedem Ansatz wurden die entsprechenden Primer hinzugegeben
Tabelle 3.10: Kolonie-PCR. Zusammensetzung des Mastermixes
Komponente Menge
dNTPs 8 µl
20×Tth-Puffer 5 µl
MgCl2 (25mM) 5 µl
Tth-Polymerase 0,5 µl
Fwd-Primer 1 µl
Rev-Primer 1 µl
DEPC-Wasser 79,5 µl
Σ 100 µl
(in der Konzentration 25 pmol/µl). Der Mastermix wurde anschließend auf 10 PCR-
Reaktionsgefaße verteilt, so dass ein Volumen von 10 µl in jedem Rohrchen vorhanden
war. Von den uber Nacht bei 37 ◦C inkubierten LB-Agarplatten wurden mit einer Pi-
pettenspitze eine einzelne Bakterienkolonie aufgenommen. Das Bakterienmaterial wurde

68 3. Material und Methoden
zuerst mit dem PCR-Ansatz in Verbindung gebracht, so dass es fur die PCR als Matrize
diente, und anschließend mit dem LB-Medium, so dass eine Vorkultivierung stattfinden
konnte. Die PCR-Reaktionsgefaße wurden in den Thermocycler verbracht und in der
Tabelle 3.11 aufgefuhrtes Programm gestartet. Gleichzeitig wurden die Vorkulturen bei
Tabelle 3.11: Kolonie-PCR. Thermische Profil
Temperatur Dauer Anzahl der Zyklen
94 ◦C ∞ 1
94 ◦C 1 min 1
94 ◦C 15 sec ⌉
54 ◦C 35 sec 19
72 ◦C 1 min ⌋
72 ◦C 5 min
4 ◦C ∞
37 ◦C und 200 rpm inkubiert. Nach der Kolonie-PCR wurden die PCR-Produkte mittels
Agarose-Gelelektrophorese (s. Kapitel 3.2.2.5) kontrolliert. Von den positiven Vorkultu-
ren wurden 10 µl zur Vermehrung in vorbereitetes LB-Medium mit Ampicillin ubertragen
und uber Nacht bei 37 ◦C und 200 rpm inkubiert.
3.2.2.9 Plasmidpraparation
Als Ausgangsmaterial fur die Plasmidpraparation diente eine entsprechende LB-Medium-
Ubernachtkultur, die mit einer einzelnen Bakterienkolonie oder einer entsprechenden Men-
ge Vorkultur (von der Kolonie-PCR) angeimpft wurde.
Die Plamidisolierung beruht auf dem Prinzip einer modifizierten alkalischen Lyse, gefolgt
von der Bindung der Plasmid-DNA an eine anionische Silica-Gel-Membran unter defi-
nierten niedrigen Salz- und pH-Bedingungen, die nach Waschschritten und Elution durch
einen Puffer wieder gelost wird. Die uber Nacht inkubierten Kulturen wurden 15 min
bei 2000× g zentrifugiert und anschließend mit dem QIAprep Spin Miniprep Kit R© gerei-
nigt. Nach der Zentrifugation wurde der Uberstand verworfen und das Pellet mit 250 µl

3.2. Methoden 69
P1-Puffer resuspendiert. Daraufhin wurde der Lysispuffer P2 hinzugegeben und durch
Schwenken mit der Zellsuspension gemischt. Mit der Zugabe von 350 µl des Puffers N3
wurden die Zellbestandteile gefallt. Nach 10 minutigem Zentrifugieren bei 10000× g wur-
de der Uberstand auf die QIAprep-Saulen uberfuhrt. Nach einem Zentrifugationsschritt
fur eine min bei 10000× g wurde der Durchfluss verworfen und die Saule mit 750 µl PE-
Puffer gewaschen. Anschließend wurde erneut fur eine min bei 10000× g zentrifugiert. Der
Durchlauf wurde verworfen. Auf die Saule wurden 50 µl EB-Puffer pipettiert und nach
1-minutiger Inkubation fur eine min bei 10000× g zenrtifugiert. Die eluierte Plasmid-
DNA wurde einer Konzentrationsmessung (s. Kapitel 3.2.2.13) und anschließend einem
Restriktionsenzymverdau unterzogen, zur Sequenzierung an die Fa. MWG–Biotech AG,
Ebersberg (s. Anhang 3.2.2.12) geschickt oder bei −20 ◦C gelagert.
3.2.2.10 Restriktionsenzymverdau
Die Linearisierung der Plasmide erforderte den Einsatz von Restriktionsenzymen. Die-
se sollten nur einmal im Vektor schneiden, nicht aber im Insert. Die passenden Enzyme
wurden ausgewahlt, indem die gesamten Sequenzen (Vektor und Insert) einer Restrikti-
onsschnittmusteranalyse unterzogen wurden (s. Anhang A.7). Anschließend wurden die
Reaktionen wie in Tabelle 3.12 dargestellt angesetzt und bei 37 ◦C uber Nacht inkubiert.
3.2.2.11 Dephosphorylierung
Eine Dephosphorylierung (Entfernung der terminalen 5′-Phosphatgruppen) von geschnit-
tener Plasmid-DNA wurde durchgefuhrt, um die nachfolgende Religation zu verhindern.
Direkt nach dem Restriktionsverdau wurde die geschnittene Plasmid-DNA (40µl) mit
vier µl der alkalischen Phosphatase sowie vier µl des entsprechenden Puffers fur 1 h bei
37 ◦C inkubiert und anschließend 15 min bei 65 ◦C gehalten, um das Enzym zu deaktivie-
ren. Danach wurde das Plasmid mit Hilfe des QIAquick PCR Purification Kit (s. 3.2.2.6)
gereinigt.

70 3. Material und Methoden
3.2.2.12 Sequenzierung
Alle Plasmide wurden zur Sequenzierung an die Firma MWG–Biotech AG, Ebersberg ver-
schickt. Dazu wurde ein µg Plasmid-DNA pro 100 zu sequenzierender Basen in ein 1,5 ml
Reaktionsgefaß verbracht und anschließend in einer Vakuumzentrifuge unter Warmezu-
fuhr getrocknet. Die Sequenzierung erfolgte mit Plasmidstandardprimern (T7 und M13).
3.2.2.13 Bestimmung der Kopienzahl der Plasmide
Die Berechnung der Kopienanzahl des Plasmids pro µl erfolgte anhand der DNA-Konzentration,
die wiederum mittels Messung der optischen Dichte (OD) bestimmt wurde. Die Messung
des OD-Wertes erfolgte zweimal bei 260 nm Wellenlange. Die Berechnung der DNA-
Konzentration erfolgte nach folgender Formel:
C[µg/ml] = OD260 × V × F
C = Konzentration der Ausgangslosung
OD260 = Optische Dichte bei 260 nm Wellenlange
Tabelle 3.12: Restriktionsenzymverdau der Plasmide mit klonierten PCR-Produkten
Plasmid DNA- Enzym Enzym- Puffer Puffer- H2O
Menge menge menge
ASPV 25µl Spe I 5µl Tango + BSA 5µl + 5µl 15µl
SHV-1 25µl EcoR V 5µl Reaction2 5µl + 5µl 15µl
Influenza-A 25µl Xmn I 2,5µl NEB2 + BSA 5µl + 2,5µl 15µl
KSPV 25µl Spe I 5µl Tango + BSA 5µl + 5µl 15µl
PCV-2 25µl Spe I 5µl Tango + BSA 5µl + 5µl 15µl
PPV 25µl Spe I 5µl Tango + BSA 5µl + 5µl 15µl
PRRSV-EU 25µl Spe I 5µl Tango + BSA 5µl + 5µl 15µl
PRRSV-NA 25µl Spe I 5µl Tango + BSA 5µl + 5µl 15µl

3.2. Methoden 71
V = Verdunnungsfaktor
F = Multiplikationsfaktor (50 fur dsDNA)
Fur die Berechnung der Kopienzahl wurde folgende Formel eingesetzt:
C[Kopien/µl] = NA ×OD260 × F × V × 10−9
L × MW
C = Konzentration der Losung in Plasmidkopien/µl
NA = 6,022×1023 ×mol−1 Avogadro-Konstante
OD260 = Optische Dichte bei 260 nm Wellenlange
V = Verdunnungsfaktor
F = Multiplikationsfaktor (50 fur dsDNA)
MW = Molekulargewicht je Basenpaar (660 g/mol)
L = Lange des Plasmids in Basen
10−9 = Umrechnungsfaktor
Nach der Berechnung der Konzentration wurde die Plasmid-Losung aliquotiert und ent-
weder in der real-time PCR eingesetzt oder bei −20 ◦C bis zur Verwendung gelagert.


4. Ergebnisse
4.1 Ergebnisse der Literaturrecherche
Sowohl fur den vorlaufigen Entwurf als auch kontinuierlich wahrend der verschiedenen
Entwicklungsphasen der PCR erfolgte eine Literaturrecherche in der Datenbank Medline1.
Zunachst wurden die Erkrankungen ermittelt, die als bedeutsame KSP Differentialdia-
gnosen viraler Genese bekannt waren. Die in der Literatur erwahnten, differentialdiagnos-
tisch relevanten Erkrankungen waren ASPV-, PCV-2-, PPV-, porzine Influenza-A-Virus-,
PRRSV- und SHV-1-Infektionen (van Oirschot, 1999b; Moennig et al., 2003). Die
Detektion dieser Erreger wurde daher fur den vorliegenden Ansatz angestrebt. In einem
weiteren Schritt wurden TaqMan-Protokolle gesucht, die fur die Detektion dieser Viren
entwickelt wurden. Diese Protokolle wurden hinsichtlich der gewahlten Genomabschnitte,
der Fragmentgroßen und der Thermoprofile ausgewertet. Die TaqMan-PCR-Protokolle,
die uber die Datenbank Medline gefunden werden konnten, sind in Tabelle 4.1 unter
Angabe der Referenz dargestellt. Die Tabelle enthalt alle bis dato publizierten Protokol-
le, obgleich fur die unmittelbare Entwicklung des vorliegenden Ansatzes nur die bis 2004
veroffentlichten Arbeiten berucksichtigt wurden. Allein das von Kleiboeker et al. (2005)
fur die Detektion und Differenzierung des PRRSV publizierte Protokoll wurde im weiteren
Verlauf der Entwicklung getestet und in den multiplex-Ansatz integriert. Fur die Detekti-
on des ASPV konnten zwei eigenstandige TaqMan-PCR-Protokolle gefunden werden, die
beide auf der Detektion eines Fragmentes des VP72-Gens beruhten. Fur das von King
et al. (2003) entwickelte Protokoll war eine Produktlange von 250 bp angegeben, diese
Information war der Publikation von Zsak et al. (2005) fur das dortige Protokoll nicht
1http://www.ncbi.nlm.nih.gov/entrez/query.fcgi
73

74 4. Ergebnisse
zu entnehmen. Die Thermoprofile dieser Protokolle wichen in einigen Punkten deutlich
voneinander ab. Das Protokoll von King et al. (2003) beinhaltete 35 Zyklen mit einem
Denaturierungsschritt bei 94 ◦C fur 30 sec, einem Annealing-Schritt bei 58 ◦C fur 60 sec
und einer Elongation bei 72 ◦C fur 45 sec. Fur das Protokoll von Zsak et al. (2005) waren
45 Zyklen mit den Schritten 95 ◦C fur 2 sec und 60 ◦C fur 60 sec angegeben.
Die fur die Detektion des SHV-1 entwickelten TaqMan-Protokolle amplifizierten hauptsach-
lich ein Fragment des Genombereichs, der fur das gB-Protein kodiert. Auch hier konnten
zwei Protokolle gefunden werden. Wahrend das von van Rijn et al. (2004) publizierte
Protokoll ausschließlich das gB-Fragment amplifizierte, war in der Publikation von Yoon
et al. (2005) auch die Amplifikation eines Fragments des gE-Gens vorgesehen. Den Publi-
kationen waren die Produktgroßen nicht zu entnehmen. Die Thermoprofile unterschieden
sich sowohl in der Dauer der einzelnen Schritte als auch der Zyklenzahl. Wahrend das Pro-
Tabelle 4.1: Ergebnisse der Literaturrecherche. Berucksichtigt wurden nur real-time
TaqMan-PCRs.
Virus Referenz
ASPV (King et al., 2003; Zsak et al., 2005)
SHV-1 (van Rijn et al., 2004; Yoon et al., 2005)
PCV-2 (Rovira et al., 2002; Brunborg et al., 2004; Olvera
et al., 2004; Chung et al., 2005)
PPV –
KSPV (Hofmann, 2003; Risatti et al., 2003; van Rijn et al.,
2004; Gaede et al., 2005; Hoffmann et al., 2005;
Risatti et al., 2005; Ophuis et al., 2006)
Influenza-A (van Elden et al., 2001; Watzinger et al., 2004; Hin-
diyeh et al., 2005; Krafft et al., 2005; Payungporn
et al., 2006)
PRRSV (Egli et al., 2001; van Rijn et al., 2004; Wasilk et al.,
2004; Revilla-Fernandez et al., 2005; Kleiboeker
et al., 2005)

4.1. Ergebnisse der Literaturrecherche 75
tokoll von van Rijn et al. (2004) 45 Zyklen mit einem Denaturierungsschritt bei 95 ◦C
fur 5 sec, einem Annealing-Schritt bei 60 ◦C fur 5 sec und einer Elongation bei 72 ◦C fur
10 sec vorsah, enthielt das Protokoll von Yoon et al. (2005) 50 Zyklen mit den Schritten
94 ◦C fur 60 sec, 60 ◦C fur 30 sec und 72 ◦C fur 60 sec.
Die TaqMan-Protokolle, die fur die Detektion des PCV-2 in der Literatur zu finden waren,
amplifizierten ausnahmslos ein Fragment aus dem ORF2 des viralen Genoms. Es konnten
vier Protokolle gefunden werden. Die Fragmentgroßen lagen bei 84 bp (Brunborg et al.,
2004), 98 bp (Olvera et al., 2004), 100 bp (Rovira et al., 2002) und 269 bp (Chung
et al., 2005). Die Thermoprofile sahen 45 (Rovira et al., 2002; Brunborg et al., 2004;
Chung et al., 2005) bzw. 40 (Olvera et al., 2004) Zyklen vor. Im Einzelnen waren die
Profile wie folgt: Das Protokoll von Brunborg et al. (2004) sah fur die 45 Zyklen einen
Denaturierungsschritt bei 95 ◦C fur 10 sec vor, der von 30 sec bei 62 ◦C gefolgt wurde.
Ahnliche Bedingungen wurden bei Olvera et al. (2004) verwendet. Die 40 Zyklen wurden
in diesem Fall mit 15 sec bei 95 ◦C und 60 sec bei 60 ◦C durchgefuhrt. Das Thermoprofil
der von Rovira et al. (2002) entwickelten PCR beinhaltete 45 Zyklen mit den Schritten
20 sec bei 95 ◦C, 20 sec bei 68 ◦C und ebenfalls 20 sec bei 70 ◦C. Das von Chung et al.
(2005) publizierte Protokoll weist ein Thermoprofil mit 45 Zyklen bei 95 ◦C fur 7 sec,
57 ◦C fur 10 sec und 72 ◦C fur 10 sec auf.
Fur das PPV konnten bis dato keine publizierten TaqMan-Protokolle gefunden werden.
Die fur die Detektion des KSPV entwickelten und publizierten TaqMan-Protokolle basie-
ren durchweg auf dem Nachweis eines Fragments aus der 5′NTR des viralen Genoms. Es
konnten der Datenbank funf eigenstandige PCR-Protokolle und ein modifiziertes Proto-
koll entnommen werden. Fur zwei der eigenstandigen Protokolle war die Produktgroße
angegeben, die bei 82 bp (Risatti et al., 2003) bzw. 93 bp (Hoffmann et al., 2005)
lag. Das von Ophuis et al. (2006) modifizierte Protokoll beruht auf den von Risatti
et al. (2003) publizierten Primern und Sonden, daher besitzt dieses Fragment ebenfalls
eine Große von 82 bp. Die Produktgroße der anderen Protokolle (Hofmann, 2003; van
Rijn et al., 2004) konnte den Publikationen nicht entnommen werden. Die verwendeten
Thermoprofile variierten in allen Parametern. Fur das von Hofmann (2003) entwickelte
Protokoll wurde ein Thermoprofil mit 50 Zyklen gewahlt, die aus einem Denaturierungs-
schritt bei 95 ◦C fur 30 sec und einer nachfolgenden Amplifikation bei 60 ◦C fur 60 sec
bestanden. Das von Risatti et al. (2003) publizierte Protokoll sah 45 Zyklen mit zwei sec

76 4. Ergebnisse
bei 95 ◦C und 30 sec bei 60 ◦C vor. Der Publikation von van Rijn et al. (2004) konnte
nur entnommen werden, dass 45 Zyklen mit einer Denaturierung bei 95 ◦C, einem An-
nealing-Schritt mit unbekannten Parametern und einer Elongation bei 72 ◦C angewendet
wurden. Das von Gaede et al. (2005) publizierte Protokoll nutzt fur die speziesspezifische
Differenzierung von Pestiviren verschiedene Primerpaare, die alle im Bereich der 5′NTR
binden. Das Primerpaar A11/A14 von McGoldrick et al. (1998) amplifiziert ein Frag-
ment von 220 bp Große. Das Pesti3/Pesti4 Primerpaar, das von Hyndman et al. (1998)
publiziert wurde, grenzt ein Genomfragment mit 171 bp Große ein. Die panpesti -Primer
324/326, die von Vilcek et al. (1994) publiziert wurden, amplifizieren ein 288 bp großes
Genomfragment. Das thermische Profil bestand aus einem Schritt fur die reverse Tran-
skription bei 50 ◦C fur 30 min, anschließender Denaturierung und Polymerasenaktivierung
bei 95 ◦C fur 15 min, gefolgt von 45 Zyklen Denaturierung bei 94 ◦C fur jeweils 20 sec
und einem Annealing-/Elongation-Schritt bei 60 ◦C fur 1 min. Das von Hoffmann et al.
(2005) beschriebene Protokoll sieht ein Thermoprofil mit 42 Zyklen vor. Diese bestehen
aus einem Denaturierungsschritt fur 15 sec bei 94 ◦C, einem Annealing der Primer fur
30 sec bei 57 ◦C und einer Elongation-Phase fur 30 sec bei 68 ◦C. Ophuis et al. (2006)
modifizierte das von Risatti et al. (2003) entwickelte Protokoll dahingehend, dass das
Thermoprofil bei gleich bleibender Zyklenzahl eine auf 15 sec verlangerte Phase bei 95 ◦C
und eine auf 60 sec verlangerte Phase bei 60 ◦C aufwies.
Fur die Detektion verschiedener Influenza-A-Viren wurde vornehmlich der fur das M-Protein
kodierende Genomabschnitt verwendet. Vier publizierte, neu entwickelte TaqMan-PCR-
Protokolle sowie ein modifiziertes Protokoll wurden hinsichtlich ihrer Parameter vergli-
chen. Mit der Ausnahme eines von Payungporn et al. (2006) entwickelten Protokolls
zur Detektion von Influenza-A-Viren des Subtyps H5N1, das neben dem Genomfragment
des M-Proteins zusatzlich die fur H5 und N1 kodierenden Genomabschnitte detektierte,
basierten die Protokolle ausschließlich auf der Detektion des M-Protein-Genomfragments.
Das von van Elden et al. (2001) entwickelte Protokoll, das auch dem von Hindiyeh
et al. (2005) modifizierten Protokoll zugrunde liegt, beinhaltet zwei Forwardprimer, was
zu unterschiedlichen Amplifikatlangen fuhrt. Die Fragmentlange betrug anhand der Lo-
kalisation der Primer 188 bp bzw. 128 bp. Das Protokoll nach Watzinger et al. (2004)
fuhrt zu einem 132 bp langen PCR-Produkt. Ein 92 bp langes Fragment wird mittels der
von Krafft et al. (2005) entwickelten PCR amplifiziert, das von Payungporn et al.

4.1. Ergebnisse der Literaturrecherche 77
(2006) publizierte PCR-Protokoll detektiert ein 191 bp langes Fragment (M-Protein). Die
von Hindiyeh et al. (2005) und Watzinger et al. (2004) entwickelten bzw. modifizier-
ten Protokolle besaßen ein identisches Thermoprofil mit 50 Zyklen, die aus 15 sec bei
95 ◦C und 60 sec bei 60 ◦C bestanden. Das Protokoll nach van Elden et al. (2001) wich
nur hinsichtlich der Zyklenzahl ab, da es nur 45 Zyklen vorsah. Ein Thermoprofil mit 50
Zyklen sah das Protokoll von Krafft et al. (2005) vor mit 15 sec bei 95 ◦C und 60 sec
bei 55 ◦C. Die von Payungporn et al. (2006) entwickelte PCR sah ein Thermoprofil mit
40 Zyklen vor, die aus einer Denaturierungsphase bei 94 ◦C fur 15 sec, einem Annealing
der Primer fur 30 sec bei 55 ◦C und einer Elongation-Phase fur 40 sec bei 72 ◦C bestanden.
Zur Detektion des PRRSV wurden bis dato funf publizierte TaqMan-Protokolle gefunden.
Sie basieren hauptsachlich auf der Detektion des ORF7 bzw. ORF6, teilweise in Kombi-
nation mit Fragmenten aus der 3′NTR. Das von Egli et al. (2001) publizierte Protokoll
zur Detektion von PRRSV-EU und PRRSV-NA fuhrte zur Amplifikation eines Fragments
aus dem ORF7. Die Lange des Amplifikats betrug 105 bp fur PRRSV-NA und 96 bp fur
PRRSV-EU. Das Thermoprofil sah 40 Zyklen vor, die aus einem Denaturierungsschritt
bei 95 ◦C fur 15 sec und einem Amplifikationsschritt bei 60 ◦C fur 60 sec bestanden. Das
von van Rijn et al. (2004) publizierte Protokoll amplifizierte ein Fragment aus ORF6 und
ORF7. Die Angaben zum Thermoprofil waren unvollstandig und entsprachen denen fur
KSPV. Die von Wasilk et al. (2004) entwickelte PCR beruhte auf einem kommerziellen
System (Tetracore Inc., Gaithersburg) und amplifizierte neben nicht weiter charakterisier-
ten Genomfragmenten ein Fragment aus der 3′NTR. Dieses Protokoll sah fur PRRSV-NA
ein Thermoprofil mit 40 Zyklen vor, die aus 30 sec bei 94 ◦C, 60 sec bei 61 ◦C und 60 sec
bei 72 ◦C bestanden. Das Profil fur PRRSV-EU bestand aus 45 Zyklen. Diese sahen 3 sec
bei 95 ◦C und 30 sec bei 60 ◦C vor. Das von Revilla-Fernandez et al. (2005) publizierte
Protokoll sah die Amplifikation eines Fragmentes aus dem ORF6 vor. Das Thermoprofil
bestand aus 45 Zyklen, die mit 15 sec bei 95 ◦C und 60 sec bei 60 ◦C durchgefuhrt wurden.
Die PCR, die von Kleiboeker et al. (2005) entwickelt wurde, beruhte fur PRRSV-EU
auf der Amplifikation eines 77 bp Fragments aus dem ORF7, fur PRRSV-NA auf der Am-
plifikation eines 114 bp Fragments aus dem ORF7 und der 3′NTR. Das thermische Profil
sah einen initialen Schritt fur die reverse Transkription bei 50 ◦C fur 30 min, gefolgt von
40 PCR Zyklen vor. Jeder Zyklus bestand aus einem Denaturierungsschritt bei 94 ◦C fur
15 sec und einem Schritt fur Annealing und Elongation bei 60 ◦C fur 15 sec.

78 4. Ergebnisse
Das anhand der Literatur gewahlte und modifizierte Profil ist in Tabelle 4.2 dargestellt.
Nach Anpassung der Zyklenzahl und -dauer wurde es in der weiteren Arbeit eingesetzt.
Die Aktivierung der Polymerase erfolgte, wie vom Hersteller empfohlen, fur 15 Minuten
bei 95 ◦C. Der Denaturierungschritt wurde auf eine Dauer von 15 sec festgelegt. Die Schrit-
te Annealing und Elongation wurden zusammengefuhrt, und erfolgten bei 60 ◦C fur eine
Minute. In der Literatur werden meistens Zyklenzahlen zwischen 40 und 50 verwendet. In
dieser Arbeit wurde ein Wert von 40 ausgewahlt.
Tabelle 4.2: TaqMan-PCR. Thermisches Profil.
Temperatur Dauer Anzahl der Zyklen Schritt
95 ◦C 15 min 1 Aktivierung der Polymerase
95 ◦C 15 sec ⌉ Denaturierung
40
60 ◦C 1 min ⌋ Annealung und Elongation
4.2 Entwicklung der multiplex real-time TaqMan PCRs
4.2.1 Ergebnisse der Sequenz-, Primer- und Sondensuche
Die Genomregionen der gewahlten Erreger sowie der internen Kontrolle, die fur eine Am-
plifikation in PCR in Frage kamen, wurden anhand der Literatur und eigener Studien
ausgewahlt. Sequenzabschnitte mit moglichst hoher Konservierung und fur die Entwick-
lung verschiedener PCR-Protokolle von anderen Autoren zur Amplifikation verwendete
Regionen wurden zunachst naher untersucht. Im nachsten Schritt wurden alle verfugba-
ren Sequenzen einem ClustalW-Alignment unterzogen und Consensus-Sequenzen erstellt.
Nach einem Vergleich der in der Sequenzdatenbank GenBank 2 verfugbaren Sequenzen
wurde fur das ASPV ein Primerpaar im Bereich des VP72-Gens ausgewahlt, mit dem ein
96 bp langes Genomfragment amplifiziert wurde. Die TaqMan-Sonde hatte eine Lange von
2http://www.ncbi.nlm.nih.gov/GenBank/

4.2. Entwicklung der multiplex real-time TaqMan PCRs 79
25 Basen. Bei der Entwicklung der pestivirenspezifischen Primer und der Sonde wurden
alle verfugbaren KSPV, BVDV und BDV-Sequenzen ausgewertet. Die ausgewahlten Pri-
mer grenzten ein 164 bp langes Fragment im Bereich der 5′NTR ein, in dem die 26 Basen
lange TaqMan-Sonde band. Fur das PPV konnte nach Auswertung der Sequenzen ein
96 bp langer Bereich des VP2-Gens ausgewahlt werden. Die hier hybridisierende TaqMan-
Sonde hatte eine Lange von 29 Basen. Im zirkularen Genom des PCV-2 wurde der ORF2
fur die Primer- und Sondensuche verwendet. Die ausgewahlten Primer amplifizierten ein
Fragment mit einer Große von 136 bp, die TaqMan-Sonde war 25 Basen lang. Im SHV-1
Genom wurde der fur das gB-Protein kodierende Bereich ausgewahlt. Die entwickelten
Primer grenzten ein 96 bp langes Fragment ein, in dem die 29 Basen lange Sonde band.
Fur das Influenza-A-Virus wurde der Bereich des M1-Proteins gewahlt. Das ausgewahlte
Primerpaar amplifizierte ein 96 bp langes Produkt, an das eine 22 Basen lange Sonde band.
Die Primer und Sonden fur die PRRSV spezifischen PCRs wurden aus der Publikation von
Kleiboeker et al. (2005) ubernommen. Ein Primerpaar amplifizierte ein 77 bp langes
Genomfragment des ORF7 europaischer PRRSV-Stamme. Bei den nordamerikanischen
Stammen wurde mit zwei Forward -Primern und einem Reverse-Primer ein 114 bp langes
Amplifikat aus dem Bereich des ORF7 und 3′NTR gebildet. Die zwei stammspezifischen
Sonden hatten jeweils eine Lange von 24 Basen.
Die Primer und Sonden wurden fur jeden Erreger so ausgewahlt, dass die Amplifikationen
mit dem vorgegebenen einheitlichen Thermoprofil ablaufen konnten.
Als interne Kontrolle wurde das β-Aktingen gewahlt. Die Auswahl erfolgte anhand einer
genomischen und der dazugehorigen mRNA-Sequenz. Dabei wurde ein Primerpaar fur die
Amplifikation sowohl genomischer DNA als auch die mRNA gewahlt. Die Produktgroße
fur die genomische DNA betrug 185 bp und fur die mRNA 90 bp (s. Abbildung 4.1). Es
wurden je eine TaqMan-Sonde entwickelt; die DNA-Sonde war 21 bp, die mRNA 23 bp
lang. Weitere Ergebnisse dazu sind in Kapitel 4.2.2.4 aufgefuhrt. Die gewahlten Primer-
und Sondensequenzen sowie die Produktlangen und Genombereiche sind in Tabelle 4.3
zusammengefasst. Alle Primer- und Sondensequenzen sind in 5′-3′ Orientierung angege-
ben.
Die nach Vorgabe der in Kapitel 3.2.2.3.2 beschriebenen Methoden entwickelten Primer
und Sonden wurden einer BLAST-Suche unterzogen (s. Kapitel 3.2.2.3.3). Dabei wurden
sie sowohl einzeln als auch in Kombination mit der entsprechenden TaqMan-Sonde un-

80 4. Ergebnisse
Abbildung 4.1: Ergebnisse der gelbasierten β-Aktin PCR. Mit ausgewahltem Primerpaar
werden je nach Ausgangsmaterial unterschiedliche Produkte amplifiziert. Bei der genomi-
schen DNA betragt die Produktgroße 185 bp, bei cDNA aus β-Aktin mRNA 90 bp. 1 -
Langenstandard, 2 - NTC, 3, 4 - DNA (Organmaterial), 5, 6, 7 - cDNA (Organmaterial
nach RNA-Extraktion und reverser Transkription).
tersucht. Bei der BLAST-Suche wurden keine uber drei bis vier Basen hinausgehenden
Sequenzidentitaten mit anderen bekannten Organismen festgestellt. Anschließend fand ei-
ne computergestutzte Ermittlung sekundarer Strukturen bei der Annealing-Temperatur
von 60 ◦C mittels des mFold-Programms (s. Kapitel 2.4.4.3) statt. Die graphische Darstel-
lung der Genomabschnitte in den Primer und Sonden banden, zeigten fur Pestiviren- sowie
Influenza-A-Primer und -sonden Sekundarstrukturen im relevanten Genomabschnitt. Die
graphische Ausgabe des mFold-Programms fur PCV-2, welches keine Sekundarstrukturen
im Bereich der 3′-Enden aufwies, ist in Abbildung 4.2 dargestellt. Eine mogliche cDNA-
Konformation fur das Pestivirusgenom ist in Abbildung 4.3 dargestellt.

4.2. Entwicklung der multiplex real-time TaqMan PCRs 81
Abbildung 4.2: Ermittlung von DNA-Sekundarstrukturen am Beispiel von PCV-2. Die
Abbildung zeigt die angenommene DNA-Konformation bei 60 ◦C. Die 3′-Enden der Primer
lagen in Abschnitten ohne sekundare Strukturen.

82 4. Ergebnisse
Abbildung 4.3: Ermittlung von DNA-Sekundarstrukturen des Pestivirusgenoms. Die
Abbildung zeigt eine mogliche cDNA-Konformation bei 60 ◦C.
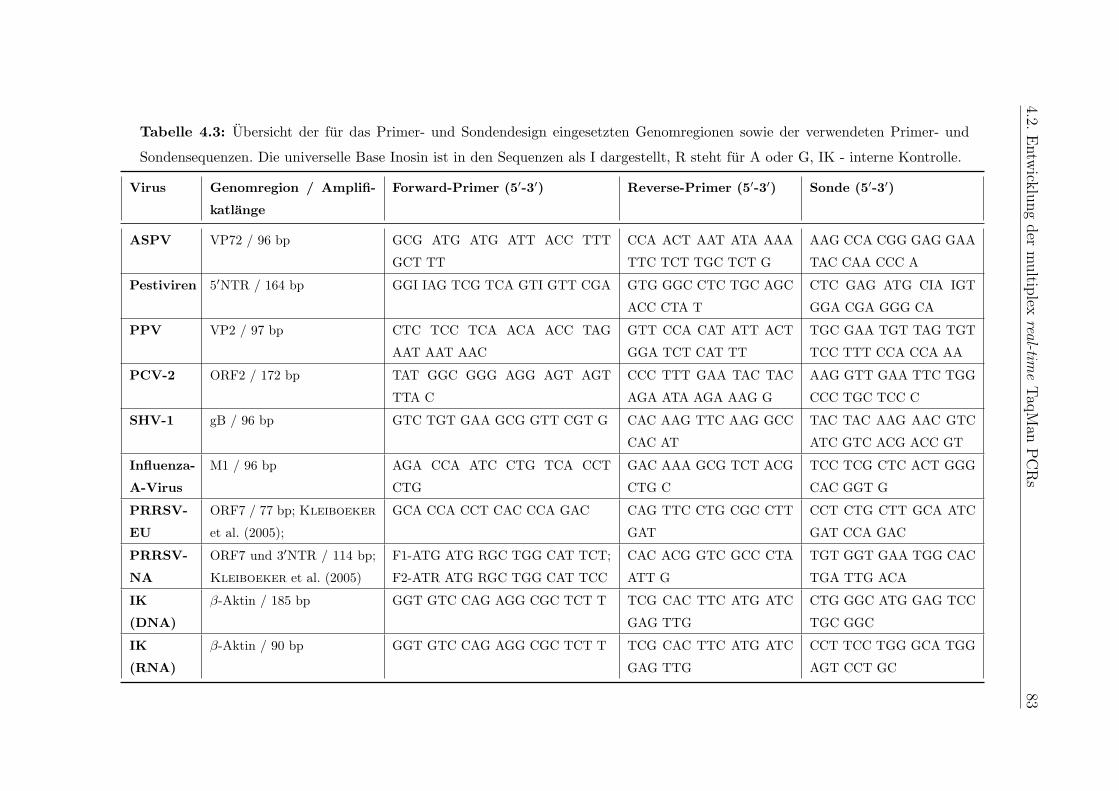
4.2.E
ntw
icklu
ng
der
multip
lexreal-tim
eTaq
Man
PC
Rs
83
Tabelle 4.3: Ubersicht der fur das Primer- und Sondendesign eingesetzten Genomregionen sowie der verwendeten Primer- und
Sondensequenzen. Die universelle Base Inosin ist in den Sequenzen als I dargestellt, R steht fur A oder G, IK - interne Kontrolle.
Virus Genomregion / Amplifi-
katlange
Forward-Primer (5′-3′) Reverse-Primer (5′-3′) Sonde (5′-3′)
ASPV VP72 / 96 bp GCG ATG ATG ATT ACC TTT
GCT TT
CCA ACT AAT ATA AAA
TTC TCT TGC TCT G
AAG CCA CGG GAG GAA
TAC CAA CCC A
Pestiviren 5′NTR / 164 bp GGI IAG TCG TCA GTI GTT CGA GTG GGC CTC TGC AGC
ACC CTA T
CTC GAG ATG CIA IGT
GGA CGA GGG CA
PPV VP2 / 97 bp CTC TCC TCA ACA ACC TAG
AAT AAT AAC
GTT CCA CAT ATT ACT
GGA TCT CAT TT
TGC GAA TGT TAG TGT
TCC TTT CCA CCA AA
PCV-2 ORF2 / 172 bp TAT GGC GGG AGG AGT AGT
TTA C
CCC TTT GAA TAC TAC
AGA ATA AGA AAG G
AAG GTT GAA TTC TGG
CCC TGC TCC C
SHV-1 gB / 96 bp GTC TGT GAA GCG GTT CGT G CAC AAG TTC AAG GCC
CAC AT
TAC TAC AAG AAC GTC
ATC GTC ACG ACC GT
Influenza-
A-Virus
M1 / 96 bp AGA CCA ATC CTG TCA CCT
CTG
GAC AAA GCG TCT ACG
CTG C
TCC TCG CTC ACT GGG
CAC GGT G
PRRSV-
EU
ORF7 / 77 bp; Kleiboeker
et al. (2005);
GCA CCA CCT CAC CCA GAC CAG TTC CTG CGC CTT
GAT
CCT CTG CTT GCA ATC
GAT CCA GAC
PRRSV-
NA
ORF7 und 3′NTR / 114 bp;
Kleiboeker et al. (2005)
F1-ATG ATG RGC TGG CAT TCT;
F2-ATR ATG RGC TGG CAT TCC
CAC ACG GTC GCC CTA
ATT G
TGT GGT GAA TGG CAC
TGA TTG ACA
IK
(DNA)
β-Aktin / 185 bp GGT GTC CAG AGG CGC TCT T TCG CAC TTC ATG ATC
GAG TTG
CTG GGC ATG GAG TCC
TGC GGC
IK
(RNA)
β-Aktin / 90 bp GGT GTC CAG AGG CGC TCT T TCG CAC TTC ATG ATC
GAG TTG
CCT TCC TGG GCA TGG
AGT CCT GC

84 4. Ergebnisse
4.2.2 Entwicklung der TaqMan PCRs
4.2.2.1 Gelbasierte PCRs
Neu entwickelte Primerpaare wurden zunachst in konventionellen, gelbasierten PCRs ge-
testet, um die korrekte Große des Amplifikats sicherzustellen und unspezifische Bindungen
der Primer zu erkennen. Fur alle PCR-Produkte konnte eine der zu erwartenden Produkt-
große entsprechende Bande im Agarosegel gezeigt werden. Fur Zellkulturuberstande bzw.
zellfreie Materialien wurden keine unspezifischen Banden festgestellt. PCR-Produkte, die
in multiplex real-time Ansatzen unter Verwendung verschiedener Organmaterialien am-
plifiziert wurden, zeigten im Agarosegel neben den spezifischen Banden unterschiedlich
ausgepragte zusatzliche Bandenmuster (nicht dargestellt).
4.2.2.2 Sequenzierung der PCR Produkte
Alle PCR-Produkte wurden in Standardvektoren kloniert und zur Absicherung der Iden-
titat zur Sequenzierung eingeschickt (Fa. MWG–Biotech AG, Ebersberg). Die Sequenzie-
rung (s. Kapitel 3.2.2.12) aller PCR-Produkte ergab korrekte Sequenzen.
4.2.2.3 Optimierung
Im Rahmen der Entwicklung der PCRs wurden die Primer- und Sondenkonzentrationen
optimiert.
4.2.2.3.1 Primertitration
Bei der Optimierung der Primerkonzentrationen sollte der fur die effektive Amplifikation
der DNA (bzw. cDNA) geeignetste Konzentrationsbereich ermittelt werden. Kriterium
war ein moglichst niedriger Ct-Wert bei hohem Fluoreszenzwert. Das passende Werkzeug
zur Ermittlung des optimalen Konzentrationsverhaltnisses stellte die SYBRTM-Green real-
time PCR (s. Kapitel 2.4.3.2) dar. Zu diesem Zwecke wurden in Doppelansatzen Primerti-
trationen (wie unter 3.2.2.4.2 beschrieben) vorbereitet und durchgefuhrt. Abbildungen 4.4
und 4.5 zeigen eine SYBRTM-Green real-time PCR mit dazugehorigen Schmelzkurven. Mit

4.2. Entwicklung der multiplex real-time TaqMan PCRs 85
den eingesetzten Primerkombinationen wurden unterschiedliche Kurvenverlaufe hinsicht-
lich der Ct - und Fluoreszenzwerte (∆Rn) erzielt (Abbildung 4.4). Da die getesteten PCRs
in multiplex-Ansatzen durchgefuhrt werden sollten, durften zum Vermeiden der gegensei-
tigen Inhibition nicht die hochsten Konzentrationen ausgewahlt werden. Es musste ein
Kompromiss zwischen moglichst niedrigen Ct - Werten und niedrigen Primerkonzentra-
tionen gefunden werden. Die in Abbildung 4.5 auf Seite 87 dargestellten Schmelzkurven
dienten der Kontrolle der Produktspezifitat und der Uberprufung der Bildung unspezi-
fischer Produkte. Bei der NTC mit einer Primerkonzentration von 900 nM kam es im
Amplifikationsgraphen zu einem Anstieg der Fluoreszenz. In der Schmelzkurvenanalyse
zeigte dieses Produkt eine Schmelztemperatur von 72,5 ◦C, das spezifische Produkt dage-
gen 75,5 ◦C.
Fur eine bessere Ubersicht wurden die gewonnenen Daten in MicrosoftR© Excel 2003 (s.
Anhang A.7) exportiert und dort analysiert. Ein Beispiel ist in Abbildungen 4.6 und 4.7
zu sehen. Die rot gestrichelte Linie begrenzt jeweils die Bereiche mit besten Ct - und
Fluoreszenzwerten. Aus diesen Bereichen wurden die niedrigsten Primerkonzentrationen
ausgewahlt. Die ermittelten Optimalwerte sind in Tabelle 4.4 (Seite 90) dargestellt. Im
Anhang A.8 auf Seite 187 sind die weiteren Ergebnisse der Primertitrationen in Tabel-
le A.1 bis A.6 zusammengefasst.

864.
Ergeb
nisse
Abbildung 4.4: Primertitration am Beispiel der ASPV SYBRTM-Green real-time PCR. Die Abbildung zeigt die Amplifikationsgra-
phen die mit verschiedenen Primerkombinationen erzielt wurden. Sie unterscheiden sich in Ct- und Fluoreszenzwerten. Die beiden
negativen Kontrollen (NTC) wurden mit 50 bzw. 900 nM Primern durchgefuhrt (s. Abbildung 3.1). Sie zeigen ebenfalls einen Anstieg
der Fluoreszenz.

4.2.E
ntw
icklu
ng
der
multip
lexreal-tim
eTaq
Man
PC
Rs
87
Abbildung 4.5: Primertitration am Beispiel der ASPV SYBRTM-Green real-time PCR. Die Abbildung zeigt die zur Abbildung 4.4
gehorenden Schmelzkurven. Die Schmelztemperatur des spezifischen Produktes betragt 75,5 ◦C. Es ist eine deutliche Bildung von
unspezifischen Produkten bei NTC mit 900 nM Primern zu sehen. Die Schmelztemperatur betragt 72,5 ◦C.
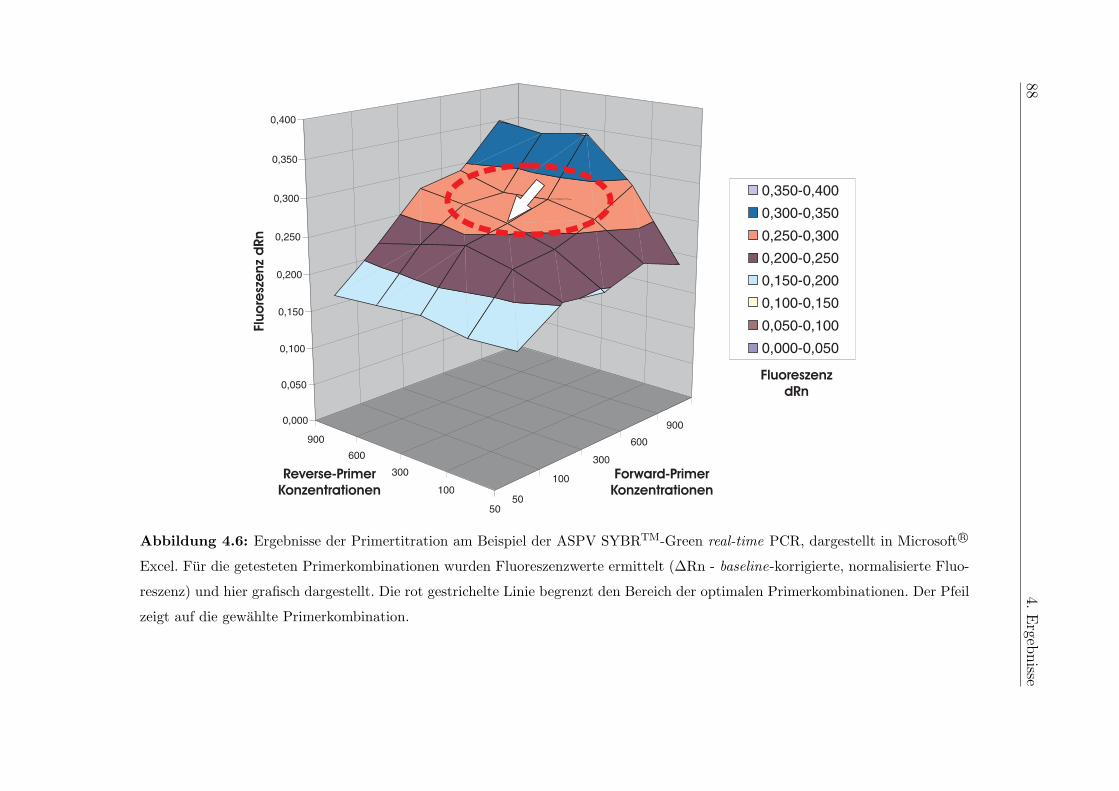
884.
Ergeb
nisse
Abbildung 4.6: Ergebnisse der Primertitration am Beispiel der ASPV SYBRTM-Green real-time PCR, dargestellt in Microsoft R©
Excel. Fur die getesteten Primerkombinationen wurden Fluoreszenzwerte ermittelt (∆Rn - baseline-korrigierte, normalisierte Fluo-
reszenz) und hier grafisch dargestellt. Die rot gestrichelte Linie begrenzt den Bereich der optimalen Primerkombinationen. Der Pfeil
zeigt auf die gewahlte Primerkombination.

4.2.E
ntw
icklu
ng
der
multip
lexreal-tim
eTaq
Man
PC
Rs
89
Abbildung 4.7: Ergebnisse der Primertitration am Beispiel der ASPV SYBRTM-Green real-time PCR, dargestellt in Microsoft R©
Excel. Fur die getesteten Primerkombinationen wurden Ct-Werte ermittelt und hier grafisch dargestellt. Die rote gestrichelte Linie
begrenzt den Bereich der optimalen Primerkombinationen. Der Pfeil zeigt auf die gewahlte Primerkombination.

90 4. Ergebnisse
4.2.2.3.2 Sondentitration
Hierbei wurden die optimalen Konzentrationen fur die jeweilige TaqMan-Sonde ermittelt.
Ziel waren niedrige Ct- und hohe ∆Rn-Werte. Die optimalen Sondenkonzentrationen sind
ebenfalls in Tabelle 4.4 festgehalten.
Tabelle 4.4: Die experimentell ermittelten optimalen Primer- und Sondenkonzentratio-
nen. Die Primer- und Sondenkonzentrationen fur die PRRSV-PCR wurden aus dem Pro-
tokoll von Kleiboeker et al. (2005) ubernommen.
Virus Fwd-Primer Rev-Primer Sonde
ASPV 300 nM 300 nM 100 nM
SHV-1 100 nM 100 nM 300 nM
PCV-2 100 nM 300 nM 200 nM
PPV 300 nM 300 nM 200 nM
”Panpesti“ 100 nM 100 nM 100 nM
Influenza-A 100 nM 300 nM 200 nM
PRRSV-NA 300 nM 300 nM 200 nM
PRRSV-EU 300 nM 300 nM 200 nM
β-Aktin mRNA 50 nM 50 nM 100 nM
β-Aktin gDNA 50 nM 50 nM 100 nM
4.2.2.4 Entwicklung interner Kontrollen
Als interne Kontrolle wurde das β-Aktingen gewahlt. Basierend auf der in GenBank 3
verfugbaren porzinen genomischen β-Aktinsequenz und unter Einbeziehung einer entspre-
chenden mRNA-Sequenz wurden ein Primerpaar und zwei TaqMan-Sonden entwickelt. Die
erste TaqMan-Sonde wurde konzipiert, um genomische DNA nachzuweisen. Diese Sonde
wurde an der Ubergangsstelle eines Introns zum Exon platziert und besaß eine komplet-
te Identitat zum gewahlten Genomabschnitt. Die zweite Sonde sollte dem Nachweis der
mRNA dienen und besaß die komplementaren Sequenzen zweier benachbarter Introns, so
3http://www.ncbi.nlm.nih.gov/GenBank/

4.2. Entwicklung der multiplex real-time TaqMan PCRs 91
Abbildung 4.8: β-Aktin Primer- und Sondenlokalisation. Dargestellt sind die Lokalisa-
tionen der Primer (beta-actin-rev-rna, beta-actin-fwd-rna) sowie zweier Sonden. Die Re-
ferenzsequenz ist die genomische β-Aktin-Sequenz des Schweines. Grau hinterlegt ist der
Bereich des Introns. Die Sonde b-actin-dna-tm dient dem Nachweis genomischer DNA und
ist mit der genomischen Sequenz identisch. Die Sonde b-actin-rna-2 ist nur mit der Sequenz
ohne Intron (also ensprechender messenger-RNA) voll identisch, mit der hier dargestellten
genomischen β-Aktin-Sequenz liegt nur eine partielle Identitat vor.
dass bei Vorliegen genomischer DNA eine vollstandige Bindung der Sonde unterbunden
wurde (s. Abbildung 4.8). Um die Spezifitat der Primer und Sonden zu uberprufen, wurden
sie in der TaqMan-PCR mit aus Organmaterial isolierten DNA, bzw. RNA nach reverser
Transkription eingesetzt. Bei der Uberprufung der TaqMan-Sonde zum Nachweis genomi-
scher DNA mit der aus mRNA erstellten cDNA wurden positive Reaktionen verzeichnet
(nicht gezeigt). Eine Differenzierung der Quelle war mit den eingesetzten Methoden nicht
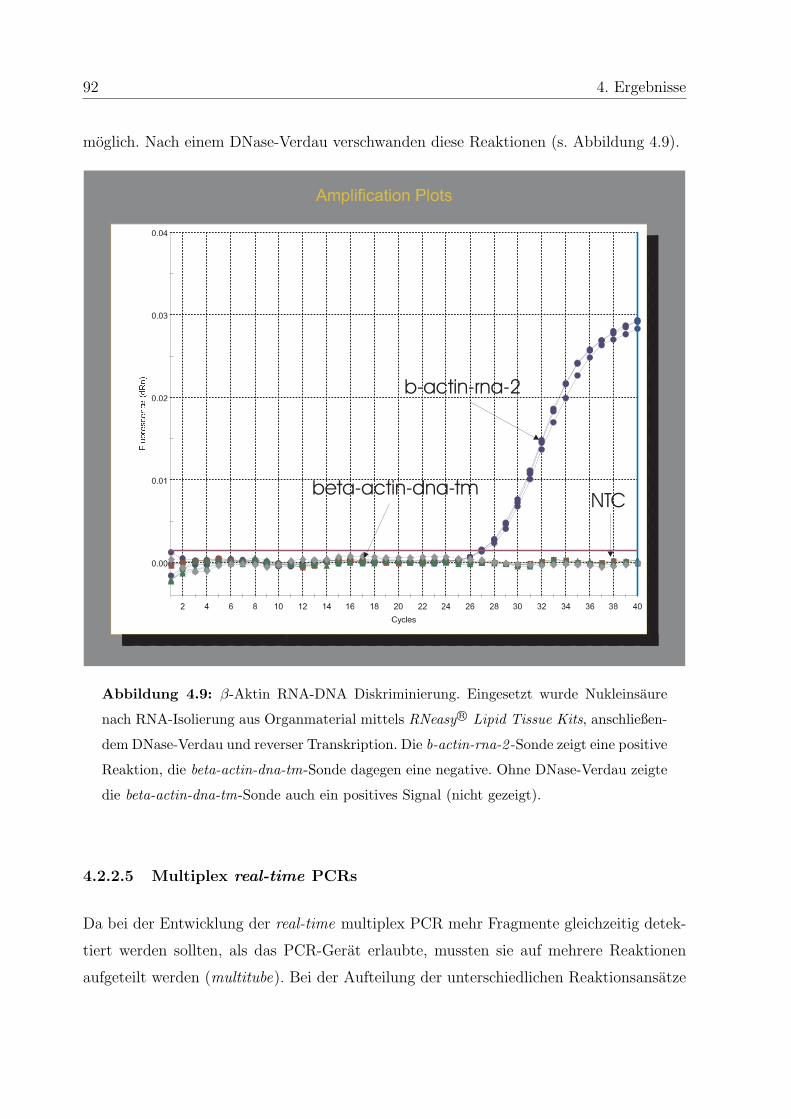
92 4. Ergebnisse
moglich. Nach einem DNase-Verdau verschwanden diese Reaktionen (s. Abbildung 4.9).
Abbildung 4.9: β-Aktin RNA-DNA Diskriminierung. Eingesetzt wurde Nukleinsaure
nach RNA-Isolierung aus Organmaterial mittels RNeasy R© Lipid Tissue Kits, anschließen-
dem DNase-Verdau und reverser Transkription. Die b-actin-rna-2 -Sonde zeigt eine positive
Reaktion, die beta-actin-dna-tm-Sonde dagegen eine negative. Ohne DNase-Verdau zeigte
die beta-actin-dna-tm-Sonde auch ein positives Signal (nicht gezeigt).
4.2.2.5 Multiplex real-time PCRs
Da bei der Entwicklung der real-time multiplex PCR mehr Fragmente gleichzeitig detek-
tiert werden sollten, als das PCR-Gerat erlaubte, mussten sie auf mehrere Reaktionen
aufgeteilt werden (multitube). Bei der Aufteilung der unterschiedlichen Reaktionsansatze

4.2. Entwicklung der multiplex real-time TaqMan PCRs 93
wurden Kriterien zugrunde gelegt, die den Eigenschaften der Viren und der Seuchensitua-
tion Rechnung tragen sollten. Zum einen erfolgte die Kombination der Erreger anhand
ihres Genoms, d. h., dass RNA-Viren untereinander und DNA-Viren untereinander kom-
biniert wurden, um einen reibungslosen Reaktionsablauf zu gewahrleisten. Zum anderen
wurde eine moglichst geringe Konkurrenz der einzelnen Primer um die Ressourcen an-
gestrebt. Ein weiteres Kriterium war die epidemiologische Situation im Sinne der Wahr-
scheinlichkeit des Auftretens der Erreger.
Die auf Grundlage dieser Kriterien gewahlte Zusammensetzung der einzelnen Ansatze ist
in Tabelle 4.5 dargestellt.
Die Kopplung der erregerspezifischen Sonden erfolgte fur alle Ansatze an die Fluores-
zenzfarbstoffe FAM und HEX, die Sonden fur die internen Kontrollen wurden mit Cy5
markiert. ROX diente in den Reaktionen als Referenzfarbstoff.
Tabelle 4.5: Zusammensetzung einzelner real-time PCR-Ansatze im Gesamtkonzept. Ref.
Dye = Referenzfarbstoff; IK = interne Kontrolle
Farbstoff Tube 1 Tube 2 Tube 3 Tube 4 Quencher
FAM ASPV PPV Panpesti PRRSV-EU BHQ-1
HEX SHV-1 PCV-2 Influenza-A PRRSV-NA BHQ-2
Cy5 − IK − IK BHQ-2
ROX Ref. Dye Ref. Dye Ref. Dye Ref. Dye −
Fur eine bessere Einschatzung der Leistung und Kinetik der PCRs wurden Standard-
kurven (s. Kapitel 2.4.4.4) erstellt und analysiert. Alle Standardkurven-PCRs wurden in
Dreifachansatzen sowohl in einzel- als auch in multiplex-PCRs mit Plasmidkonzentratio-
nen von 107 bis 100 Kopien pro Ansatz durchgefuhrt. Zur Auswertung wurden die Stufen
von 106 bis 101 Kopien pro Ansatz herangezogen.
Die multiplex-Ansatze zeigten ausnahmslos eine geringere Effizienz als die entsprechenden
PCRs im Einzelansatz. Im Einzelnen wurde fur die ASPV-spezifische PCR eine Effizienz
von 91,0 % im einzel- und von 82,4 % im multiplex-Ansatz mit SHV-1 ermittelt. Die
SHV-1 spezifische PCR zeigte eine Effizienz von 96,7 % in der einzel-, bzw. von 80,4 % in
der multiplex-PCR mit ASPV.
Fur die PCV-2 und PPV-spezifischen PCRs betrugen die Effizienzen in den jeweiligen

94 4. Ergebnisse
einzel-PCRs 98,8 % (PCV-2) bzw. 99,5 % (PPV). Kombiniert in einer multiplex-PCR be-
trugen die jeweiligen Effizienzen 85,6 % bzw. 90,4 %. Bei der Ermittlung der Effizienzen der
Pestiviren- bzw. Inluenza-A-spezifischen PCRs konnten Einzeleffizienzen von 92,9 % und
98,8 % festgestellt werden. Im multiplex-Ansatz betrugen die Effizienzen 90,8 % (Pestivi-
ren) bzw. 69,9 % (Influenza). Eine zusammengefasste Darstellung der Ergebnisse befindet
sich im Anhang A.8.
4.3 Validierung
4.3.1 Sensitivitat
Gemaß des Validierungsschemas der OIE sollte das Detektionslimit bzw. die analyti-
sche Sensitivitat der PCR in Endpunktverdunnungen anhand der Anzahl nachweisba-
rer Genomaquivalente bzw. infektioser Einheiten (KID50, PFU) bestimmt werden. Wo
moglich, wurden Endpunktverdunnungen auch in Zellkultur nach Art einer Virustitration
getestet. Fur jedes Virus wurde die analytische Sensitivitat des Ansatzes in einzel-PCRs
in Genomaquivalenten bestimmt. Dazu wurden in Standardvektoren einklonierte PCR-
Produkte logarithmisch verdunnt und in einzel- und multiplex TaqMan real-time PCRs
eingesetzt. Die Verdunnungsstufen enthielten 107 bis 100 Kopien pro PCR-Ansatz. Fur
alle einzel-PCRs konnte ausnahmslos ein Detektionslimit von 101 Kopien/Ansatz festge-
stellt werden. Die Ergebnisse sind in Tabellen A.7 bis A.13 im Anhang A.8 dargestellt.
Parallel wurden fur das porzine Influenza-A-Virus, PRRSV, PPV, SHV-1 und KSPV End-
punktverdunnungen einer Virussuspension (Zellkulturuberstand) bekannten Titers ange-
legt und sowohl in Zellkultur in Form einer Virustitration als auch in PCR-Ansatzen
untersucht. Dabei wurde die Sensitivitat des zellkulturellen Systems mit der Nachweis-
grenze der vorliegenden PCR verglichen und der theoretische Virustiter der logarithmi-
schen Verdunnungsreihe der tatsachliche KID50 der entsprechenden Losung zugeordnet.
Zu diesem Zweck wurden die logarithmisch angelegten Verdunnungsstufen in zwei Ali-
quots geteilt. Fur die Virustitrationen wurde ein Aliquot wiederum in logarithmischen
Verdunnungsreihen auf permissive Zellen verimpft. Die Berechnung des Virustiters der
Verdunnungsstufe erfolgte hier in KID50 pro 100µl. Fur den Vergleich der Methoden

4.3. Validierung 95
wurde diese Zahl auf das in der PCR eingesetzte Volumen von 6µl umgerechnet. Das
zweite Aliquot wurde der Nukleinsaureextraktion zugefuhrt und in der PCR eingesetzt.
Die PCRs wurden in Dreifachansatzen sowohl qualitativ (positiv/negativ) als auch quan-
titativ (Genomaquivalente) bewertet. Die Ergebnisse der beiden Untersuchungsmethoden
wurden in Relation zueinander gesetzt. Im Einzelnen waren die Ergebnisse wie folgt: In
der SHV-1 spezifischen PCR konnte eine Virussuspension mit 60,5 KID50/6µl als positiv
getestet werden, was 1,37 Genomaquivalenten entsprach. Die PPV-PCR ergab ein posi-
tives Signal in der Stufe 7 der logarithmischen Verdunnungsreihe, was einer KID50/6µl
von 0,74 bzw. 1,17 Genomaquivalenten entsprach. Die letzte Verdunnungsstufe, die in der
PCR zum Nachweis der Pestiviren als positiv getestet wurde, hatte einen KID50/6µl-Wert
von 0,60 oder 7,18 Genomaquivalenten. In der Influenza-A-spezifischen PCR konnte eine
Virussuspension mit 0,06 KID50 (umgerechnet auf 6µl) als positiv getestet werden, was
192 Genomaquivalenten entsprach. Es konnten außerdem drei weitere Verdunnungsstu-
fen als positiv getestet werden. Die Konzentrationen lagen hier bei 19,5, 2,1 und 0,81
Genomaquivalenten. Zur Bestimmung der analytischen Sensitivitat der beiden PRRSV-
PCRs wurden je ein amerikanischer und europaischer Impfstamm des Virus (PRRSV-EU,
bzw. PRRSV-NA) in Zellkultur vermehrt und titriert. Die PRRSV-EU real-time PCR
wies in der Verdunnungsstufe mit auf 6µl umgerechneten 0,6 KID50 26,4 Genomaquiva-
lente nach, die PRRSV-NA PCR entsprechend bei 0,06 KID50 373 Genomaquivalente. Bei
beiden real-time PCRs konnte jeweils eine weitere Verdunnungsstufe der logarithmischen
Endpunktverdunnungsreihe als positiv detektiert werden. Die PRRSV-NA-PCR ergab
13,5, die PRRSV-EU-PCR 1,07 Genomaquivalente. Beispieldarstellungen befinden sich in
den Abbildungen 4.10 und 4.11. Weitere Versuchsergebnisse befinden sich im Anhang A.8.
4.3.2 Spezifitat
Beim Design der Primer und Sonden wurde eine BLAST-Suche durchgefuhrt, die eine
Interaktion mit anderen Erregern moglichst ausschließen sollte. Diese Suche ergab fur al-
le ausgewahlten Primer und Sonden spezifische Identitaten mit dem jeweiligem Erreger
(Ergebnisse nicht dargestellt).
Des Weiteren wurden Tests mit Virusisolaten und verschiedenen Organmaterialien (s.
Kapitel 3.1.2.1) durchgefuhrt. Bei der Entwicklung und Validierung der pestivirusspezifi-

96 4. Ergebnisse
Abbildung 4.10: Bestimmung der Nachweisgrenze der PPV-PCR mittels Endpunktbe-
stimmung. In diesem Balkendiagramm sind die in logarithmischen Verdunnungsstufen (V1
bis V8) einer Virussuspension bekannten Titers ermittelten Genomaquivalente dargestellt
(Standardabweichungen sind als Balken eingetragen). Zur besseren Darstellbarkeit wur-
den die Werte logarithmiert. Diese Werte wurden mit den in Zellkultur ermittelten und
auf das PCR-Probenvolumen umgerechneten Virustitern korreliert. Die Virustiter sind als
logKID50/6 µl oberhalb der Kennzeichnung der Verdunnungsstufen angegeben.
schen PCR wurden Virussuspensionen und virushaltiges Organmaterial von experimentell
infizierten Tieren untersucht. Es konnten die eingesetzten KSPV-Isolate aus den phylo-
genetischen Gruppen 1.1-1.3, 2.1-2.3 sowie 3.4 positiv getestet werden. Zudem waren
BVDV-1-, BVDV-2-, BDV- sowie eine pestivirushaltige Virussuspension in der real-time
PCR positiv. Lediglich das von einem experimentell mit BDV Gifhorn 10754 infizierten
Schwein stammende Organmaterial war negativ. Die Ergebnisse sind in Tabelle A.7 auf
Seite 194 abgebildet.
Fur das Primer- und Sondendesign der PCV-2-spezifischen PCR wurde die typspezifische
Region des ORF2 gewahlt (s. Kapitel 2.3.6.1). Eine Uberprufung der Kreuzreaktivitat mit
PCV-1 erfolgte mittels PCV-1-Plasmiden (freundlicherweise zur Verfugung gestellt von
Frau Dr. Mankertz, Robert-Koch-Institut, Berlin). Es wurde gezeigt, dass keine Kreuzre-
aktionen vorlagen. Außerdem wurden die als PCV-2-positiv deklarierten Proben korrekt

4.3. Validierung 97
Abbildung 4.11: Bestimmung der Nachweisgrenze der Influenza-A-PCR mittels End-
punktbestimmung. In diesem Balkendiagramm sind die in logarithmischen Verdunnungs-
stufen (V1 bis V8) einer Virussuspension bekannten Titers ermittelten Genomaquivalente
dargestellt (Standardabweichungen sind als Balken abgetragen). Zur besseren Darstellbar-
keit wurden die Werte logarithmiert. Diese Werte wurden mit den in Zellkultur ermittelten
und auf das PCR-Ansatzvolumen umgerechneten Virustitern korreliert. Die Virustiter sind
als logKID50/6 µl oberhalb der Kennzeichnung der Verdunnungsstufen angegeben.
als positiv erkannt, sowie die PCV-2 negativen als solche bestatigt (nicht gezeigt).
Im Rahmen der Spezifitatsprufung wurden zwolf Isolate beim Schwein vorkommender
Subtypen des Influenza-A-Virus (s. Tabelle 3.2) und drei weitere als positiv erkannt (s.
Tabelle A.8).
Die vier Plasmide mit klonierten Sequenzen des VP72-Proteins des ASPV, die im Rahmen
dieser Arbeit zur Verfugung standen, wurden alle in der real-time PCR erkannt (nicht ge-
zeigt).
Uber 20 SHV-1-Isolate, welche aus verschiedenen Regionen Deutschlands sowie aus Frank-
reich, Osterreich und Tschechien stammten, konnten in die Uberprufung einbezogen wer-
den (s. Kapitel tab:SHV1Isolate). In der real-time PCR wurden sie alle positiv getestet
(s. Tabelle A.9).
Die im Kapitel 3.1.2.1 aufgefuhrten PPV-Isolate wurden ebenfalls in der hier entwickel-

98 4. Ergebnisse
ten PCR erkannt. Außerdem wurden zwei von 33 getesteten klinischen Proben (s. Kapi-
tel 4.3.3) als positiv detektiert. Die in das Gesamtkonzept aufgenommene real-time PCR
zur Detektion und Differenzierung von europaischen und nordamerikanischen PRRSV-
Stammen (Kleiboeker et al., 2005) wurde ohne Modifikationen der Primer- bzw. Son-
densequenzen ubernommen. Im Rahmen der Validierung dieser PCRs wurden zwei PRRSV-
Impfstamme (MLV und DV) eingesetzt, beide wurden spezifisch als positiv erkannt. Dabei
bestanden keine Kreuzreaktionen zwischen den beiden PCRs (nicht gezeigt).
4.3.3 Klinische Proben
Zusatzlich zu den oben aufgefuhrten, im Rahmen der Spezifitatsstudien untersuchten
Virusisolaten und Organmaterialien, wurden im Rahmen der weiteren PCR-Validierung
experimentell gewonnene Proben aus dem EURL sowie klinische Proben aus der Routi-
nediagnostik der Außenstelle fur Epidemiologie der TiHo Hannover in Bakum getestet.
Alle Untersuchungen fanden mit der endgultigen Version des multiplex multitube Ansat-
zes statt. Die Auswahl wurde aus dem vorhandenen Probenmaterial zufallig getroffen.
Es wurden insgesamt 17 Proben untersucht, die von Schweinen stammten, die am EURL
experimentell mit verschiedenen KSPV- bzw. BDV-Stammen infiziert und anschließend
mit den Routinemethoden des EURL zum Virus- und Antikorpernachweis untersucht
worden waren. Die Tiere dieser Versuchsvorhaben stammten aus dem Lehr- und For-
schungsgut Ruthe und waren im Vorfeld der Versuche ausschließlich auf Pestivirenfreiheit
untersucht worden. Ein Teil des Probenmaterials stammte von diesen Probennahmen vor
der Infektion und war somit vorberichtlich KSPV, BDV und BVDV negativ. Eine Probe
mit der Bezeichnung ZA4 stammte aus einem KSPV-Ausbruch in Sudafrika im Jahr 2005.
In dieser Probe wurde vorberichtlich KSPV mittels PCR und Virusisolierung nachgewie-
sen. In Tabelle 4.6 sind die Ergebnisse der Untersuchungen der oben aufgefuhrten Proben
zusammenfasst. Alle vorberichtlich KSPV-positiven Tiere konnten in der multiplex PCR
erkannt werden, wobei das Probenmaterial eines mit CSF0309 (Stamm Kanagawa) infi-
zierten Schweins nicht eindeutig beurteilt werden konnte, da es sowohl schwach positive
Ergebnisse mit hohen Ct-Werten als auch negative Resultate ergab. Alle Proben, die vor
der Infektion entnommen worden waren, konnten korrekt als negativ bestatigt werden.

4.3. Validierung 99
Die Detektion des BDV-Stamms Gifhorn 10754 in der Probe eines vorberichtlich positiven
Schweins gelang nicht. In Organmaterialien von drei Tieren konnte PCV-2 nachgewiesen
werden. Die Tests fur alle anderen Viren verliefen negativ.
Die 33 Proben aus der Außenstelle fur Epidemiologie waren im Vorfeld auf verschiede-
ne porzine Viren und andere Pathogene untersucht worden. Erwartungsgemaß wurden
hier weder ASPV, SHV-1 noch Pestiviren festgestellt (s. Tabelle 4.7). 21 von 33 Proben
(64 %) wurden in der PCV-2-spezifischen PCR positiv getestet. Im Vergleich mit den Er-
gebnissen der Untersuchungen der Außenstelle fur Epidemiologie, die fur 16 der Proben
vorlagen, zeigte sich, dass es acht Abweichungen von den dort erhobenen Befunden gab.
Die Abweichungen betrafen v. a. dort als fraglich befundete Proben. Funf dieser fraglichen
Proben wurden in der hier verwendeten PCR als positiv detektiert, zwei als negativ. Eine
Probe wurde in Bakum negativ, mit der hier entwickelten PCR jedoch positiv getestet.
Die PPV-spezifische PCR detektierte zwei der 33 Proben als positiv (6 %), ein Vergleich
mit Ergebnissen anderer Untersuchungsstellen war in diesem Fall nicht moglich. Neun
von 33 Proben (27 %) wurden mit der entwickelten Influenza-A-spezifischen PCR positiv
getestet. Ein Vergleich mit den Ergebnissen der Außenstelle fur Epidemiologie war fur
drei Proben moglich. Es zeigte sich eine Abweichung insofern, dass eine dort als positiv
getestete Probe im vorliegenden Ansatz negativ war. Die im Rahmen der vorliegenden
Arbeit verwendeten PRRSV-spezifischen PCRs ergaben drei (9 %) positive Ergebnisse
fur PRRSV-EU und vier positive Ergebnisse fur PRRSV-NA (12 %). Der Vergleich mit
Befunden aus Bakum war fur zwei PRRSV-EU-Befunde und einen PRRSV-NA-Befund
moglich. Fur PRRSV-EU ergaben sich keine Abweichungen, die PRRSV-NA-Befunde wi-
chen voneinander ab. Wahrend die Probe in Bakum als positiv befunden wurde, zeigte
sie in der vorliegenden PCR ein negatives Ergebnis.
Die oben genannten Ergebnisse schließen Proben mit ein, deren Ergebnisse fur mehr als
ein Virus positiv ausfielen. Insgesamt wurden mit der hier entwickelten PCR 12 Proben
(36 %) doppelt positiv und eine Probe (3 %) dreifach positiv getestet. Die dreifach positi-
ve Proben gab positive Ergebnisse fur PCV-2, Influenza-A-Virus und PRRSV-NA. Diese
Probe war auch in Bakum auf PCV-2 und Influenza-A-Virus positiv getestet worden,
ein Ergebnis fur PRRSV-NA lag fur einen Vergleich nicht vor. Bei den doppelt positiven
Proben ergaben vier Proben (12 %) positive Ergebnisse fur PCV-2 und Influenza-A-Virus,
zwei Proben (6 %) positive Ergebnisse fur PCV-2 und PRRSV-NA, zwei Proben (6 %) po-

100 4. Ergebnisse
sitive Ergebnisse fur PCV-2 und PPV, drei Proben (9 %) positive Ergebnisse fur PCV-2
und PRRSV-EU und eine Probe die Ergebniskombination PRRSV-NA und Influenza-A-
Virus.
Ein Vergleich mit den Ergebnissen aus Bakum war fur die doppelt positiven Proben nur
teilweise moglich, da nicht alle Erreger, die mit der vorliegenden PCR detektiert wurden,
auch dort in die Tests einbezogen wurden. Im Detail wurde eine vorberichtlich PCV-
2 fragliche Probe in der multiplex-PCR PCV-2 und Influenza-A-Virus positiv getestet.
Eine andere, vorberichtlich PCV-2 und Influenza-A-Virus positive Probe wurde mit der
vorliegenden PCR als PCV-2 und PRRSV-NA positiv, jedoch Influenza-A-Virus negativ
detektiert. Eine weitere, vorberichtlich PCV-2 positive Probe wurde hier zusatzlich PPV
positiv getestet. Das Ergebnis einer PCV-2 und Influenza-A-Virus positiven Probe konnte
bestatigt werden. Fur eine PCV-2 und Influenza-A-Virus positive Probe lag aus Bakum
ausschließlich ein PRRSV-EU negatives Ergebnis vor, das mit der hier verwendeten PCR
bestatigt werden konnte. Eine vorberichtlich PCV-2 fragliche Probe wurden im vorliegen-
den Ansatz PCV-2 und PRRSV-EU positiv detektiert. In einer weiteren PCV-2 positiven
Probe konnte neben diesem Erreger auch PRRSV-NA gefunden werden.

4.3.V
alidieru
ng
101
Tabelle 4.6: Ergebnisse der Untersuchung KSPV-positiver sowie -negativer Proben. Die Proben stammten von Schweinen aus dem
Lehr- und Forschungsgut Ruthe, die im Rahmen verschiedener Tierversuche am EURL eingesetzt wurden. Die mit 0 dpi (Tage post
infectionem) gekennzeichneten Proben sind als vorberichtlich pestivirennegativ anzusehen. Unter Isolat sind jeweils die eingesetzten
Pestiviren-Isolate aufgefuhrt.
Probe Nr.: Isolat ASPV SHV-1 PCV-2 PPV Pestiviren Influenza-A PRRSV-EU PRRSV-NA
ZA4 CSF0849 − − + − + − − −
287 CSF0849 − − − − + − − −
284 CSF0849 − − + − + − − −
282 CSF0905 − − + − + − − −
272 CSF0634 − − − − + − − −
270 CSF0647 − − − − + − − −
266 CSF0309 − − − − − − − −
237 CSF0695 − − − − + − − −
217 CSF0634 − − − − + − − −
217 (0 dpi) negativ − − − − − − − −
216 (0 dpi) negativ − − − − − − − −
209 CSF0905 − − − − + − − −
208 CSF0905 − − − − + − − −
100 (0 dpi) negativ − − − − − − − −
99 (0 dpi) negativ − − − − − − − −
80 (0 dpi) negativ − − − − − − − −
219 BDV-Gifhorn − − − − − − − −
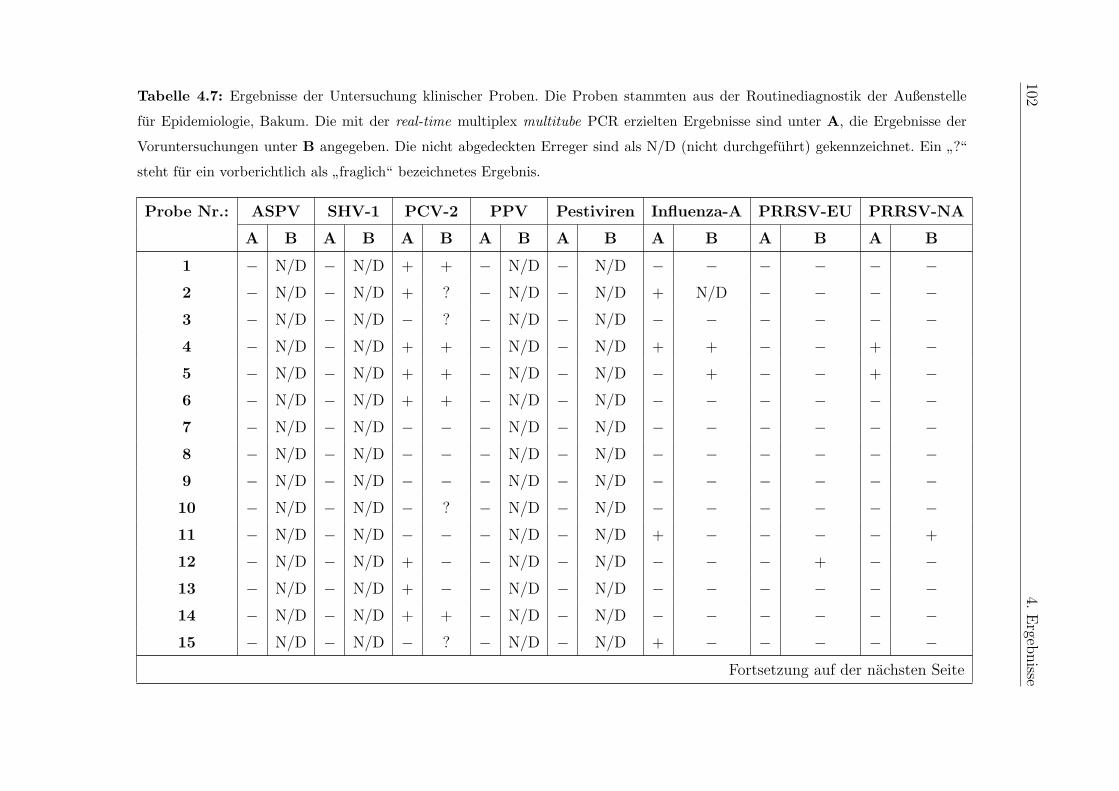
1024.
Ergeb
nisse
Tabelle 4.7: Ergebnisse der Untersuchung klinischer Proben. Die Proben stammten aus der Routinediagnostik der Außenstelle
fur Epidemiologie, Bakum. Die mit der real-time multiplex multitube PCR erzielten Ergebnisse sind unter A, die Ergebnisse der
Voruntersuchungen unter B angegeben. Die nicht abgedeckten Erreger sind als N/D (nicht durchgefuhrt) gekennzeichnet. Ein”?“
steht fur ein vorberichtlich als”fraglich“ bezeichnetes Ergebnis.
Probe Nr.: ASPV SHV-1 PCV-2 PPV Pestiviren Influenza-A PRRSV-EU PRRSV-NA
A B A B A B A B A B A B A B A B
1 − N/D − N/D + + − N/D − N/D − − − − − −
2 − N/D − N/D + ? − N/D − N/D + N/D − − − −
3 − N/D − N/D − ? − N/D − N/D − − − − − −
4 − N/D − N/D + + − N/D − N/D + + − − + −
5 − N/D − N/D + + − N/D − N/D − + − − + −
6 − N/D − N/D + + − N/D − N/D − − − − − −
7 − N/D − N/D − − − N/D − N/D − − − − − −
8 − N/D − N/D − − − N/D − N/D − − − − − −
9 − N/D − N/D − − − N/D − N/D − − − − − −
10 − N/D − N/D − ? − N/D − N/D − − − − − −
11 − N/D − N/D − − − N/D − N/D + − − − − +
12 − N/D − N/D + − − N/D − N/D − − − + − −
13 − N/D − N/D + − − N/D − N/D − − − − − −
14 − N/D − N/D + + − N/D − N/D − − − − − −
15 − N/D − N/D − ? − N/D − N/D + − − − − −
Fortsetzung auf der nachsten Seite
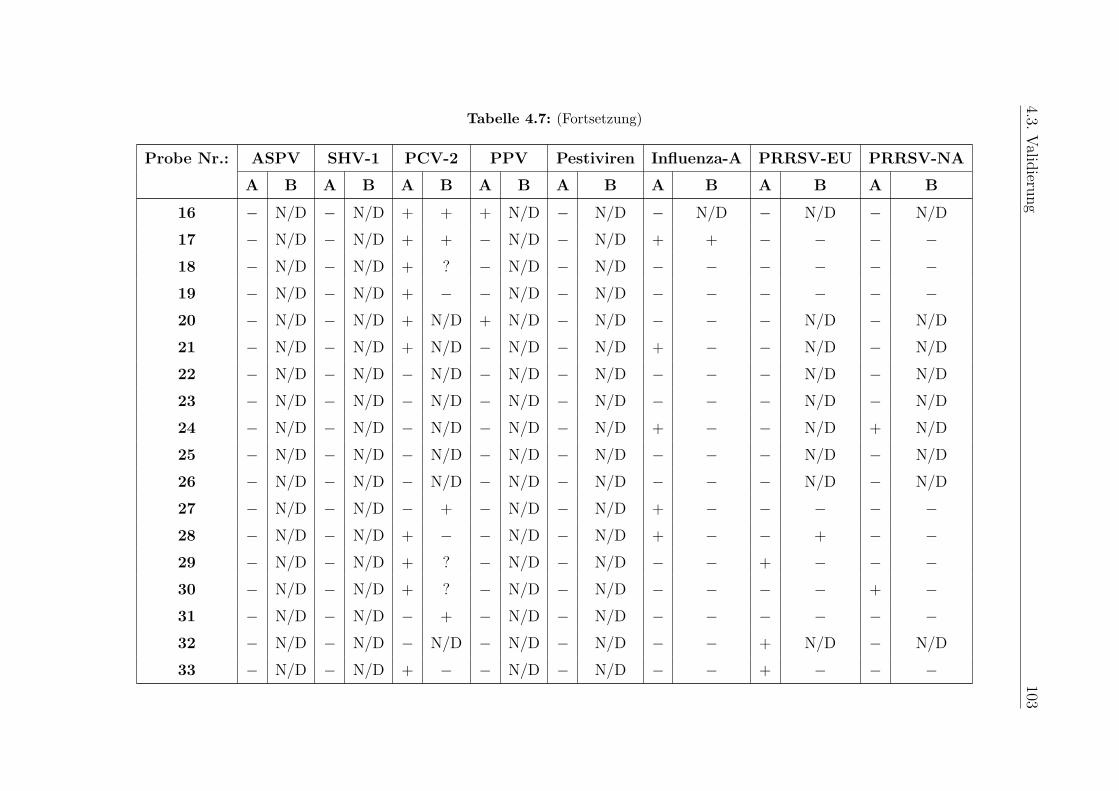
4.3.V
alidieru
ng
103
Tabelle 4.7: (Fortsetzung)
Probe Nr.: ASPV SHV-1 PCV-2 PPV Pestiviren Influenza-A PRRSV-EU PRRSV-NA
A B A B A B A B A B A B A B A B
16 − N/D − N/D + + + N/D − N/D − N/D − N/D − N/D
17 − N/D − N/D + + − N/D − N/D + + − − − −
18 − N/D − N/D + ? − N/D − N/D − − − − − −
19 − N/D − N/D + − − N/D − N/D − − − − − −
20 − N/D − N/D + N/D + N/D − N/D − − − N/D − N/D
21 − N/D − N/D + N/D − N/D − N/D + − − N/D − N/D
22 − N/D − N/D − N/D − N/D − N/D − − − N/D − N/D
23 − N/D − N/D − N/D − N/D − N/D − − − N/D − N/D
24 − N/D − N/D − N/D − N/D − N/D + − − N/D + N/D
25 − N/D − N/D − N/D − N/D − N/D − − − N/D − N/D
26 − N/D − N/D − N/D − N/D − N/D − − − N/D − N/D
27 − N/D − N/D − + − N/D − N/D + − − − − −
28 − N/D − N/D + − − N/D − N/D + − − + − −
29 − N/D − N/D + ? − N/D − N/D − − + − − −
30 − N/D − N/D + ? − N/D − N/D − − − − + −
31 − N/D − N/D − + − N/D − N/D − − − − − −
32 − N/D − N/D − N/D − N/D − N/D − − + N/D − N/D
33 − N/D − N/D + − − N/D − N/D − − + − − −


5. Diskussion
5.1 Gesamtkonzept
Die KSP gehort zu den wichtigsten viralen Erkrankungen der Hausschweine weltweit
und besitzt eine hohe wirtschaftliche Bedeutung (Edwards, 2000). Die soziookonomi-
schen Folgen eines großen KSP-Ausbruchs wie z. B. 1997 in den Niederlanden zeigen, von
welch herausragender Bedeutung eine fruhe, schnelle und verlassliche Diagnose fur die
Eindammung und Bekampfung des Seuchengeschehens ist (Murray und McCutcheon,
1999; Elbers et al., 2004). Eine moderne und effiziente Labordiagnostik ist daher so-
wohl fur die Uberwachung der Schweinepopulation in”Friedenszeiten“ als auch fur die
fruhe Erkennung eines Ausbruchs essentiell (Moennig, 2000). Da die klinischen Sym-
ptome und pathologisch-anatomischen Befunde einer KSPV-Infektion sehr variabel und
haufig unspezifisch sind (Depner et al., 1997b; Moennig, 1992; Elbers et al., 2002),
kommen viele andere Erkrankungen unterschiedlicher Genese als Differentialdiagnosen in
Betracht. Zu den viralen Differentialdiagnosen gehoren ASPV-, PCV-2-, PPV-, porzine
Influenza-Virus-, PRRSV- sowie SHV-1-Infektionen (van Oirschot, 1999b; Moennig
et al., 2003). Ein nicht unerhebliches Problem fur die fruhe Erkennung eines Ausbruchs
ist die psychologische Hemmschwelle fur Landwirte und Tierarzte, einen KSP-Verdacht
zu außern, da sofort weitreichende Konsequenzen fur den betroffenen Betrieb die Folge
sind. Eine Nachweismethode, die schnell und sicher eine mogliche KSPV-Infektion erfasst
und die wichtigsten viralen Differentialdiagnosen abklart, konnte ein wichtiges Binde-
glied darstellen. Zu den schnellsten Methoden des Virusgenomnachweises gehort die PCR
(Belak, 2005). Die technischen Entwicklungen der letzten Jahre eroffnen die Moglichkeit,
Genomfragmente mehrerer Pathogene gleichzeitig in einer multiplex PCR nachzuweisen.
105

106 5. Diskussion
Multiplex Ansatze konnen in der Differentialdiagnostik wichtiger Erreger eine zeitsparen-
de Alternative darstellen, die auch den personellen Aufwand minimieren, da viele Schrit-
te automatisierbar sind (Elnifro et al., 2000). In der Humanmedizin finden derartige
Ansatze z. B. fur die Detektion durch Blutkonserven ubertragbarer Erkrankungen An-
wendung (Candotti et al., 2004).
Ziel der vorliegenden Arbeit war es daher, die Umsetzbarkeit dieses Konzeptes fur die
KSP-Differentialdiagnostik zu uberprufen und eine moderne und schnelle Nachweisme-
thode differentialdiagnostisch relevanter viraler Pathogene in Abgrenzung zu KSPV bzw.
Pestiviren zu entwickeln. Die technische Umsetzung sollte als multiplex multitube real-
time RT-PCR erfolgen, die neben einem Pestivirusnachweis die oben bereits erwahnten
viralen Erreger (ASPV, PCV-2, Influenza-A-Virus, PRRSV-EU, PRRSV-US, SHV-1) ein-
schloss.
5.2 Auswahl der Nachweismethode
Fur die Umsetzung eines multiplex PCR-Ansatzes stehen generell sowohl die konven-
tionelle, gel-basierte als auch die real-time PCR zur Verfugung. Real-time PCRs bie-
ten im Vergleich zur gel-basierten PCR eine erheblich hohere Sensitivitat und Spezifitat
(Bustin, 2002). Ein weiterer Vorteil ist die Zeitersparnis durch Wegfall der Gelelektropho-
rese (Higuchi et al., 1993). Um eine moglichst rasche Diagnose bzw. Differentialdiagnose
zu gewahrleisten, wurde daher fur diese Arbeit der real-time PCR der Vorzug gegeben.
Die Anzahl der in Frage kommenden differentialdiagnostisch relevanten Erreger machten
einen multitube Ansatz notwendig, da die Moglichkeiten der Farbstoffdetektion gerates-
pezifischen Limitierungen unterlag. Es standen vier Kanale zur Erfassung der Fluoreszenz
zu Verfugung. Da ein Referenzfarbstoff sowie eine interne Kontrolle in jedem Lauf mit-
gefuhrt werden sollten, waren nur zwei Kanale verfugbar. Aus diesem Grund wurden zwei
Erreger in einem Ansatz mit den entsprechenden Referenzen und Kontrollen kombiniert.
Im vorliegenden Fall war die Limitierung der Anzahl der Nachweise pro Ansatz durchaus
ein Vorteil, da durch gezielte Kombination der Viren einerseits der Verlust von Sensiti-
vitat minimiert, und andererseits den Eigenschaften der Viren Rechnung getragen werden
konnte. Die Verwendung eines einheitlichen Thermoprofils macht es moglich, das Testsys-

5.2. Auswahl der Nachweismethode 107
tem beliebig um weitere relevante Erreger zu erweitern.
Die Detektionsmoglichkeiten fur eine real-time PCR umfassen spezifische und unspezifi-
sche DNA-Nachweismethoden (s. Kapitel 2.4.3.2 und 2.4.3.3). Die Zielstellung einer mul-
tiplex PCR schloss von vornherein unspezifische DNA-Detektionsmethoden aus (wie z. B.
SYBRTM-Green real-time PCR). Eine gut etablierte, robuste und weit verbreitete Tech-
nik stellt die TaqMan real-time PCR dar (Terry et al., 2002). Neben dieser Technik
besteht die Moglichkeit Molecular Beacons bzw. das PriProET System einzusetzen. Von
der ersten Moglichkeit wurde z. B. im Rahmen des EU Projektes QLK2-CT-2000-004861
fur den Nachweis der Viren des respiratorischen Clusters (ASPV, KSPV, PRRSV, SHV-1,
PPV) Gebrauch gemacht.
Die Molecular Beacon Technik stellt eine hochspezifische Methode dar. Fur wissenschaft-
lichen Fragestellungen (z. B. SNP single nucleotide polymorphism) ist die hohe Spezifitat
dieses Systems erwunscht, um Punktmutationen nachzuweisen. Fur den Einsatz in der
Diagnostik, in der zudem RNA-Viren, deren Mutationsrate relativ hoch ist, zu detek-
tieren sind, konnte eben dies zu erheblichen Nachweisproblemen fuhren. Zudem ist das
Design der Sonden sehr aufwendig und die Synthese teuer (Bustin, 2002). Aus dem im
Internet publizierten Bericht zu den Ergebnissen des oben genannten EU-Projekts2 geht
hervor, dass diese Problematik ein nicht unerhebliches Hindernis bei der Entwicklung einer
KSPV- bzw. PRRSV-spezifischen PCR darstellte. Eine erhebliche Anzahl Beacons und
Primer musste entwickelt werden, um eine akzeptable Losung zu finden. Die Detektion
der Genotypen konnte fur das KSPV nur uber die Integration mehrerer Beacons in einem
Ansatz gewahrleistet werden. Im Gegensatz dazu ist die TaqMan Technik wesentlich ro-
buster. Diese Technik erlaubte die Kombination der Vorteile einer real-time PCR mit der
Moglichkeit, mehrere Genfragmente gleichzeitig spezifisch zu detektieren (Bustin und
Nolan, 2004a).
Im Rahmen des EU-Projektes wurden PriProET Protokolle fur die Viren des hamorrhagi-
schen und vesikularen Clusters entwickelt. Die Methodik ist ebenfalls sensitiv und robust
und scheint den Ergebnissen des erwahnten Projektes nach, auch fur die Diagnostik geeig-
net. Zum Zeitpunkt des Entwurfes der vorliegenden Arbeit war diese Technik noch nicht
sehr verbreitet bzw. fur veterinarmedizinische Fragestellungen etabliert. Es wurde daher
1http://www.multiplex-eu.org2http://www.multiplex-eu.org/results.pdf, Stand 15.01.2006

108 5. Diskussion
auf die fur die gewahlte Fragestellung erprobte Technik zuruckgegriffen.
5.3 Literaturrecherche und Auswahl der Erreger
In der Entwurfsphase der PCRs wurden die differentialdiagnostisch relevanten Erkrankun-
gen viraler Genese in der Literatur gesucht. Die fur diesen Ansatz ausgewahlten viralen
Erreger sind die wichtigsten virusbedingten Differentialdiagnosen der KSP (Moennig
et al., 2003) und zeigen ein ahnliches Spektrum an Symptomen (s. Kapitel 2.3). Der
hier entwickelte Testansatz sollte nur virale Erreger einschließen, somit wurden wichtige
Differentialdiagnosen bakterieller Genese nicht berucksichtigt. Zu diesen Erkrankungen
gehoren u. a. Rotlauf, E. coli -, Salmonellen-, Pasteurellen- und Leptospireninfektionen
(Moennig et al., 2003). Die Konzeption des Systems ließe es zu, diese spater zu in-
tegrieren. Diese Option unterstreicht die Vorteile des vereinheitlichten Ansatzes. In der
Fragestellung, die bei der Auswahl der Erreger zugrunde gelegt wurde, unterscheidet sich
das vorliegende Konzept deutlich von der des EU-Projektes QLK2-CT-2000-00486. Ob-
gleich das Erregerspektrum sehr ahnlich ist, wurde die Auswahl nicht anhand der gleichen
Kriterien getroffen. Wahrend in der vorliegenden Arbeit ausschließlich das klinische Bild
im Sinne der differentialdiagnostischen Relevanz fur die KSP ausschlaggebend war, wur-
de im Rahmen des EU Projektes die okonomische Relevanz in den Vordergrund gestellt.
Dies fuhrte dazu, dass im vorliegenden Ansatz auch das porzine Influenza-A-Virus und
das PCV-2 eingeschlossen wurden, wahrend diese Viren im Gegensatz zum MKS-Virus,
VSV und SVDV im Spektrum des letzteren Ansatzes nicht vertreten waren.
Im Rahmen des Entwurfes und Entwicklung des vorliegenden PCR-Ansatzes erfolgten
Literaturrecherchen in der Datenbank Medline3. Ziel dieser Suche war es, anhand der
publizierten, TaqMan-basierten PCR-Protokolle eine Vorauswahl der Genomregionen zu
treffen und ein Thermoprofil zu erarbeiten. Mit Ausnahme des von Kleiboeker et al.
(2005) entwickelten Protokolls zur Detektion und Differenzierung der europaischen und
nordamerikanischen PRRSV-Stamme, welches in den multiplex-Ansatz integriert wurde,
wurden die publizierten Protokolle ausschließlich als Anhaltspunkt fur die eigenstandige,
3http://www.ncbi.nlm.nih.gov/entrez/query.fcgi

5.3. Literaturrecherche und Auswahl der Erreger 109
auf einen multiplex-Ansatz abgestimmte Entwicklung genommen. Die Auswertung der
gefundenen TaqMan-PCR-Protokolle hinsichtlich der gewahlten Genomabschnitte, der
Fragmentgroßen und der Thermoprofile wurde in den Entwurf der PCR einbezogen.
Die Thermoprofile, die fur die gefundenen TaqMan-Protokolle eingesetzt wurden, wichen
in einigen Punkten deutlich voneinander ab. Die Zyklenzahl lag generell zwischen 35
(King et al., 2003) und 50 (Yoon et al., 2005; Hofmann, 2003; Watzinger et al.,
2004; Hindiyeh et al., 2005; Krafft et al., 2005) Zyklen. Fur die vorliegende Arbeit
wurde nach ersten Vorversuchen eine Zyklenzahl von 40 festgesetzt. Da es bekannt ist,
dass die TaqMan-Sonden im Laufe der PCR degradieren, und es zu einem schwachen An-
stieg der Fluoreszenz kommen kann, sind diese Werte kritisch zu beurteilen.
Unter den publizierten Thermoprofilen gab es solche, die einen separaten Elongation-
Schritt bevorzugten, und andere, die Annealing der Primer und Elongation simultan
durchfuhrten. Die gewahlten Produktlangen erlaubten die Wahl eines Thermoprofils ohne
getrennten Elongation-Schritt, daher wurden Annealing und Elongation in der vorlie-
genden Arbeit zusammengefasst. Insgesamt sieben der 24 Protokolle wurden mit einem
Denaturierungschritt uber 15 sec bei 95 ◦C und einer nachfolgenden Amplifikation bei
60 ◦C fur 60 sec durchgefuhrt (Olvera et al., 2004; Ophuis et al., 2006; van Elden
et al., 2001; Watzinger et al., 2004; Hindiyeh et al., 2005; Egli et al., 2001; Revilla-
Fernandez et al., 2005). Dabei wurden fast alle Virusspezies abgedeckt (PCV-2, KSPV,
Influenza-A-Virus, PRRSV). Weitere Protokolle wiesen ahnliche Spezifikationen auf (van
Rijn et al., 2004; Hofmann, 2003; Krafft et al., 2005). Aufgrund der breit gefacherten
Anwendung dieses Protokolls fur fast alle Virusspezies, die fur den vorliegenden Ansatz
in Frage kamen, wurde das oben genannte Thermoprofil fur die weitere Entwicklung aus-
gewahlt und erwies sich im Folgenden als praktikabel.
5.3.1 Auswahl der Genomregionen
Fur die Auswahl der Genomregionen wurden nach Moglichkeit konservierte Bereiche
gewahlt, die in der Literatur als etablierte Regionen fur den PCR-Nachweis publiziert
waren. Durch ein Alignment aller bis dato verfugbaren Sequenzen wurde die Umsetzbar-
keit fur das aktuelle System uberpruft und entsprechende Bereiche ausgewahlt.
Fur die Detektion des ASPV ist im OIE Manual of Standards for Diagnostic Tetsts and

110 5. Diskussion
Vaccines (CHAPTER 2.1.12) u. a. die von King et al. (2003) publizierte TaqMan-PCR
empfohlen. Sowohl dieses Protokoll als auch das von Zsak et al. (2005) publizierte PCR-
Protokoll basieren auf der Detektion eines Genomfragmentes des VP72-Strukturproteins
des ASPV. Das von Hjertner et al. (2005) publizierte Protokoll zur Detektion des ASPV
beruht hingegen auf einem Fragment des VP73. Bei diesem Protokoll handelt es sich je-
doch nicht um ein TaqMan-Protokoll, sondern um ein so genanntes Invader R© Assay. Der
Empfehlung des OIE Manual of Standards folgend, wurde fur die vorliegende Arbeit ein
Genomfragment des VP72 gewahlt. Dieses Genomfragment weist eine fast 100 %ige Nu-
kleotididentitat unter den ASPV-Stammen auf (Yu et al., 1996) und ist daher fur die
Detektion aller bekannten Stamme des ASPV geeignet.
Die in der Literatur verfugbaren real-time PCR-Protokolle zur Detektion des SHV-1 al-
leine bzw. in Kombination mit anderen Viren verwenden entweder die Genomsequenz des
gB-Proteins (van Rijn et al., 2004; Yoon et al., 2005), oder wie die im Rahmen des EU-
Projektes QLK2-CT-2000-00486 entwickelten Molecular Beacons die fur das gD-Protein
kodierende Sequenz. Das gB-Genomfragment wird auch im OIE Manual of Standards
als mogliches, hochkonserviertes PCR-Ziel erwahnt. Die Anzahl der fur ein weiterfuhren-
des Alignment verfugbaren Sequenzen war stark limitiert, zeigte jedoch eine hohe Se-
quenzidentitat im genannten Abschnitt. Daher wurde dieser Bereich auch fur die SHV-1-
spezifische PCR im Rahmen dieser Arbeit zugrunde gelegt.
Bei der Entwicklung der PCV-2-spezifischen PCR wurden 136 Sequenzen ausgewertet.
Der ausgewahlte Genombereich des ORF2 wurde in mehreren Publikationen als geeignet
beschrieben (Kim und Chae, 2003; Brunborg et al., 2004). Einerseits liegt hier ein ho-
her Konservierungsgrad der Genomsequenz vor, andererseits ist er fur die Differenzierung
zwischen PCV-1 und PCV-2 geeignet (Larochelle et al., 1999). Dies konnte sowohl im
ClustalW-Alignment als auch in weiteren Tests bestatigt werden.
PPV-spezifische TaqMan-PCR-Protokolle waren zum Zeitpunkt der Entwicklung nicht
publiziert. Die gewahlte Genomregion des VP2 erwies sich im Alignment der 45 verfugba-
ren Sequenzen als hoch konserviert und wurde fur das Primer- und Sondendesign ver-
wendet. In der Literatur bis dato publizierte PCR-Protokolle zur Detektion von PPV
amplifizieren hauptsachlich ein Genomfragment dieser Region.
Bei der Entwicklung der Influenza-A PCR kam in erster Linie der fur das Matrix-Protein
kodierende Genomabschnitt in Frage. Dieser Abschnitt ist hochkonserviert (> 80 %) un-

5.3. Literaturrecherche und Auswahl der Erreger 111
ter verschiedenen Influenza-A Subtypen (Stone et al., 2004). Die in der Literatur publi-
zierten TaqMan-PCR-Protokolle, die nicht zur Differenzierung unterschiedlicher Subty-
pen konzipiert wurden, nutzen ebenfalls diese hochkonservierte Genomregion zum Nach-
weis unterschiedlicher Influenza-A-Viren (van Elden et al., 2001; Watzinger et al.,
2004). Im ClustalW-Alignment der verfugbaren Sequenzen wurde die hohe Konservie-
rung bestatigt und dieser Bereich daher fur den Test verwendet.
Gangige Genomfragmente fur den Nachweis porziner Pestiviren liegen im Bereich der
5′NTR, des E2-Gens und des NS5B-Gens (Lowings et al., 1996; Bjorklund et al.,
1999; Risatti et al., 2003; Uttenthal et al., 2003; Gaede et al., 2005; Hoffmann
et al., 2005; Ophuis et al., 2006). Diese Bereiche werden auch fur phylogenetische Ver-
gleiche unterschiedlicher KSPV-Stamme im Rahmen molekular-epidemiologischer Studi-
en herangezogen (Frias-Lepoureau und Greiser-Wilke, 2002). Das im Rahmen des
EU-Projektes QLK2-CT-2000-00486 entwickelte PriProET System fur den Nachweis von
KSPV basiert auf Primern, die von Uttenthal et al. (2003) publiziert wurden. Die
pestivirenspezifischen Primer grenzen ein Genomfragment im Bereich des 5′NTR und
des NPRO-Gens ab. Das OIE Manual of Diagnostic tests empfiehlt die Regionen der
5′NTR und des E2-Gens. Da die meisten TaqMan-basierten Systeme die 5′NTR verwen-
den (Risatti et al., 2005; Hoffmann et al., 2005; Ophuis et al., 2006), wurde dieses
Genomfragment auch in der vorliegenden Arbeit verwendet. Beim Primer- und Sondende-
sign wurden 1000 verfugbare Sequenzen aller Pestiviren (BVDV-1, BVDV-2, BDV, KSPV)
ausgewertet und eine hochkonservierte Region fur das Design ausgewahlt.
Fur die PRRSV-spezifischen PCRs wurde das von Kleiboeker et al. (2005) publizierte
Protokoll ubernommen. Fur die PRRSV-EU-Stamme basierte der Ansatz auf einem Ge-
nomfragment des ORF7, fur PRRSV-NA auf Fragmenten des ORF7 und der 3′NTR.
Die auf Grundlage der oben genannten Daten entwickelten Primer und Sonden wurden in
einem ersten Schritt in einzelnen PCR-Ansatzen getestet. Um die Identitat des Amplifi-
kats zu verifizieren, wurden die Produkte dieser PCRs zur Sequenzierung eingeschickt. Da
fur alle PCR-Produkte korrekte Sequenzen nachgewiesen wurden, konnten diese Primer
und Sonden fur die weitere Entwicklung verwendet werden.

112 5. Diskussion
5.4 Aufteilung des multitube PCR-Ansatzes
Bei der Aufteilung der zu detektierenden Erreger auf die Tubes bot sich an, RNA-Viren
und DNA-Viren zu trennen, da sie in gleicher und somit passender Anzahl vorlagen. Fur
die Funktion der RT-PCRs zur Detektion der RNA-Viren konnten somit beste Reaktions-
bedingungen geschaffen und eine Interferenz mit DNA vermieden werden. Zudem konnte
die Entwicklung der internen Kontrolle spezifisch fur RNA bzw. DNA durchgefuhrt wer-
den. Die Konzeption der einzelnen multiplex Ansatze unterschied sich wesentlich von
der Konzeption der Ansatze im Rahmen des oben genannten EU Projektes. Wahrend in
der vorliegenden Arbeit virusspezifische und epidemiologische Kriterien zugrunde gelegt
und in ein einheitliches Testprofil eingepasst wurden, erfolgte die dortige Kombination in
symptomatischen Clustern mit unterschiedlichen Methoden. Die hier gewahlte Kombina-
tion der vereinheitlichten Ansatze erfolgte aus folgenden Grunden: Die DNA-Viren ASPV
und SHV-1 wurden in einem Ansatz kombiniert, da eine sichere und sensitive Detekti-
on dieser anzeigepflichtigen Erkrankungen von großer Bedeutung ist. Ein gleichzeitiges
Auftreten beider Erkrankungen in einem Schwein ist hochst unwahrscheinlich, daher ist
davon auszugehen, dass der PCR zur Detektion des in der Probe vorhandenen Erregers
alle Ressourcen (dNTPs, Polymerase, MgCl2) zur Verfugung stehen, und daher beste Re-
aktionsbedingungen gegeben sind.
Im zweiten Ansatz wurden die DNA-Viren PPV und PCV-2 kombiniert. PCV-2 ist ein
weit verbreitetes Virus, das v. a. Faktorenerkrankungen auslost und dessen Detektion
hauptsachlich in Zusammenhang mit einer Bestimmung der Viruslast aussagekraftig wird.
Fur eine verlassliche Diagnose ist es somit sinnvoll, eine Standardverdunnungsreihe mit-
zufuhren und die Viruslast quantitativ zu errechnen. Der Ansatz liess diese Option zu.
PPV ist ebenfalls weit verbreitet. Der Krankheitskomplex fuhrt zu schweren wirtschaftli-
chen Schaden. Im vorliegenden Ansatz wurden PPV daher durch die Wahl eines kurzeren
Amplifikationsfragmentes gunstigere Reaktionsbedingungen eingeraumt.
Die Ansatze drei und vier enthielten die RNA-Viren. Im dritten Ansatz wurde der Pesti-
virusnachweis mit der Influenza-A-spezifischen PCR kombiniert. Bei der Entwicklung der
PCR wurde dem pestiviruspezifischen Nachweis der Vorzug gegeben, um eine moglichst
umfassende Erfassung der beim Schwein vorkommenden KSPV-und Pestivirusisolate zu
gewahrleisten. Da die Bestatigung eines Primarausbruchs in Hausschweinebestanden auf

5.5. Nukleinsaureisolierung 113
der Grundlage des Kapitels VI der Entscheidung der Kommission 2002/106/EC4 (Diagno-
sehandbuch) in mindestens zwei Tests zum Genomnachweis erfolgen muss, ist es moglich,
den in der Entscheidung geforderten KSPV-spezifischen Nachweis in einem zweiten Schritt
durchzufuhren. Da ruminante Pestiviren nur selten zu einer nachweisbaren Viramie im
Schwein fuhren (Paton et al., 1994), ist davon auszugehen, dass positive Genomnach-
weise fast ausschließlich in Korrelation mit KSPV-Infektionen auftreten. Dem Pestivi-
rusnachweis konnten in diesem Falle aufgrund der beschrankten Moglichkeiten in der
Primer- und Sondenauswahl keine gunstigeren Reaktionsbedingungen eingeraumt wer-
den. Allerdings hatte es sich gezeigt, dass in einem multiplex-Ansatz die Effizienz der
pestivirusspezifischen-PCR konstanter bleibt. Ein gemeinsames Auftreten der beiden Er-
reger ist nicht auszuschließen. Sollte es daher zu einem gleichzeitigen Vorliegen der beiden
Erreger in einer Probe kommen, ist die Amplifikation der Pestiviren effizienter. In den
Fallen einer solitaren Influenzainfektion kommen diese Einschrankungen nicht zum Tra-
gen.
Da fur die Detektion und Differenzierung der PRRSV-Stamme ein etabliertes Protokoll
ubernommen wurde, war die Umsetzung in einem Ansatz (Ansatz 4) sinnvoll, um die fur
dieses Protokoll optimierten Bedingungen beizubehalten.
5.5 Nukleinsaureisolierung
Im Bestreben, moglichst einfach umzusetzende, standardisierte und unter den Bedingun-
gen unterschiedlicher Labore reproduzierbare Techniken zu nutzen, wurden, wo moglich,
kommerzielle Kits eingesetzt. Fur den Erfolg einer PCR ist die Qualitat der Nukleinsaure
von entscheidender Bedeutung, daher erfolgte die Isolierung der Nukleinsauren mit stan-
dardisierten kommerziellen Kits (s. Kapitel 3.2.2.1). Bis Mitte 2005 befanden sich diverse
Extraktionssysteme auf dem Markt. Die mit Abstand am weitesten verbreitete Methode
basiert auf dem Einsatz von Silica-Gel Membranen. Wie Scheibner (2000) feststellte,
ist der RNeasyR© Lipid Tissue Kit fur die RNA-Extraktion aus Organmaterial am bes-
ten geeignet. Fur die Isolierung von DNA stehen unterschiedliche kommerzielle Kits zur
4http://europa.eu.int/eur-lex/pri/en/oj/dat/2003/l_324/l_32420031211en00550056.pdf

114 5. Diskussion
Verfugung. Neben einer getrennten Isolierung von RNA und DNA besteht die Moglich-
keit einer simultanen Extraktion, fur die ebenfalls kommerzielle Kits existieren. Diese
sind allerdings nur fur zellfreies Material ausgelegt, was ihren Einsatz in der Diagnostik
einschrankt. Außerdem sah das Gesamtkonzept eine getrennte Handhabung von DNA-
bzw. RNA-Viren vor, so dass aus diesen Grunden auf eine simultane DNA- und RNA-
Extraktion verzichtet wurde.
5.6 Optimierung der PCRs
5.6.1 Optimierung der Reaktionsbedingungen
Jede neu entwickelte PCR bedarf einer Optimierung sowohl der physikalischen als auch
der chemischen Reaktionsbedingungen. Zu den physikalischen Bedingungen gehort in ers-
ter Linie das thermische Profil. Nach der Festlegung der Primersequenzen erfolgt eine
Anpassung der Annealing-Temperatur.
Da das Gesamtkonzept einen auf vier Tubes verteilten Ansatz bei einheitlichem Ther-
moprofil vorsah, wurden die physikalischen Reaktionsbedingungen fur jede PCR nicht
individuell variiert. Dadurch war notwendig, moglichst alle Primer und Sonden fur die
definierten Reaktionsbedingungen zu entwerfen. Primer- und Sondensequenzen mussten
hinsichtlich ihrer Schmelztemperatur an das ausgewahlte Thermoprofil angepasst werden.
Weitere Optimierungsmoglichkeiten bestehen generell bei den chemischen Reaktionsbe-
dingungen. Da es sich bei diesem Ansatz um ein sehr heterologes System handelte, waren
sie entsprechend beschrankt. Es wurde folglich entschieden, ein einheitliches, kommerziell
erhaltliches PCR-Mastermix zu verwenden. Der ausgewahlte QuantiTect R© Multiplex PCR
Kit wurde speziell fur multiplex real-time PCR konzipiert und hat sich wahrend der PCR
Validierung als geeignet erwiesen. Der große Vorteil kommerzieller Mastermixe besteht in
weitgehend konstanter Qualitat und Ubertragbarkeit der Ergebnisse.

5.6. Optimierung der PCRs 115
5.6.2 Optimierung der Primerkonzentrationen
Die Ermittlung des fur eine effektive Amplifikation geeigneten Konzentrationsbereichs er-
folgte mittels SYBRTM-Green real-time PCR in Doppelansatzen einer Primertitration.
Die Auswahlkriterien waren ein moglichst niedriger Ct-Wert bei hohen Fluoreszenzwer-
ten, da dies fur eine hohe Effizienz der PCR spricht (Nolan, 2004).
Fur den vorliegenden multiplex PCR-Ansatz war die Optimierung der Primer- und Son-
denkonzentrationen von besonders großer Bedeutung, da auf diese Weise gegenseitige In-
hibitionen und Interaktionen minimiert werden konnten (Elnifro et al., 2000). Es wurde
die kleinstmogliche Primerkonzentration ausgewahlt, die akzeptable Ct- und Fluoreszen-
zwerte (∆Rn) zeigte. Fur alle PCR-Ansatze konnte mittels einer graphischen Darstellung
der Titrationsergebnisse in MicrosoftR© Office Excel in Konzentrationsbereich gefunden
werden, der den Anforderungen entsprach.
Die Produktspezifitat und die Bildung unspezifischer Produkte wurden mittels einer NTC
mit unterschiedlichen Primerkonzentrationen getestet. Primerkonzentrationen von 900 nM
fuhrten zur Bildung eines unspezifischen Produktes. Es ist bekannt, dass hohe Primerkon-
zentrationen die Bildung von Primer-Dimeren und unspezifischen Produkten begunstigen
(Bustin, 2000). Im vorliegenden Fall war das Produkt jedoch anhand der Schmelztem-
peratur sicher vom spezifischen Produkt zu unterscheiden.
5.6.3 Optimierung der Sondenkonzentrationen
In gleicher Weise wurden die optimalen Sondenkonzentrationen bestimmt. Ziel war es,
niedrige Ct- und hohe ∆R-Werte zu kombinieren, da dies mit einer guten Funktion der
Sonde verbunden ist (Peters et al., 2004). Fur alle PCR-Ansatze konnten Konzentrati-
onsbereiche gefunden werden, die eine gute Funktion der Sonde gewahrleisteten.
5.6.4 Real-time PCR Effizienzen
Da die ermittelten Ct- und Fluoreszenzwerte alleine keine Auskunft uber die Effizienz und
Kinetik der PCR gaben, wurden Standardkurven erstellt und analysiert, die Aussagen zu
diesen Parametern ermoglichten (Higuchi et al., 1993; Bustin, 2000). Zur Erstellung

116 5. Diskussion
der Standardkurve wurden die Ct-Werte einer Standardverdunnungsreihe linearisierter
Plasmide ermittelt und gegen ihre logarithmische Genomaquivalentzahl aufgetragen. Die
Ansatze wurden dreifach sowohl in Einzel- als auch in multiplex-Ansatzen getestet. Die
Neigung der Standardkurve korrelierte mit der Effizienz der PCR (Bustin, 2000).
Fur einzel-PCRs werden Effizienzen zwischen 90 und 100 % angestrebt, fur multiplex-
Ansatze liegen sie in der Regel etwas darunter (Brisson et al., 2004). Fur alle einzel-
Ansatze konnten Effizienzen in diesem Effizienzbereich erzielt werden, und waren somit
zufriedenstellend. Die multiplex-Ansatze zeigten ausnahmslos niedrigere Effizienzen, wo-
bei die Reaktionen fur PPV und Pestiviren uber der fur einzel-Ansatze angestrebten
Effizienzmarke von 90 % blieben. Die Ansatze fur ASPV, SHV-1 und PCV-2 zeigten Effi-
zienzen von 82,4, 80,4 bzw. 85,6 %. Es ist davon auszugehen, dass diese Effizienzen fur eine
diagnostische PCR ausreichend sind, da bei 40 PCR-Zyklen davon auszugehen ist, dass
eine positive klinische Probe detektiert wurde. Die Influenza-A-spezifische PCR zeigte
eine Effizienz von knapp 70 %, die als grenzwertig anzusehen ist.
5.7 Auswahl der internen Kontrollen
Ublicherweise werden so genannte Housekeeping-Genes zur Etablierung interner Kontrol-
len verwendet, da diese kontinuierlich in allen Korperzellen exprimiert werden. Wahrend
fur Genexpressionsstudien eine Normalisierung der RNA-Expression anhand einer inter-
nen Referenz mit moglichst geringer Variabilitat sehr wichtig ist (Dheda et al., 2004),
sind fur eine nicht-quantitative Methode andere Kriterien von Bedeutung. Der interne
Standard in einer nicht-quantitativen real-time PCR dient dem Nachweis der erfolgrei-
chen Nukleinsaureextraktion und Amplifikation5. Wahrend zu diesem Zweck die absolute
Expression von untergeordneter Bedeutung ist, spielt die kontinuierliche Expression eine
wichtige Rolle.
Die getrennte Handhabung der RNA- und DNA-Viren machte die Entwicklung entspre-
chender interner Kontrollen notwendig. Im Bestreben, die internen Kontrollen auf der
Grundlage des gleichen Housekeeping-Genes zu etablieren, wurde GenBank nach entspre-
chenden Sequenzen durchsucht. Kriterien der Auswahl waren, dass sowohl eine mRNA als
5Greiser-Wilke personliche Mitteilung

5.8. Auswahl der Isolate und Organproben 117
auch entsprechende genomische Sequenz mit ausgewiesenen Introns und Exons verfugbar
waren. β-Aktin entsprach diesen Kriterien und ist auch im OIE Manual of Standards als
mogliche interne Kontrolle angegeben (Anonym, 2005a).
Im Rahmen der Uberprufung der internen Kontrollen wurde festgestellt, dass ein positives
Signal fur genomische DNA in Testlaufen zu verzeichnen war, die aus Organmaterial ex-
trahierte RNA als Probenmaterial beinhalteten. Somit bestand die Moglichkeit, dass die
extrahierte RNA eine DNA-Kontamination aufwies (s. Kapitel 4.2.2.4). Diese Moglich-
keit wird auch vom Hersteller in der Gebrauchsanweisung zum Extraktionskit eingeraumt
(Qiagen, RNeasy R©). Die Empfehlung zur Behebung des Problems ist ein DNase-Verdau.
Tatsachlich konnte im vorliegenden Fall festgestellt werden, dass nach einem Verdau keine
genomische DNA mehr nachweisbar war. In Anbetracht der Tatsache, dass es sich hier
um eine diagnostische PCR handelt, erscheint eine geringfugige Kontamination mit DNA
vernachlassigbar. Vielmehr spielt in diesem Zusammenhang eine robuste, qualitativ hoch-
wertige und konstante RNA-Extraktion eine entscheidende Rolle.
5.8 Auswahl der Isolate und Organproben
Bei der Auswahl der Virusisolate wurde versucht, die genetische Vielfalt der Erreger abzu-
decken und auf diese Weise die Detektion moglichst aller Stamme des betreffenden Virus
abzusichern. Bei der Auswahl klinischer Proben sollte der epidemiologischen Situation
Rechnung getragen werden. Die Verfugbarkeit fast aller Viren und klinischen Proben war
jedoch limitiert.
Das Arbeiten mit ASPV-haltigen Materialien war aufgrund der biologischen Sicherheits-
stufe unter den gegebenen Bedingungen nicht moglich. Alle Tests wurden daher mit in
Standardplasmide klonierten Sequenzen des VP72-Proteins durchgefuhrt. Die bereits oben
erwahnte fast 100 %ige Identitat der ASPV-Stamme im Bereich dieses Proteins lasst die
aus dieser Limitierung moglicherweise erwachsenden Probleme in den Hintergrund treten.
Uber 20 SHV-1-Isolate aus den Bestanden des nationalen Referenzlabors fur AK am
Friedrich-Loeffler-Institut (Wusterhausen) konnten hier getestet werden. Da Deutschland
seit Februar 2003 offiziell AK-frei ist (Teuffert et al., 2005), spiegelten diese Isolate

118 5. Diskussion
hauptsachlich die historische Situation in der deutschen, franzosischen und tschechischen
Schweinepopulationen wider. Da aktuell in der Schweinepopulationen in anderen Landern
zirkulierende SHV-1-Stamme nicht in die Untersuchung einbezogen werden konnten, ist
nicht mit absoluter Sicherheit davon auszugehen, dass alle Stamme erkannt wurden. Die
relativ hohe Konservierung der gewahlten Genomregion und die geringe Mutationsrate
des Virus legen dieses jedoch nahe.
Fur die Validierung der PPV-spezifischen PCR stand nur eine geringe Anzahl an Isolaten
zur Verfugung. Der Stamm NADL-2 ist gut charakterisiert und wird aufgrund seiner At-
tenuierung als Impfstamm eingesetzt (Prikhod’ko et al., 2003; Bergeron et al., 1996).
Es kann nicht eingeschatzt werden, ob die anderen Isolate die genetische Diversitat hin-
reichend wiedergeben. Jedoch wurde anhand des Alignments verfugbarer Sequenzen eine
hohe Konservierung des in der PCR nachzuweisenden VP2-Proteins gezeigt.
Vorberichtlich PCV-2-positive Seren und Organmaterial wurden fur die Uberprufung der
PCV-2-PCR verwendet. Da die Proben aus der aktuellen Routinediagnostik verschiedener
Untersuchungsamter stammten, ist davon auszugehen, dass die epidemiologische Situati-
on hinreichend wiedergegeben wurde. Da PCV-2-Infektionen in der Schweinepopulation
weit verbreitet sind, und Krankheitssymptome vornehmlich von der Viruslast abhangen
(Krakowka et al., 2000; Liu et al., 2000), ist eine quantitative Erfassung des Erregers
ratsam (McNeilly et al., 2002).
Alle verfugbaren Genotypen des KSPV (exkl. Gruppe 3) wurden durch gut charakteri-
sierte Isolate aus der Virusbank des EURL reprasentiert, die neben historischen Stammen
auch Feldisolate aus den letzten Jahren beinhaltet. Das Organmaterial stammte aus-
schließlich von experimentell infizierten Tieren, wurde jedoch ausgewahlt, um eine großtmogli-
che Vielfalt einzuschließen. Die Genotypen 2.3 und 2.1 gehoren zu den aktuell in wei-
ten Teilen der Erde zirkulierenden KSPV-Stammen (Paton und Greiser-Wilke, 2003;
Sandvik et al., 2005). Die epidemiologische Aktualitat der Isolate war somit ebenfalls
gegeben. Die am EURL zu diesem Zeitpunkt verfugbaren BDV-Isolate wurden in die Un-
tersuchungen einbezogen, wobei BDV-Stamme ovinen und porzinen Ursprungs vorhanden
waren. Organmaterial experimentell mit BDV infizierter Schweine konnte ebenfalls unter-
sucht werden.
Fur die Uberprufung und Validierung der PCR zur Detektion des porzinen Influenza-A-
Virus wurden zwolf Isolate unterschiedlicher Subtypen verwendet. Diese Isolate beinhal-

5.9. Validierung 119
teten die Subtypen H1N1, H3N2 und H1N2 und somit alle in der Schweinepopulation
kozirkulierenden Virusstamme (Reeth et al., 2004).
Fur die Umsetzung der PRRSV-spezifischen PCR wurde auf das Protokoll von Kleiboeker
et al. (2005) zuruckgegriffen, welches die Differenzierung der EU- und NA-Stamme des
PRRSV erlaubte. Zur Uberprufung in Zusammenhang mit der vorliegenden Arbeit wurden
die Impfstamme MLV (PRRSV-NA) und DV (PRRSV-EU) eingesetzt. Fur den Stamm
DV wurde gezeigt, dass er in der ORF5 Region eine uber 86 %ige Identitat auf Nukleotid-
ebene mit aktuellen Feldisolaten aufweist (Harder et al., 2004). Die fur die ORF5-Region
gezeigte Identitat und die geringe Variabilitat des ORF7 (Gall et al., 1998) legen nahe,
dass europaische PRRSV-Stamme durch den Impfstamm DV gut reprasentiert wurden.
Die in der vorliegenden Arbeit genutzten Genomregionen lagen allerdings im ORF7 und
der 3′NTR.
5.9 Validierung
Fur eine neu entwickelte PCR ist es von großer Bedeutung, ihre Leistungsfahigkeit und
Grenzen zu ermitteln. Dabei ist sie einerseits anhand von Validierungskriterien zu testen
und andererseits mit den bisher etablierten Methoden zur Diagnostik der gewahlten Erre-
ger zu vergleichen. Zu den Validierungskriterien gehoren die analytische Sensitivitat und
Spezifitat der Methode. Beide Kriterien wurden soweit wie moglich, nach OIE-Vorgaben
berucksichtigt. Fur den Einsatz in der Diagnostik sollte die neue PCR gleichwertig oder
besser als der zellkulturelle Nachweis sein. Hinsichtlich der analytischen Sensitivitat wur-
de in verschiedenen Untersuchungen gezeigt, dass TaqMan real-time RT-PCRs im Ver-
gleich zur Virusisolierung oder der konventionellen, gelbasierten einfachen oder nested
RT-PCR als sensitiver bzw. zumindest gleichwertig zu beurteilen sind (Kearns et al.,
2002; Locatelli et al., 2000; McGoldrick et al., 1998, 1999; Smith et al., 2001). Fur
die vorliegenden PCRs konnte dies ebenfalls bestatigt werden.

120 5. Diskussion
5.9.1 Sensitivitat
Die analytische Sensitivitat ist definiert als die kleinste Menge eines Pathogens, die von
Null unterschieden werden kann (Anonym, 2004b). Zur Bestimmung werden Genomaqui-
valente und infektiose Einheiten wie KID50 oder PFU herangezogen. Im Rahmen des vor-
liegenden Projektes erfolgte die Bestimmung der Nachweisgrenze in Genomaquivalenten
und, wo moglich, in Korrelation mit einer zellkulturell ermittelten Angabe zum Virustiter
in KID50. Genomaquivalente lassen sich in der quantitativen real-time PCR auf der Basis
von in Vektoren klonierter PCR-Produkte aus Standardkurven ermitteln (Bustin, 2000).
Fur ASPV und PCV-2 stand nur die Moglichkeit zur Verfugung, das Detektionslimit an-
hand der nachweisbaren Genomaquivalentzahl zu bestimmen. Im Falle des ASPV reichte
die Sicherheitsstufe des Labors nicht aus, um mit vermehrungsfahigem Virus zu arbeiten.
PCV-2 konnte nicht in Zellkultur vermehrt werden. Es wurden mehrere Versuche unter-
nommen, das Virus aus nachweislich positivem Organmaterial in Zellkultur (PK(15)A)
anzuzuchten und zu titrieren, welche jedoch scheiterten. Fur das PCV-2 ist bekannt, dass
die Anzucht in Zellkultur haufig nicht moglich ist (T. Harder, personliche Mitteilung).
Fur beide PCRs wurde mit in Vektoren klonierten PCR-Produkten eine Nachweisgrenze
von 10 Kopien pro Ansatz ermittelt.
Die Sensitivitatsprufung im Falle des SHV-1, des PPV, der Pestiviren, des Influenza-A-
Virus sowie beider PRRSV-Stamme erfolgte mittels titrierter Virussuspensionen, welche
als Ausgangsmaterial fur die PCRs eingesetzt wurden. Fur den direkten Vergleich wurde
das Probenvolumen der Virustitration korrigiert und die KID50 fur 6µl angegeben. Dieser
Vergleich wurde gewahlt, um die infektiosen Einheiten mit der Zahl der Genomaquiva-
lente zu korrelieren.
Ausschließlich gegenuber der SHV-1 spezifischen PCR zeigte die Virusisolierung bzw.
-titration eine hohere Sensitivitat. Hier konnte eine Virussuspension mit 60,5 KID50/6µl
von der PCR als positiv getestet werden, was 1,37 Genomaquivalenten entsprach. Der zell-
kulturelle Nachweis gelang auch fur die darauf folgenden zwei Verdunnungsstufen. Dies
konnte auf das G/C-reiche Genom des SHV-1 zuruckzufuhren (Klupp et al., 2004b), das
ein”schwieriges“ Ziel fur eine PCR darstellt. Daten zum Virustiter in klinischem Mate-
rial sind nicht publiziert; diese hangen jedoch stark vom gewahlten Organmaterial und
dem Zeitpunkt der Probennahme im Infektionsgeschehen ab. Legt man den hohen Titer

5.9. Validierung 121
des Zellkulturuberstandes als Anhaltspunkt an, ist davon auszugehen, dass eine PCR, die
einen Virustiter von 101,78 KID50 im Probenvolumen sicher erkennt, fur diagnostische
Zwecke ausreichend ist. Betrachtet man die Ergebnisse dieser Sensitivitatsprufung genau-
er, zeigt sich, wie der Tabelle A.14 zu entnehmen ist, dass die Stufe 5 der Verdunnungsreihe
bereits negativ war. Allerdings wurde in der nachst hoheren Verdunnungsstufe eine deut-
lich positive Reaktion verzeichnet. Dies konnte mit einer ungleichmaßigen Ausverdunnung
der DNA erklart werden, zumal einer der dreifach-Ansatze positiv war. Es ist davon aus-
zugehen, dass die oben genannte Sensitivitat nicht die maximale, sondern die minimale
Leistungsfahigkeit des vorliegenden Ansatzes reprasentiert.
Die PPV-PCR ergab ein positives Signal in der Stufe 7 der logarithmischen Verdunnungs-
reihe, und konnte somit 0,74 KID50/6µl bzw. 1,17 Genomaquivalente sicher nachweisen.
In diesem Fall erwiesen sich PCR und zellkultureller Nachweis als annahernd gleichwer-
tig.
Die Sensitivitatsprufung der pestivirusspezifischen PCR wurde ebenfalls anhand einer
titrierten Virussuspension durchgefuhrt. Die letzte Verdunnungsstufe, die als positiv ge-
testet wurde, hatte einen KID50/6µl-Wert von 0,60 oder 7,18 Genomaquivalenten. Es
ist somit auch in diesem Fall davon auszugehen, dass die Sensitivitat der PCR mit der
des Goldstandards in der KSPV-Diagnostik, der Virusisolierung (Moennig, 2000), ver-
gleichbar ist. Im Rahmen des EU Projektes QLK2-CT-2000-00486 konnte die gelbasierte
KSPV-multiplex-PCR eine Nachweisgrenze von 0,32 KID50 erreichen. Aus der Publikation
geht nicht hervor, auf welches Volumen sich diese Angabe bezieht. Da in Virustitrationen
eine Toleranz von 1/2 log-Stufen eingeraumt wird, ware sie mit der in der vorliegenden
Arbeit ermittelten Sensitivitat vergleichbar.
Fur das Influenza-A-Virus wurde die letzte positive Reaktion im zellkulturellen System bei
Verdunnungsstufe 4 verzeichnet, wobei auch der Ausgangstiter der Virussuspension mit
105,25 KID50/100µl relativ niedrig war. Die PCR konnte drei weitere Verdunnungsstufen
als positiv detektieren und war somit wesentlich sensitiver. Die in diesen Verdunnungs-
stufen detektierten Konzentrationen lagen bei 19,5, 2,1 und 0,81 Genomaquivalenten. Es
ist nicht auszuschließen, dass die suboptimalen Wachstumseigenschaften des Influenza-
A-Virus in der Zellkultur einen Einfluss auf das Ergebnis hatten. Zur Bestimmung der
analytischen Sensitivitat der beiden PRRSV-PCRs wurden je ein amerikanischer und eu-
ropaischer Stamm des Virus (PRRSV-EU, bzw. PRRSV-NA) in Zellkultur vermehrt und

122 5. Diskussion
anschließend titriert. Der zellkulturelle Nachweis dieses Virus ist schwierig und die Ver-
wendung der MARC-145 Zelllinie war dem OIE Manual of Standards zufolge suboptimal
fur den Nachweis europaischer PRRSV-Stamme. Fur den verwendeten Impfstamm DV
traten jedoch keine Probleme auf und auch der PRRSV-NA Stamm ließ sich problemlos
anzuchten. Wie im OIE Manual of Standards vorgeschlagen, erfolgte der zellkulturelle
Nachweis uber zwei Passagen. Die PRRSV-EU real-time PCR wies in der Verdunnungs-
stufe mit auf 6µl umgerechneten 0,6 KID50 26,4 Genomaquivalente nach, die PRRSV-NA
PCR entsprechend bei 0,06 KID50 373 Genomaquivalente. Bei beiden real-time PCRs
konnte jeweils eine weitere Verdunnungsstufe positiv detektiert werden. Die PRRSV-NA
ergab 13,5 die PRRSV-EU 1,07 Genomaquivalente. Auch in diesem Fall war die real-time
PCR dem zellkulturellen Nachweis uberlegen. Generell ist die Ubertragbarkeit dieser Aus-
sagen auf klinisches Organmaterial unterschiedlicher Qualitat und Art durchaus kritisch
zu betrachten, da hier die PCR jedoch auch bei klinischen Proben gute Ergebnisse zeigte,
ist eine akzeptables Detektionslimit auch fur PRRSV anzunehmen.
5.9.2 Spezifitat
Die analytische Spezifitat ist definiert als Fahigkeit eines diagnostischen Tests, einen Er-
reger von anderen Erregern zu unterscheiden. Zu diesem Zweck werden i. d. R. genetisch
verwandte Pathogene analysiert und klinisches Material von Tieren untersucht, die mit
Erregern infiziert sind, die sich in der klinischen Symptomatik der nachzuweisenden Er-
krankung ahneln (Anonym, 2004b).
Die zu detektierenden Erreger besaßen nur zum Teil genetische Verwandte, die im Schwein
nachweisbar waren. Das ASPV ist der einzige Vertreter im Genus Asfivirus der Familie
Asfarviridae (Dixon et al., 2000). Eine gewisse genetische Verwandtschaft besteht zu
Iridoviren (Murphy et al., 1999), die jedoch ausschließlich eine Rolle bei Fischen und
Amphibien spielen. Probleme mit genetisch verwandten Viren waren somit fur das ASPV
nicht zu erwarten.
Das SHV-1 gehort zur Subfamilie der Alphaherpesvirinae (Klupp et al., 2004b), welche
u. a. auch veterinarmedizinisch bedeutsame bovine, equine, caprine, canine, feline und
einige aviare Herpesviren sowie das Herpes simplex Virus des Menschen enthalt. Da die
Herpesviren speziesspezifisch sind (Modrow et al., 2003), ist eine Interaktion der Detek-

5.9. Validierung 123
tion des SHV-1 mit genetisch verwandten Erregern nicht anzunehmen.
Nur die Subfamilie Parvovirinae der Familie Parvoviridae enthalt Viren, die Vertebraten
im weitesten Sinne betreffen (Murphy et al., 1995). Nur das Genus Parvovirus enthalt
veterinarmedizinisch bedeutsame Erreger (Modrow et al., 2003). Neben dem porzinen
Parvovirus enthalt es Erreger wichtiger Erkrankungen der Katzen, Hunde, Nerze, Nager,
Ganse und Enten. Die phylogenetische Analyse der Parvoviren zeigt, dass das porzine
Parvovirus ein relativ eigenstandiger Zweig im phylogenetischen Baum ist. Die nachste
Verwandtschaft besteht auf Ebene der Genomsequenz zu felinen und caninen Parvoviren
(CPV-2) sowie zum Parvovirus der Waschbaren (Murphy et al., 1995). Fur keines die-
ser Parvoviren ist ein Nachweis im Schwein bekannt. Daher ist auch hier nicht von einer
Interferenz in der PCR auszugehen.
Fur die Analyse der Spezifitat der Circovirus-PCR war die enge Verwandtschaft des PCV-
2 zum PCV-1 von großer Bedeutung. Wahrend das PCV-1 nicht mit einem Krankheits-
bild in Verbindung gebracht wird, ist das PCV-2, wie bereits erwahnt, mit verschiedenen
Krankheitsbildern assoziiert (PMWS, PDNS, PRDC). Es konnte mit Plasmiden, die das
PCV-1-Genom enthielten, gezeigt werden, dass keine Kreuzreaktivitat zwischen diesen
beiden PCV-Typen in der PCR bestand. Andere Vertreter der Familie Circoviridae bzw.
wie das psittacine beak and feather virus spielen fur das Schwein keine Rolle (Todd et al.,
2001a,b).
Die PCR zur Detektion der porzinen Influenza wurde typspezifisch, d. h., fur Influenza-
A-Viren etabliert. Bei den Influenza-A-Viren handelt es sich um RNA-Viren mit einem
segmentierten Genom (Modrow et al., 2003). Dieses Charakteristikum bringt es mit sich,
dass neben den momentan in der Schweinepopulation kozirkulierenden Subtypen durch
Reassortment mit aviaren oder humanen Influenzastammen neue Stamme mit anderen
Subtypenkombinationen entstehen konnten (Reeth et al., 2004). Eine typspezifische De-
tektion ist somit sinnvoll. Da sowohl human- als auch veterinarmedizinisch ausschließlich
Influenza-A-Viren eine Rolle spielen (Truyen, 2003), konnte auf die Integration von In-
fluenza B und C in dieser Untersuchung verzichtet werden.
Die Wahl der so genannten Panpesti-Sonde zum Nachweis der KSP brachte es mit sich,
dass per se die nahe verwandten Viren der BVD und BD ebenfalls detektiert wurden.
Aus epidemiologischer Sicht ist diese Wahl moglich, eine Spezifitatsprufung konnte da-
her jedoch nur ausserhalb des Genus Pestivirus erfolgen. Die Familie Flaviviridae enthalt

124 5. Diskussion
keine weiteren Vertreter, die im Schwein zu erwarten waren. Probleme in der Detektion
sind somit unwahrscheinlich. Allerdings bleibt anzumerken, dass im Falle einer positiven
Reaktion eine weitere Differenzierung erfolgen muss. Eine weitere Abklarung ist jedoch oh-
nehin notwendig,da es die rechtlichen Grundlagen nicht gestatten (Council Directive
2001/89/EC), die Diagnose der anzeigepflichtigen Seuchen auf einen einzelnen positiven
Befund zu grunden.
Das PRRSV gehort zum Genus Arterivirus in der Familie der Arteriviridae (Cavanagh,
1997; Meulenberg et al., 1993b). Die verwandten Spezies des equinen Arteritis-Virus
und des Laktatdehydrogenase-Virus sind fur das Schwein aufgrund des beschrankten
Wirtsspektrums nicht von Bedeutung. Fur die Spezifitat der PCR war die Diskriminie-
rung der europaischen und amerikanischen Stamme von Bedeutung. Es konnte bestatigt
werden, dass, wie von Kleiboeker et al. (2005) gezeigt wurde, eine spezifische Differen-
zierung zwischen den Stammen stattfindet.
Da es sich um eine differentialdiagnostische PCR handelte, waren von vornherein Erre-
ger integriert, die mit einem ahnlichen Krankheitsbild einhergingen. Bei der Prufung der
Isolate und klinischen Proben konnte gezeigt werden, dass zwischen den verschiedenen Er-
regern keine Kreuzreaktivitat bestand. Die verfugbaren positiven und negativen Proben
wurden von allen PCRs korrekt erkannt. Allerdings reichte die Anzahl der Proben nicht
aus, um eine vollstandige Validierung entsprechend der OIE-Richtlinien durchzufuhren.
Um die Aussage betreffend der Spezifitat zu erhohen, mussten weitere Untersuchungen mit
Probenmaterial zusatzlicher differentialdiagnostisch in Frage kommender Erreger durch-
gefuhrt werden.
Die BLAST-Suche ergab fur alle entwickelten Primer und TaqMan-Sonden eine spezifi-
sche Bindung. Es wurden ausnahmslos geringfugige Identitaten (maximal 3-4 Basen) mit
fremden Sequenzen festgestellt.
Trotz der praktischen Limitierungen in der Uberprufung der Testspezifitat ist auf Grund-
lage der Ergebnisse aus dem Testen der Isolate und klinischen Proben sowie unter Zuhil-
fenahme der BLAST-Suche davon auszugehen, dass die gewahlten Genomregionen spezi-
fisch detektiert werden. Es ist jedoch ratsam die Prufung der analytischen Spezifitat mit
anderen Erregern fortzufuhren.

5.9. Validierung 125
5.9.3 Klinische Proben
Zur Uberprufung der diagnostischen Sensitivitat und Spezifitat sieht das OIE Manual of
Standards die Testung klinischer Proben bekannten Hintergrunds vor. Aufgrund der Limi-
tierung des zur Verfugung stehenden Untersuchungsmaterials war die Prufung klinischer
Proben nur in geringem Umfang moglich, zudem bestanden nur fur einen Teil der Proben
direkte Vergleichsmoglichkeiten hinsichtlich der Ergebnisse. Somit konnen die erhobenen
Befunde nur als Anhaltspunkte angesehen werden.
Neben 33 Proben aus der Routinediagnostik der Außenstelle fur Epidemiologie der TiHo
Hannover standen 17 Proben aus dem EURL zur Verfugung. Das Probenmaterial, das
aus dem EURL stammte, beinhaltete sowohl Proben, die von Tieren stammten, die ex-
perimentell mit verschiedenen KSPV-Stammen bzw. einem BDV-Stamm infiziert waren
als auch negatives Material, welches vor der Infektion von verschiedenen Tieren gewon-
nen wurde. Die KSPV-infizierten Tiere wurden mit der Ausnahme eines Tieres korrekt
erkannt. Ein Tier, welches mit BDV Gifhorn infiziert wurde, konnte nicht detektiert wer-
den. Die nicht detektierte Probe stammte von einem vorberichtlich mit dem KSPV-Stamm
CSF0309 infizierten Tier (Tier 266). In den Untersuchungen, die mit den Routinemetho-
den des EURLs durchgefuhrt worden waren, reagierte es positiv in real-time RT-PCR und
Virusisolierung. Dieses Tier konnte mit der vorliegenden PCR nicht schlussig detektiert
werden, da schwach positive Reaktionen bei hohen Ct-Werten neben negativen Laufen
auftraten. Eine nachfolgende Titration des nachgewiesenen Virus ergab einen niedrigen
Titer von 100,75 bzw. 101 KID50/ml Serum. Der Nachweis im Antigen-ELISA gelang nicht.
Diese Befunde legen nahe, dass mit einem niedrigen Virustiter auch in der vorliegenden
Probe gerechnet werden musste. Die dargestellten Ergebnisse des EURL wurden mit frisch
gewonnenem Material erzielt. Es ist nicht auszuschließen, dass durch die Lagerung bzw.
den Gefrier-Tau-Zyklus eine Abbau der Nukleinsaure stattfand, die zu einem Absinken
unter die Nachweisgrenze des vorliegenden Testsystems fuhrte.
Im Falle des vorberichtlich BDV-positiven Materials, das durch die vorliegende PCR nicht
detektiert werden konnte, ist zu bedenken, dass sich BDV in Schweinen nur selten (Paton
et al., 1994) und nicht zu hohen Titern vermehrt, so dass anzunehmen ist, dass das Ma-
terial nur geringe Virusmengen enthielt. Dieses Tier war mit den Routinemethoden des
EURL, real-time RT-PCR und Virusisolierung auf permanenten Schafthymuszellen, po-

126 5. Diskussion
sitiv gestestet worden. Allerdings zeigte das Organmaterial dieses Tieres auch hier hohe
Ct-Werte (Ct 35,3 bzw. 35,5) und negative Ergebnisse in der Virusisolierung auf porzinen
Zellen.
In drei Proben konnte zusatzlich PCV-2 nachgewiesen werden. Diese Ergebnisse konnten
jedoch nicht mit vorberichtlichen Befunden korreliert werden, da die Voruntersuchungen
am EURL ausschließlich die Detektion von Pestiviren umfasste. Aus serologischen Unter-
suchungen ist bekannt, dass PCV-2 ubiquitar vorkommt und nicht unbedingt mit einem
Krankheitsbild korreliert sein muss (Rose et al., 2002). Die serologische Pravalenz wird
weltweit mit nahezu 100 % fur Mastschweine angenommen (Segales und Domingo,
2002). Somit war eine Detektion bei einem Teil der kommerziell bezogenen Tiere wahr-
scheinlich.
Bei der Uberprufung der Proben aus dem EURL wurden keine anderen Viren nachge-
wiesen. Dies spricht dafur, dass die multiplex-PCR die gewahlten Erreger spezifisch und
ohne hohe Hintergrundaktivitat detektiert. Die Tatsache, dass alle vorberichtlich negati-
ven Tiere bzw. Proben als negativ erkannt wurden, lasst den Schluss zu, dass auf dieser
limitierten Probengrundlage eine gute diagnostische Spezifitat erzielt wurde.
Im weiteren Verlauf der Validierung konnten 33 klinische Proben getestet werden. Diese
stammten aus der Routinediagnostik der Außenstelle fur Epidemiologie der TiHo Hanno-
ver in Bakum und waren im Vorfeld auf verschiedene porzine Viren und andere Pathoge-
ne untersucht worden. Ergebnisse dieser Untersuchungen lagen nur fur einen Teil dieser
Proben vor und schlossen i. d. R. nicht alle Viren ein, die mit dem vorliegenden multiplex-
Ansatz detektiert wurden. Die Bewertung der Ergebnisse gestaltete sich somit sehr schwie-
rig und konnte nur anhand der zur Verfugung stehenden Ergebnisse aus Bakum verglei-
chend erfolgen. Die Untersuchung zufallig ausgewahlter Proben zeigte, dass der Nach-
weis der anzeigepflichtigen Tierseuchen ASPV und SHV-1 negativ verlief. Auch der Test
auf pestivirale Erkrankungen verlief negativ. Da im fraglichen Zeitraum weder KSPV-,
SHV-1- noch ASPV-Infektionen in der deutschen Hausschweinepopulation nachgewiesen
wurden (Handistatus der OIE, http://www.oie.int/hs2/report.asp?lang=en, Stand
am 15.12.2005), war von diesen Ergebnissen auszugehen. PCV-2 konnte in 21 der 33 Pro-
ben detektiert werden. Nicht selten lagen Mehrfachinfektionen vor. Fur 16 dieser Proben
lagen auch Ergebnisse aus der Außenstelle fur Epidemiologie vor, die fur einen Vergleich
herangezogen werden konnten. Es zeigte sich, dass es zu Abweichungen in den Ergebnis-

5.9. Validierung 127
sen kam, wobei diese v. a. fur von der Außenstelle fur Epidemiologie fraglich beurteilte
Proben deutlich wurden.
Da die Kriterien, die zur Beurteilung der Ergebnisse als fraglich fuhrten, nicht bekannt
waren, ist eine Bewertung dieser Abweichungen nicht moglich. Funf der fraglichen Proben
wurden mit der vorliegenden PCR positiv getestet, zwei negativ. Eine Probe, die von der
Außenstelle fur Epidemiologie negativ getestet wurde, wurde in der vorliegenden PCR
wiederholt positiv detektiert. Generell ist festzuhalten, dass PCV-2 in der Schweinepopu-
lation weit verbreitet ist und v. a. in Kombination mit anderen Erregern und bei hoher
Viruslast bei der Entstehung von Krankheiten beteiligt sein kann (Allan et al., 2000;
Krakowka et al., 2001; Choi und Chae, 2000; Kim und Chae, 2003). Der Nachweis
allein musste also nicht mit einem Krankheitsgeschehen vergesellschaftet sein.
Da keine PPV-Ergebnisse zum Vergleich vorlagen, waren die positiven Befunde fur zwei
der 33 Proben nicht zu verifizieren. Die PPV-positiven Befunde traten in Kombination
mit PCV-2 auf. Wie oben bereits erwahnt, sind Mehrfachinfektionen in Zusammenhang
mit PCV-2 haufig. PPV ist dabei ein oftmals mit PCV-2 assoziierter Erreger, der die
Infektion mit PCV-2 verstarkt (Ellis et al., 1999; Allan et al., 1999; Kennedy et al.,
2000).
Neun Proben wurden positiv auf Influenza-A detektiert, wobei sechs Mehrfachinfektio-
nen vorlagen. Nur drei dieser Ergebnisse konnten vergleichend ausgewertet werden. Zwei
Mischinfektionen mit PCV-2 wurden in beiden Nachweissystemen detektiert, wobei mit
der vorliegenden multiplex-PCR fur eine Probe auch ein positives Signal fur PRRSV-NA
detektiert wurde. Eine Probe, die vorberichtlich Influenza-A-Virus positiv war, wurde ne-
gativ getestet. Auf Grundlage der vorliegenden Informationen zu dieser Probe und den
verwendeten Nachweistechniken konnte die Ursache dieser Abweichung nicht geklart wer-
den.
In drei (PRRSV-EU) bzw. vier (PRRSV-NA) Proben wurde PRRSV detektiert. Zwei
PRRSV-EU-Befunde konnten mit Ergebnissen aus Bakum verglichen werden und erwie-
sen sich als identisch. Die Ergebnisse des PRRSV-NA-spezifischen Nachweises wichen fur
die einzige vergleichend auswertbare Probe voneinander ab, wobei die fragliche Probe
vorberichtlich positiv war, jedoch in der vorliegenden PCR negativ getestet wurde. Diese
Probe zeigte in der vorliegenden PCR ein positives Ergebnis fur Influenza-A.

128 5. Diskussion
5.10 Schlussfolgerung und Ausblick
Die hier entwickelten real-time PCR-Protokolle wurden nach Maßgabe der OIE Richtlinien
zur Validierung und Qualitatskontrolle zur Diagnose infektioser Erkrankungen eingesetz-
ter PCR-Methoden etabliert und teilweise validiert. Die erste Stufe des Validierungssche-
mas, die Uberprufung der Umsetzbarkeit, wurde erfolgreich abgeschlossen und die PCRs
daraufhin optimiert und standardisiert. Wahrend die Untersuchung der analytischen Sen-
sitivitat und der Reproduzierbarkeit erfolgreich abgeschlossen werden konnten, waren die
Moglichkeiten der Spezifitatsprufung auf der Grundlage des limitierten Probenmateri-
als nur teilweise moglich. Die zweite Stufe der Validierung wurde somit in weiten Teilen
abgeschlossen, bedarf jedoch einer Fortfuhrung unter Praxisbedingungen an klinischen
Proben.
Die dritte Stufe der Validierung sieht die Bestimmung der diagnostischen Sensitivitat
(D-SN) und Spezifitat (D-SP) sowie der Wiederholbarkeit und Reproduzierbarkeit des
Testsystems vor. Ein Vergleich mit etablierten”Goldstandards“ sollte im Rahmen dieser
Stufe ebenfalls erfolgen. Die Bestimmung der D-SN und D-SP sollte anhand einer großen
Probenanzahl bekannten Hintergrunds vorgenommen werden, die i. d. R. nicht verfugbar
ist. Im Rahmen dieser Arbeit konnte diese Stufe der Testvalidierung daher nicht hin-
reichend durchgefuhrt werden. Die OIE Richtlinien zur Validierung geben an, dass diese
Stufe in vielen Fallen im praktischen Einsatz parallel zum Goldstandard erfolgen kann. Es
sollte daher eine weitere Testung und Validierung des PCR-Protokolls in der Routinedia-
gnostik anhand des verfugbaren klinischen Materials durchgefuhrt werden. Der parallele
Einsatz des Goldstandards ist nicht fur alle Erreger durchfuhrbar, konnte jedoch in den
meisten Fallen erreicht werden. Der Vergleich mit dem Goldstandard, der Virusisolierung
in Zellkultur, wurde fur die meisten Protokolle bei der Prufung der analytischen Sensi-
tivitat durchgefuhrt und zeigte eine den Goldstandard ubertreffende bzw. in Einzelfallen
gleichwertige Sensitivitat. Im Rahmen dieser Arbeit wurden soweit wie moglich sowohl
klinische Proben als auch unterschiedliche Isolate der entsprechenden Viren getestet, die
die Detektionssicherheit der gewahlten Genomfragmente trotz vorhandener Limitierungen
absichern sollten. Die unter diesen Umstanden moglichen Testschritte wurden erfolgreich
durchgefuhrt und ergaben zufriedenstellende Ergebnisse. Um eine weitere Validierung
entsprechend der OIE-Richtlinien zu gewahrleisten, sollten weitere Untersuchungen mit

5.10. Schlussfolgerung und Ausblick 129
klinischem Material folgen.
Trotz der Notwendigkeit der weiteren Validierung kann der etablierte PCR-Ansatz bereits
in der vorliegenden Form einen wichtigen Beitrag zur Differentialdiagnose der KSP leis-
ten. Neben der hohen Sensitivitat des Testsystems und der schnellen Verfugbarkeit einer
verlasslichen Diagnose bietet dieser Ansatz auch den erheblichen Vorteil einer moglichen
Erweiterung um zusatzliche, z. B. bakterielle Erreger. Da alle PCR-Protokolle unter glei-
chen Bedingungen durchgefuhrt werden, ist es moglich, sie beliebig zu erweitern. Außer-
dem besteht die Moglichkeit, diesen Ansatz auf Krankheitskomplexe anderer Tierspezies
zu ubertragen. Die vorliegende PCR ist aufgrund ihrer einfachen Handhabung und Um-
setzbarkeit fur die Routinediagnostik besser geeignet als Systeme, die auf der Detektion
unter Zuhilfenahme unterschiedlichster Methoden beruhen. Ein Nachteil des vorliegenden
Konzeptes ist die Tatsache, dass sich aufgrund des”Panpesti“-Nachweises eine weitere
Differenzierung im Falle einer positiven Reaktion anschließen muss. Da es die rechtlichen
Grundlagen nicht gestatten (Council Directive 2001/89/EC), die Diagnose der an-
zeigepflichtigen Seuchen auf einen einzelnen positiven Befund zu grunden, ist jedoch eine
weitere Abklarung ohnehin notwendig. Die vorliegende Methode ist als ein Screeningtest
anzusehen. Sollten sich Verdachtsmomente ergeben, die auf eine anzeigepflichtige Seuche,
wie KSP, AK oder ASP hindeuten, mussen weitere Tests eingeleitet werden, die den Ver-
dacht bestatigen oder entkraften.


6. Zusammenfassung
Andreas Gavrilenko
Entwicklung einer real-time multiplex multitube RT-PCR zur Differentialdia-
gnostik der klassischen Schweinepest
Die Klassische Schweinepest (KSP) ist eine wirtschaftlich bedeutende, anzeigepflichti-
ge Tierseuche, deren Ausbruche weltweit enorme wirtschaftliche Verluste verursachen.
Die Erkrankung wird durch das Virus der Klassischen Schweinepest (KSPV), ein RNA-
Virus, das zum Genus Pestivirus der Virusfamilie Flaviviridae gehort, verursacht. Da das
klinische Bild einer KSPV-Infektion sehr variabel und haufig unspezifisch ist, kommen
viele andere Erkrankungen viraler, bakterieller und nicht-infektioser Genese differenti-
aldiagnostisch in Frage. Zu den viralen Differentialdiagnosen gehoren die Afrikanische
Schweinepest (ASP), porzine Circovirusinfektionen (PCV-2), porzine Parvovirusinfektio-
nen (PPV), porzine Influenza, Infektionen mit dem porzinen respiratorischen und repro-
duktiven Syndrom Virus (PRRSV) und die Aujeszkysche Krankheit (SHV-1 Infektionen).
Eine klinische Diagnose ist aufgrund der Vielzahl moglicher Differentialdiagnosen unmoglich
und rechtlich als alleiniges Entscheidungskriterium der Seuchenfeststellung nicht zulassig.
Sowohl fur die routinemaßige Uberwachung der Schweinepopulation als auch zur schnellen
und verlasslichen Detektion und Kontrolle eines KSP-Ausbruchs ist daher eine moderne
und effiziente Labordiagnostik von herausragender Bedeutung.
Zu den schnellsten und empfindlichsten Methoden des Virusgenomnachweises gehort die
Polymerasekettenreaktion (PCR), die es in so genannten multiplex Ansatzen zudem ge-
stattet, verschiedene Erreger gleichzeitig, und bei Nutzung fluoreszierend markierter Son-
131

132 6. Zusammenfassung
den, in Echtzeit (”real-time“) zu detektieren.
Im Rahmen der vorliegenden Arbeit wurde auf der Basis einer multiplex real-time PCR
eine moderne und schnelle Nachweismethode differentialdiagnostisch relevanter viraler
Erreger zu Pestivirusinfektionen entwickelt. Aufgrund der Anzahl der zu detektierenden
Erregergenome erfolgte die Umsetzung in einem so genannten multitube Ansatz. Die Kom-
bination der Erreger in den Ansatzen erfolgte unter Berucksichtigung der erregerspezifi-
schen Eigenschaften und der Relevanz des Erregers.
Fur die PCR Ansatze wurden folgende Kombinationen gewahlt: ASPV und SHV-1, PPV
und PCV-2, porzine Influenza und Pestiviren sowie die beiden Stamme des PRRSV. Als
interne Kontrolle wurde das housekeeping gene beta-Aktin gewahlt. Nach der Entwick-
lung spezifischer Primerpaare und TaqMan-Sonden, wurden die entsprechenden real-time
PCRs zunachst einzeln und dann in den vorgesehenen Kombination getestet und opti-
miert. Eine bereits publizierte real-time PCR zum Nachweis und zur Diskriminierung
amerikanischer und europaischer PRRSV-Stammen (Kleiboeker et al., 2005) wurde
ubernommen und in das Gesamtkonzept integriert. Alle PCR-Reaktionen wurden mit ei-
nem einheitlichen Thermoprofil durchgefuhrt. Die Validierung der neu etablierten PCRs
erfolgte nach OIE Kriterien.
Die analytische Sensitivitat der PCRs wurde in infektiosen Einheiten bzw. Genomaqui-
valenten bestimmt und erwies sich im Vergleich zu dem Virusnachweis in Zellkultur als
uberlegen bzw. in Einzelfallen als gleichwertig. Die Spezifitat der Primer und TaqMan-
Sonden wurde mittels einer umfangreichen BLAST-Suche sichergestellt. Klinische Proben,
welche die genetische Variabiltat der Erreger und die epidemiologische Situation wider-
spiegelten, wurden in die Testung der PCRs einbezogen.
Der entwickelte PCR-Ansatz bietet die Moglichkeit einer schnellen und sicheren labordia-
gnostischen Differentialdiagnose der KSP. In der gewahlten Konzeption konnte er zudem
ein wichtiges Bindeglied darstellen, wenn es um den Ausschluss der KSP ohne konkre-
ten Seuchenverdacht geht. Das vorliegende Konzept unter Verwendung eines einheitlichen
Thermoprofils bietet zudem den Vorteil, dass das Testsystem beliebig um weitere relevante
Erreger erweitert werden kann, ohne die restlichen Ansatze zu beeinflussen.

7. Summary
Andreas Gavrilenko
Development of realtime multiplex multitube RT-PCR for differential diagno-
sis of classical swine fever
Classical swine fever (CSF) is an economically important viral disease of domestic pigs
worldwide. It is notifiable to the OIE. The causative agent, classical swine fever virus
(CSFV), is an enveloped RNA-virus which belongs to the family Flaviviridae, genus Pes-
tivirus.
The clinical picture is highly variable and often unspecific. For this reason, many other
viral, bacterial and non-infectious diseases have to be considered as differential diagnoses.
The diseases caused by viruses are African swine fever (ASF), porcine circovirus (PCV-2)
infections, porcine parvovirus (PPV) infections, porcine influenza, porcine reproductive
and respiratory syndrome (PRRS), and Aujeszky´s disease (caused by suid herpesvirus
type one, SHV-1). Diagnosis based on clinical signs alone is neither possible nor legitima-
te. As a consequence, a fast and reliable laboratory diagnosis is of utmost importance,
both for surveillance and in case of an CSF outbreak. Polymerase chain reaction (PCR)
is one of the fastest and most sensitive methods for the detection of viral genomes. The
use of fluorescent labelled probes enables detection of multiple targets using multiplex
real-time PCR.
In this project a rapid and reliable test based on real-time multiplex reverse transcription
(RT) PCR for differential diagnosis of CSFV was developed. Due to limitations of simul-
taneously detectable fluorescent dyes, the assay was designed as multitube test. The com-
bination of pathogens within the assay was designed under consideration of pathogens’-
133

134 7. Summary
specific characteristics and their relevance.
The following combinations were selected: ASFV and SHV-1, PPV and PCV-2, porcine
Influenza and Pestiviruses, as well as PRRSV strains. Beta-actin housekeeping gene was
chosen as internal control. After designing specific primers and TaqMan probes, the real-
time RT-PCRs were tested individually and subsequently optimized in the determined
combinations. An already published real-time PCR for the detection and discrimination
of American and European PRRSV strains (Kleiboeker et al., 2005) was integrated
in the assay. For all PCR reactions a uniform thermal profile was developed. The assay
was validated according to OIE validation criteria. The analytical sensitivity of the PCRs
was determined and expressed as infectious units and/or genome equivalents. It proved
to be superior or at least equivalent (for single PCRs) in comparison to virus isolation
in cell culture. Specificity of primers and probes was assured by means of an extensive
BLAST search. Clinical samples reflecting the genetic variability of the pathogens and
the epidemiological situation, were included in the evaluation of the PCRs.
The developed multiplex PCR provides a fast and reliable tool for differential diagnosis
of CSF. The selected concept could be an important link in terms of exclusion of CSF
without concrete epidemic suspicion. Furthermore, the uniform thermal profile offers the
opportunity to include additional relevant PCR targets without affecting the other PCRs.

Literaturverzeichnis
Aguero, M., J. Fernandez, L. Romero, M. C. Sanchez, M. Arias und J. M.
Sanchez-Vizcaıno (2003): Highly sensitive PCR assay for routine diagnosis of African
swine fever virus in clinical samples. J. Clin. Microbiol. 41, Nr. 9, 4431–4434
Aguero, M., J. Fernandez, L. J. Romero, M. J. Zamora, C. Sanchez, S. Belak,
M. Arias und J. M. Sanchez-Vizcaıno (2004): A highly sensitive and specific gel-
based multiplex RT-PCR assay for the simultaneous and differential diagnosis of African
swine fever and Classical swine fever in clinical samples. Vet. Res. 35, Nr. 5, 551–563
Allan, G., B. Meehan, D. Todd, S. Kennedy, F. McNeilly, J. Ellis, E. G.
Clark, J. Harding, E. Espuna, A. Botner und C. Charreyre (1998): Novel
porcine circoviruses from pigs with wasting disease syndromes. Vet. Rec. 142, Nr. 17,
467–468
Allan, G. M., F. McNeilly, J. P. Cassidy, G. A. Reilly, B. Adair, W. A.
Ellis und M. S. McNulty (1995): Pathogenesis of porcine circovirus; experimental
infections of colostrum deprived piglets and examination of pig foetal material. Vet.
Microbiol. 44, Nr. 1, 49–64
Allan, G. M., S. Kennedy, F. McNeilly, J. C. Foster, J. A. Ellis, S. J.
Krakowka, B. M. Meehan und B. M. Adair (1999): Experimental reproducti-
on of severe wasting disease by co-infection of pigs with porcine circovirus and porcine
parvovirus. J. Comp Pathol. 121, Nr. 1, 1–11
Allan, G. M., F. McNeilly, S. Kennedy, B. Meehan, J. Ellis und S. Krakowka
(2000): Immunostimulation, PCV-2 and PMWS. Vet. Rec. 147, Nr. 6, 170–171
135

136 Literaturverzeichnis
Altschul, S. F. und E. V. Koonin (1998): Iterated profile searches with PSI-BLAST–a
tool for discovery in protein databases. Trends Biochem. Sci 23, Nr. 11, 444–447
Andreyev, V. G., R. D. Wesley, W. L. Mengeling, A. C. Vorwald und K. M.
Lager (1997): Genetic variation and phylogenetic relationships of 22 porcine reproduc-
tive and respiratory syndrome virus (PRRSV) field strains based on sequence analysis
of open reading frame 5. Arch Virol 142, Nr. 5, 993–1001
Anonym (2004a): Classical Swine Fever. In: O. B. S. Commission (Hrsg.) Manual of
Diagnostic Tests and Vaccines for Terrestrial Animals. 5 Aufl., Office International des
Epizooties, Bd. 1, book chapter 2.1.13., S. 244–257
Anonym (2004b): Principles of validation of diagnostic assays for infectious diseases. In:
O. B. S. Commission (Hrsg.) Manual of Diagnostic Tests and Vaccines for Terrestrial
Animals. 5 Aufl., Office International des Epizooties, book chapter I .1.13.
Anonym (2004c): Validation and quality control of polymerase chain reaction methods
used for the diagnosis of infectious diseases. In: O. B. S. Commission (Hrsg.) Manual
of Diagnostic Tests and Vaccines for Terrestrial Animals. 5 Aufl., Office International
des Epizooties, book chapter I .1.14.
Anonym (2005a): Manual of Diagnostic Tests and Vaccines for Terrestrial Animals.
Office International des Epizooties, Paris, 5th Aufl.
Anonym (2005b): Quantifizierungstrategien mit dem LightCycler PCR Analysesystem.
Pamphlet
Anonym (2005c): Technical Disease Cards. African swine fever. In: Manual of Diagnostic
Tests and Vaccines for Terrestrial Animals. Office International des Epizooties, Bd. 1
Arnauld, C., O. Legeay, Y. Laurian, R. Thiery, M. Denis, P. Blanchard und
A. Jestin (1998): Development of a PCR-based method coupled with a microplate
colorimetric assay for the detection of Porcine Parvovirus and application to diagnosis
in piglet tissues and human plasma. Mol. Cell Probes 12, Nr. 6, 407–416
Aujeszky, A. (1902): Ueber eine neue Infektions Krankheit bei Haustieren. Zentralblatt
fur Bakteriologie, Parasitenkunde, Infektionskrankheiten und Hygiene, Erste Abteilung
Originale 32, 353–357

Literaturverzeichnis 137
Bachmann, P. A., B. E. Sheffy und J. T. Vauhan (1975): Experimental in utero
infection of fetal pigs with a porcine parvovirus. Infect Immun. 12, Nr. 3, 455–460
Baldino, F. J., M. F. Chesselet und M. E. Lewis (1989): High-resolution in situ
hybridization histochemistry. Methods Enzymol. 168, 761–777
Baltimore, D. (1970): Viral RNA-dependent DNA polymerase in virions of RNA tumor
viruses. Nature 226, 1209–1211
Bartenschlager, R. und V. Lohmann (2000): Replication of hepatitis C virus. J
Gen Virol 81, Nr. Pt 7, 1631–1648
Basto, A. P., R. S. Portugal, R. J. Nix, C. Cartaxeiro, F. Boinas, L. K.
Dixon, A. Leitao und C. Martins (2005): Development of a nested PCR and its
internal control for the detection of African swine fever virus (ASFV) in Ornithodoros
erraticus. Arch Virol
Bastos, A. D., M. L. Penrith, C. Cruciere, J. L. Edrich, G. Hutchings,
F. Roger, E. Couacy-Hymann und R. Thomson (2003): Genotyping field strains
of African swine fever virus by partial p72 gene characterisation. Arch Virol 148, Nr. 4,
693–706
Becher, P., M. Konig, D. J. Paton und H. J. Thiel (1995): Further characterization
of border disease virus isolates: evidence for the presence of more than three species
within the genus pestivirus. Virology 209, Nr. 1, 200–206
Becher, P., M. Orlich und H. J. Thiel (1998): Complete genomic sequence of border
disease virus, a pestivirus from sheep. J. Virol. 72, Nr. 6, 5165–5173
Bej, A. K., M. H. Mahbubani, R. Miller, J. L. DiCesare, L. Haff und R. M.
Atlas (1990): Multiplex PCR amplification and immobilized capture probes for detec-
tion of bacterial pathogens and indicators in water. Mol Cell Probes 4, Nr. 5, 353–365
Belak, S. (2005): The molecular diagnosis of porcine viral diseases: a review. Acta Vet.
Hung. 53, Nr. 1, 113–124
Belak, S. und P. Thoren (2001): Molecular diagnosis of animal diseases: some expe-
riences over the past decade. Expert. Rev Mol Diagn 1, Nr. 4, 434–443

138 Literaturverzeichnis
Belak, S., E. Rivera, A. Ballagi-Pordany, W. Hanzhong, F. Widen und
T. Soos (1998): Detection of challenge virus in fetal tissues by nested PCR as a test
of the potency of a porcine parvovirus vaccine. Vet. Res. Commun. 22, Nr. 2, 139–146
Ben-Porat, T., R. A. Veach und S. Ihara (1983): Localization of the regions of
homology between the genomes of herpes simplex virus, type 1, and pseudorabies virus.
Virology 127, Nr. 1, 194–204
Benfield, D. A., E. Nelson, J. E. Collins, L. Harris, S. M. Goyal, D. Robison,
W. T. Christianson, R. B. Morrison, D. Gorcyca und D. Chladek (1992):
Characterization of swine infertility and respiratory syndrome (SIRS) virus (isolate
ATCC VR-2332). J Vet Diagn Invest 4, Nr. 2, 127–133
Benfield, D. A., J. E. Collins, S. A. Dee, P. G. Hallbur, H. S. Joo, K. M.
Lager, W. L. Mengeling, M. P. Murtaugh, K. D. Rossow, G. W. Stevenson
und J. J. Zimmerman (1999): Porcine reproductive and respiratory syndrome. In:
B. E. Straw, W. L. Mengeling, S. D. D’Allaire und D. J. J. Taylor (Hrsg.)
Diseases of Swine. 8 Aufl., Iowa State University Press, S. 201–232
Bensaude, E., J. L. Turner, P. R. Wakeley, D. A. Sweetman, C. Pardieu,
T. W. Drew, T. Wileman und P. P. Powell (2004): Classical swine fever virus
induces proinflammatory cytokines and tissue factor expression and inhibits apopto-
sis and interferon synthesis during the establishment of long-term infection of porcine
vascular endothelial cells. J. Gen. Virol. 85, Nr. Pt 4, 1029–1037
Bente, D. A. (2003): Evaluierung konventioneller und real-time RT-PCR-Protokolle fur
die spezifische Diagnose des Virus der klassischen Schweinepest. Dissertation, Institut
fur Virologie, Tierarztliche Hochschule Hannover
Bergeron, J., J. Menezes und P. Tijssen (1993): Genomic Organization and Map-
ping of Transcription and Translation Products of the Nadl-2 Strain of Porcine Parvo-
virus. Virology 197, Nr. 1, 86–98
Bergeron, J., B. Hebert und P. Tijssen (1996): Genome organization of the Kresse
strain of porcine parvovirus: Identification of the allotropic determinant and comparison
with those of NADL-2 and field isolates. Journal of Virology 70, Nr. 4, 2508–2515

Literaturverzeichnis 139
Berns, K. I. (1990): Parvovirus replication. Microbiol Rev 54, Nr. 3, 316–329
Bikour, M. H., E. Cornaglia, J. M. Weber und Y. Elazhary (1994): Antigenic
characterization of an H3N2 swine influenza virus isolated from pigs with proliferative
and necrotizing pneumonia in Quebec. Can J Vet Res 58, Nr. 4, 287–290
Bjorklund, H., P. Lowings, T. Stadejek, S. Vilcek, I. Greiser-Wilke,
D. Paton und S. Belak (1999): Phylogenetic comparison and molecular epidemiology
of classical swine fever virus. Virus Genes 19, Nr. 3, 189–195
Bouma, A., J. A. Stegeman, B. Engel, E. P. de Kluijver, A. R. Elbers und
M. C. de Jong (2001): Evaluation of diagnostic tests for the detection of classical
swine fever in the field without a gold standard. J. Vet. Diagn. Invest 13, Nr. 5,
383–388
Brisson, M., S. Hall, R. K. Hamby, R. Park und H. K. Srere (2004): Optimization
of Single and Multiplex Real-Time PCR. In: S. A. Bustin (Hrsg.) A-Z of Quantitative
PCR. International University Line, book chapter 18, S. 619–642
Brown, H. S., D. R. Bishop und C. R. West (1986): A methodology for assessing
mutagenic hazards of chemicals. Toxicol. Ind. Health 2, Nr. 3, 163–182
Brown, I. H., R. J. Manvell, D. J. Alexander, P. Chakraverty, V. S. Hinshaw
und R. G. Webster (1993): Swine influenza outbreaks in England due to a new H1N1
virus. Vet Rec 132, Nr. 18, 461–462
Brunborg, I. M., T. Moldal und C. M. Jonassen (2004): Quantitation of porcine
circovirus type 2 isolated from serum/plasma and tissue samples of healthy pigs and
pigs with postweaning multisystemic wasting syndrome using a TaqMan-based real-time
PCR. J. Virol. Methods 122, Nr. 2, 171–178
Buhk, H. J., I. Tischer und M. A. Koch (1985): Cloning and Sequencing of the Porci-
ne Circovirus (Pcv) Genome. Zentralblatt fur Bakteriologie Mikrobiologie und Hygiene
Series A-Medical Microbiology Infectious Diseases Virology Parasitology 260, Nr. 4,
465–465

140 Literaturverzeichnis
Bustin, S. A. (2000): Absolute quantification of mRNA using real-time reverse trans-
cription polymerase chain reaction assays. J. Mol. Endocrinol. 25, Nr. 2, 169–193
Bustin, S. A. (2002): Quantification of mRNA using real-time reverse transcription PCR
(RT-PCR): trends and problems. J. Mol. Endocrinol. 29, Nr. 1, 23–39
Bustin, S. A. und I. A. McKay (1999): The product of the primary response gene
BRF1 inhibits the interaction between 14-3-3 proteins and cRaf-1 in the yeast trihybrid
system. DNA Cell Biol 18, Nr. 8, 653–661
Bustin, S. A. und T. Nolan (2004a): Chemistries. In: S. A. Bustin (Hrsg.) A-Z of
Quantitative PCR. International University Line, book chapter 6, S. 217–278
Bustin, S. A. und T. Nolan (2004b): Data analysis and Interpretation. In: S. A.
Bustin (Hrsg.) A-Z of Quantitative PCR. International University Line, book chap-
ter 11, S. 439–492
Bustin, S. A. und T. Nolan (2004c): Pitfalls of quantitative real-time reverse-
transcription polymerase chain reaction. J. Biomol. Tech. 15, Nr. 3, 155–166
Bustin, S. A. und T. Nolan (2004d): Primers and Probes. In: S. A. Bustin (Hrsg.)
A-Z of Quantitative PCR. International University Line, book chapter 7, S. 281–328
Calsamiglia, M., J. Segales, J. Quintana, C. Rosell und M. Domingo (2002):
Detection of porcine circovirus types 1 and 2 in serum and tissue samples of pigs with
and without postweaning multisystemic wasting syndrome. J Clin Microbiol 40, Nr. 5,
1848–1850
Candotti, D., J. Temple, S. Owusu-Ofori und J. P. Allain (2004): Multiplex real-
time quantitative RT-PCR assay for hepatitis B virus, hepatitis C virus, and human
immunodeficiency virus type 1. J Virol Methods 118, Nr. 1, 39–47
Cao, S., H. Chen, J. Zhao, J. Lu, S. Xiao, M. Jin, A. Guo, B. Wu und Q. He
(2005a): Detection of porcine circovirus type 2, porcine parvovirus and porcine pseu-
dorabies virus from pigs with postweaning multisystemic wasting syndrome by multiplex
PCR. Vet Res Commun 29, Nr. 3, 263–269

Literaturverzeichnis 141
Cao, S., C. Y. Zhang und G. H. Yi (2005b): [Recombinant analysis of classical swine
fever virus]. Yi. Chuan Xue. Bao. 32, Nr. 1, 52–56
Cartwright, S. F. und R. A. Huck (1967): Virus Isolated in Association with Herd
Infertility, Abortions and Stillbirths in Pigs. Vet Rec 81, 196–197
Cartwright, S. F., M. Lucas und R. A. Huck (1969): A small haemagglutinating
porcine DNA virus. I. Isolation and properties. J Comp Pathol 79, Nr. 3, 371–377
Cavanagh, D. (1997): Nidovirales: a new order comprising Coronaviridae and Arterivi-
ridae. Arch Virol 142, Nr. 3, 629–633
Cay, B., G. Chappuis, C. Coulibaly, Z. Dinter, S. Edwards, I. Greiser-Wilke,
M. Gunn, P. Have, G. Hess, N. Juntti und . (1989): Comparative analysis of
monoclonal antibodies against pestiviruses: report of an international workshop. Vet
Microbiol 20, Nr. 2, 123–129
Chae, C. (2004): Postweaning multisystemic wasting syndrome: a review of aetiology,
diagnosis and pathology. Vet. J. 168, Nr. 1, 41–49
Chen, J. M., Z. L. Wang, Z. C. Pang, C. Y. Sun, Y. X. Sun, C. P. Song und
Y. Yi (2005): [Design and rapid evaluation of a TaqMan assay for the detection of
influenza A viruses]. Zhonghua Shi Yan. He. Lin. Chuang. Bing. Du Xue. Za Zhi. 19,
Nr. 1, 80–83
Cheville, N. F. und W. L. Mengeling (1969): The pathogenesis of chronic hog
cholera (swine fever). Histologic, immunofluorescent, and electron microscopic studies.
Lab. Invest. 20, Nr. 3, 261–274
Choi, C. und C. Chae (1999): In-situ hybridization for the detection of porcine circovirus
in pigs with postweaning multisystemic wasting syndrome. J. Comp Pathol. 121, Nr. 3,
265–270
Choi, C. und C. Chae (2000): Distribution of porcine parvovirus in porcine circovirus
2-infected pigs with postweaning multisystemic wasting syndrome as shown by in-situ
hybridization. J. Comp Pathol. 123, Nr. 4, 302–305

142 Literaturverzeichnis
Choi, C. und C. Chae (2002): Localization of classical swine fever virus in male gonads
during subclinical infection. J. Gen. Virol. 83, Nr. Pt 11, 2717–2721
Choi, C., C. Chae und E. G. Clark (2000): Porcine postweaning multisystemic was-
ting syndrome in Korean pig: detection of porcine circovirus 2 infection by immunohi-
stochemistry and polymerase chain reaction. J. Vet. Diagn. Invest 12, Nr. 2, 151–153
Christopher-Hennings, J., E. A. Nelson, R. J. Hines, J. K. Nelson, S. L.
Swenson, J. J. Zimmerman, C. L. Chase, M. J. Yaeger und D. A. Benfield
(1995): Persistence of porcine reproductive and respiratory syndrome virus in serum
and semen of adult boars. J Vet Diagn Invest 7, Nr. 4, 456–464
Christopher-Hennings, J., E. A. Nelson, J. K. Nelson, K. D. Rossow, J. L.
Shivers, M. J. Yaeger, C. C. Chase, R. A. Garduno, J. E. Collins und D. A.
Benfield (1998): Identification of porcine reproductive and respiratory syndrome virus
in semen and tissues from vasectomized and nonvasectomized boars. Vet Pathol 35,
Nr. 4, 260–267
Christopher-Hennings, J., L. D. Holler, D. A. Benfield und E. A. Nelson
(2001): Detection and duration of porcine reproductive and respiratory syndrome vi-
rus in semen, serum, peripheral blood mononuclear cells, and tissues from Yorkshire,
Hampshire, and Landrace boars. J. Vet. Diagn. Invest 13, Nr. 2, 133–142
Chung, W. B., W. H. Chan, H. C. Chaung, Y. Lien, C. C. Wu und Y. L. Huang
(2005): Real-time PCR for quantitation of porcine reproductive and respiratory syn-
drome virus and porcine circovirus type 2 in naturally-infected and challenged pigs. J.
Virol. Methods 124, Nr. 1-2, 11–19
Collett, M. S., D. K. Anderson und E. Retzel (1988): Comparisons of the pesti-
virus bovine viral diarrhoea virus with members of the flaviviridae. J Gen Virol 69 (
Pt 10), 2637–2643
Collins, J. E., D. A. Benfield, W. T. Christianson, L. Harris, J. C. Hennings,
D. P. Shaw, S. M. Goyal, S. McCullough, R. B. Morrison, H. S. Joo und .
(1992): Isolation of swine infertility and respiratory syndrome virus (isolate ATCC VR-

Literaturverzeichnis 143
2332) in North America and experimental reproduction of the disease in gnotobiotic
pigs. J Vet Diagn Invest 4, Nr. 2, 117–126
Conzelmann, K. K., N. Visser, P. van Woensel und H. J. Thiel (1993): Molecular
characterization of porcine reproductive and respiratory syndrome virus, a member of
the arterivirus group. Virology 193, Nr. 1, 329–339
Cropper, M., H. W. Dunne, A. D. Leman, A. L. Starkey und D. C. Hoefling
(1976): Prevalence of antibodies to porcine enteroviruses and porcine parvovirus in body
fluids of fetal pigs from small vs large litters. J Am Vet Med Assoc 168, Nr. 3, 233–235
Cutler, R. S., T. W. Molitor, A. D. Leman und R. E. Werdin (1983): Farm
studies of porcine parvovirus infection. J Am Vet Med Assoc 182, Nr. 6, 592–594
Dahle, J. und B. Liess (1995): Comparative study with cloned classical swine fever virus
strains ALFORT and GLENTORF: Clinical, pathological, virological and serological
findings in weaner pigs. Wien. Tierarztl. Mschr. 82, 232–238
Darbyshire, J. H. (1960): A serological relationship between swine fever and mucosal
disease of cattle. Vet. Rec. 72, 331
Davison, A. J. und N. M. Wilkie (1983): Location and orientation of homologous
sequences in the genomes of five herpesviruses. J Gen Virol 64 (Pt 9), 1927–1942
de A. Diaz, J. I. Nunez, L. Ganges, M. Barreras, M. T. Frias und F. Sobrino
(1998): An RT-PCR assay for the specific detection of classical swine fever virus in
clinical samples. Vet. Res. 29, Nr. 5, 431–440
de Schweinitz, E. A. und M. Dorset (1904): New facts concerning the etiology of
hog cholera. US Department of Agriculture, 20th Ann. Rep. Bai S. 157–162
Dea, S., N. Sawyer, R. Alain und R. Athanassious (1995): Ultrastructural cha-
racteristics and morphogenesis of porcine reproductive and respiratory syndrome virus
propagated in the highly permissive MARC-145 cell clone. Adv Exp Med Biol 380,
95–98

144 Literaturverzeichnis
Dee, S. A., M. Torremorell, K. Rossow, C. Mahlum, S. Otake und K. Faaberg
(2001): Identification of genetically diverse sequences (ORF 5) of porcine reproductive
and respiratory syndrome virus in a swine herd. Can J Vet Res 65, Nr. 4, 254–260
Deng, R. und K. V. Brock (1992): Molecular cloning and nucleotide sequence of a
pestivirus genome, noncytopathic bovine viral diarrhea virus strain SD-1. Virology
191, Nr. 2, 867–869
Depner, K. R., V. Moennig und B. Liess (1997a): Aktuelle Fragen zur klassischen
Schweinepest beim Wildschwein. Amtstierarztlicher Dienst und Lebensmittelkontrolle
I/97, 44–47
Depner, K. R., V. Moennig und B. Liess (1997b): Epidemiologische Aspekte der
Infektionsbiologie der klassischen Schweinepest. Prakt. Tierarzt 78, Nr. coll.vet.XXVII,
63–67
Dheda, K., J. F. Huggett, S. A. Bustin, M. A. Johnson, G. Rook und A. Zumla
(2004): Validation of housekeeping genes for normalizing RNA expression in real-time
PCR. Biotechniques 37, Nr. 1, 112–119
Dixon, L. K., D. L. Rock und E. Vinuela (1995): African swine fever-like viruses. In:
F. A. Murphy, C. M. Fauquet, D. H. L. Bishop, S. A. Ghabrial, A. W. Jarvis,
G. P. Martinelli, M. A. Mayo und M. D. Summers (Hrsg.) Virus taxonomy. Sixth
report of the international committee on taxonomy of viruses. Springer Verlag, S. 92–94
Dixon, L. K., J. V. Costa, J. M. Escribano, D. L. Rock, E. Vinuela und P. J.
Wilkinson (2000): Virus taxonomy. Classification and nomenclature of viruses. Report
Donaldson-Wood, C. R., H. S. Joo und R. H. Johnson (1977): The effect on
reproductive performance of porcine parvovirus infection in a susceptible pig herd. Vet
Rec 100, Nr. 12, 237–239
Done, S. H., D. J. Paton und M. E. C. White (1996): Porcine reproductive and
respiratory syndrome (PRRS): A review, with emphasis on pathological, virological
and diagnostic aspects. British Veterinary Journal 152, 153–174

Literaturverzeichnis 145
Easterday, B. C. und V. S. Hinshaw (1992): Swine influenza. In: A. D. Leman,
B. E. Straw, W. L. Mengeling, S. D. D’Allaire und D. J. J. Taylor (Hrsg.)
Diseases of Swine. Iowa State Press, S. 349–357
Echeverria, M. G., M. R. Pecoraro, N. B. Pereyra, C. L. Pidone, C. M.
Galosi, M. E. Etcheverrigaray und E. O. Nosetto (2000): Rapid diagnosis
of pseudorabies virus infection in swine tissues using the polymerase chain reaction
(PCR). Rev Argent Microbiol 32, Nr. 3, 109–115
Edwards, M. C. und R. A. Gibbs (1994): Multiplex PCR: advantages, development,
and applications. PCR Methods Appl 3, Nr. 4, S65–S75
Edwards, S. (2000): Survival and inactivation of classical swine fever virus [In Process
Citation]. Vet. Microbiol. 73, Nr. 2-3, 175–181
Edwards, S., R. H. Newman und H. White (1991): The virulence of British isolates
of bovid herpesvirus 1 in relationship to viral genotype. British Veterinary Journal
147, 216–231
Egli, C., B. Thur, L. Z. Liu und M. A. Hofmann (2001): Quantitative TaqMan (R)
RT-PCR for the detection and differentiation of European and North American strains
of procine reproductive and respiratory syndrome virus. Journal of Virological Methods
98, Nr. 1, 63–75
Elbers, A. R., A. Bouma und J. A. Stegeman (2002): Quantitative assessment of
clinical signs for the detection of classical swine fever outbreaks during an epidemic.
Vet. Microbiol. 85, Nr. 4, 323–332
Elbers, A. R., J. H. Vos, A. Bouma und J. A. Stegeman (2004): Ability of ve-
terinary pathologists to diagnose classical swine fever from clinical signs and gross
pathological findings. Prev. Vet. Med. 66, Nr. 1-4, 239–246
Elbers, K., N. Tautz, P. Becher, D. Stoll, T. Rumenapf und H. J. Thiel
(1996): Processing in the pestivirus E2-NS2 region: identification of proteins p7 and
E2p7. J. Virol. 70, Nr. 6, 4131–4135

146 Literaturverzeichnis
Ellis, J., L. Hassard, E. Clark, J. Harding, G. Allan, P. Willson,
J. Strokappe, K. Martin, F. McNeilly, B. Meehan, D. Todd und D. Haines
(1998): Isolation of circovirus from lesions of pigs with postweaning multisystemic was-
ting syndrome. Can. Vet. J. 39, Nr. 1, 44–51
Ellis, J., S. Krakowka, M. Lairmore, D. Haines, A. Bratanich, E. Clark,
G. Allan, C. Konoby, L. Hassard, B. Meehan, K. Martin, J. Harding,
S. Kennedy und F. McNeilly (1999): Reproduction of lesions of postweaning mul-
tisystemic wasting syndrome in gnotobiotic piglets. J. Vet. Diagn. Invest 11, Nr. 1,
3–14
Ellis, J., E. Clark, D. Haines, K. West, S. Krakowka, S. Kennedy und G. M.
Allan (2004): Porcine circovirus-2 and concurrent infections in the field. Vet. Micro-
biol. 98, Nr. 2, 159–163
Ellis, J. A., A. Bratanich, E. G. Clark, G. Allan, B. Meehan, D. M. Haines,
J. Harding, K. H. West, S. Krakowka, C. Konoby, L. Hassard, K. Martin
und F. McNeilly (2000): Coinfection by porcine circoviruses and porcine parvovirus
in pigs with naturally acquired postweaning multisystemic wasting syndrome. J. Vet.
Diagn. Invest 12, Nr. 1, 21–27
Elnifro, E. M., A. M. Ashshi, R. J. Cooper und P. E. Klapper (2000): Multiplex
PCR: optimization and application in diagnostic virology. Clin Microbiol Rev 13, Nr. 4,
559–570
Enzmann, P. J. und H. Rehberg (1977): The structural components of hog cholera
virus. Z. Naturforsch. C 32, Nr. 5-6, 456–458
Forman, A. J., C. Lenghaus, G. G. Hogg und C. J. Hale (1977): Association of
a parvovirus with an outbreak of foetal death and mummification in pigs. Aust Vet J
53, Nr. 7, 326–329
Forsberg, R., T. Storgaard, H. S. Nielsen, M. B. Oleksiewicz, P. Cordioli,
G. Sala, J. Hein und A. Botner (2002): The genetic diversity of European type
PRRSV is similar to that of the North American type but is geographically skewed
within Europe. Virology 299, Nr. 1, 38–47

Literaturverzeichnis 147
Fouchier, R. A., T. M. Bestebroer, S. Herfst, K. L. van Der, G. F.
Rimmelzwaan und A. D. Osterhaus (2000): Detection of influenza A viruses from
different species by PCR amplification of conserved sequences in the matrix gene. J
Clin Microbiol 38, Nr. 11, 4096–4101
Fraser, G. und S. P. Ramachandran (1969): Studies on the virus of Aujeszky’s
disease. I. Pathogenicity for rats and mice. J Comp Pathol 79, Nr. 4, 435–444
Frey, H. R. (2003): Aujeszkysche Krankheit (AK). In: Virusinfektionen bei Haus- und
Nutztieren. 2nd Aufl., Schlutersche GmbH & Co. KG, book chapter 4.3, S. 74–76
Frias-Lepoureau, M. T. und I. Greiser-Wilke (2002): An Update on Classical
Swine Fever Virus Molecular Epidemiology. In: A. Morilla, K. J. Yoon und J. J.
Zimmerman (Hrsg.) Trends in Emerging Viral Infections of Swine. Iowa State Press,
book chapter 5.5, S. 165–172
Gaede, W., R. Reiting, H. Schirrmeier, K. R. Depner und M. Beer (2005):
[Detection and species-specific differentiation of pestiviruses using real-time RT-PCR].
Berl Munch. Tierarztl. Wochenschr. 118, Nr. 3-4, 113–120
Gall, A. L., O. Legeay, H. Bourhy, C. Arnauld, E. Albina und A. Jestin
(1998): Molecular variation in the nucleoprotein gene (ORF7) of the porcine reproduc-
tive and respiratory syndrome virus (PRRSV). Virus Res 54, Nr. 1, 9–21
Gao, S. J. und P. S. Moore (1996): Molecular approaches to the identification of
unculturable infectious agents. Emerg. Infect Dis 2, Nr. 3, 159–167
Gonzague, M., C. Plin, L. Bakkali-Kassimi, A. Boutrouille und C. Cruciere
(2002): Development of an internal control for the detection of the African swine fever
virus by PCR. Mol Cell Probes 16, Nr. 3, 237–242
Grace, M. B., C. B. McLeland, S. J. Gagliardi, J. M. Smith, I. W. E. Jackson
und W. F. Blakely (2003): Development and assessment of a quantitative reverse
transcription-PCR assay for simultaneous measurement of four amplicons. Clin. Chem.
49, Nr. 9, 1467–1475

148 Literaturverzeichnis
Gradil, C. M., M. J. Harding und K. Lewis (1994): Use of polymerase chain reaction
to detect porcine parvovirus associated with swine embryos. Am J Vet Res 55, Nr. 3,
344–347
Greiser-Wilke, I., K. Depner, J. Fritzemeier, L. Haas und V. Moennig (1998):
Application of a computer program for genetic typing of classical swine fever virus
isolates from Germany. J. Virol. Methods 75, Nr. 2, 141–150
Greiser-Wilke, I., B. Zimmermann und S. Dreier (2005): Genetic typing of CSF vi-
rus isolates using the CSF database of the European Community Reference Laboratory.
Unpublished Work
Gruber, A., K. R. Depner und B. Liess (1995): Experimental infection of weaner
pigs with a field isolate of Hog cholera/Classical Swine Fever Virus derived from a
recent outbreak in Lower Saxony. II. Pathological findings. Wien. Tierarztl. Mschr.
82, 179–184
Gustafson, D. P. (1970): Introduction to viral diseases of dairy cattle. J Dairy Sci 53,
Nr. 5, 611–614
Haas, L. (2003): Porzine Circovirus-Infektion (PCV-2-Infektion). In: Virusinfektionen
bei Haus- und Nutztieren. 2nd Aufl., Schlutersche GmbH & Co. KG, book chapter 4.12,
S. 92–94
Hamel, A. L., L. L. Lin und G. P. Nayar (1998): Nucleotide sequence of porcine
circovirus associated with postweaning multisystemic wasting syndrome in pigs. J.
Virol. 72, Nr. 6, 5262–5267
Handel, K., H. Kehler, K. Hills und J. Pasick (2004): Comparison of reverse
transcriptase-polymerase chain reaction, virus isolation, and immunoperoxidase assays
for detecting pigs infected with low, moderate, and high virulent strains of classical
swine fever virus. J. Vet. Diagn. Invest 16, Nr. 2, 132–138
Harder, T. C., E. grosse Beilage, T. Pabst und P. H. Hubert (2004): Porci-
ne Respiratory and Reproductive Syndrome Virus - Detection and Differentiation of

Literaturverzeichnis 149
Vaccine-like Viruses of European (Strain DV) and American Genotypes in Field Samp-
les. In: Proceedings of the 18th IPVS Congress. IPVS Veranstaltungs GmbH, Bd. 1, S.
74–74
Harding, J. C. S. (2004): The clinical expression and emergence of porcine circovirus
2. Veterinary Microbiology 98, Nr. 2, 131–135
Harding, J. C. S., E. G. Clark, J. H. Strokappe, P. I. Wilson und J. A.
Ellis (1998): Post-weaning multisystemic wasting syndrome: epidemiology and clinical
presentation. Swine Health Prod. 6, 249–254
Harms, P. A., S. D. Sorden, P. G. Halbur, S. R. Bolin, K. M. Lager,
I. Morozov und P. S. Paul (2001): Experimental reproduction of severe disease
in CD/CD pigs concurrently infected with type 2 porcine circovirus and porcine repro-
ductive and respiratory syndrome virus. Vet Pathol 38, Nr. 5, 528–539
Heid, C. A., J. Stevens, K. J. Livak und P. M. Williams (1996): Real time quan-
titative PCR. Genome Research 6, Nr. 10, 986–994
Higuchi, R., G. Dollinger, P. S. Walsh und R. Griffith (1992): Simultaneous
amplification and detection of specific DNA sequences. Biotechnology (N. Y) 10, Nr. 4,
413–417
Higuchi, R., C. Fockler, G. Dollinger und R. Watson (1993): Kinetic PCR
analysis: real-time monitoring of DNA amplification reactions. Biotechnology (N. Y)
11, Nr. 9, 1026–1030
Hindiyeh, M., V. Levy, R. Azar, N. Varsano, L. Regev, Y. Shalev, Z. Gross-
man und E. Mendelson (2005): Evaluation of a multiplex real-time reverse transcrip-
tase PCR assay for detection and differentiation of influenza viruses A and B during
the 2001-2002 influenza season in Israel. J Clin Microbiol 43, Nr. 2, 589–595
Hjertner, B., B. Meehan, J. McKillen, F. McNeilly und S. Belak (2005):
Adaptation of an Invader assay for the detection of African swine fever virus DNA. J.
Virol. Methods 124, Nr. 1-2, 1–10

150 Literaturverzeichnis
Hoffmann, B., M. Beer, C. Schelp, H. Schirrmeier und K. Depner (2005):
Validation of a real-time RT-PCR assay for sensitive and specific detection of classical
swine fever. J. Virol. Methods
Hofmann, M. A. (2003): Construction of an infectious chimeric classical swine fever
virus containing the 5 ’ UTR of bovine viral diarrhea virus, and its application as a
universal internal positive control in real-time RT-PCR. Journal of Virological Methods
114, Nr. 1, 77–90
Hofmann, M. A., K. Brechtbuhl und N. Stauber (1994): Rapid characterization
of new pestivirus strains by direct sequencing of PCR-amplified cDNA from the 5’
noncoding region. Arch. Virol. 139, Nr. 1-2, 217–229
Holland, P. M., R. D. Abramson, R. Watson und D. H. Gelfand (1991): Detec-
tion of Specific Polymerase Chain-Reaction Product by Utilizing the 5’-3’ Exonuclease
Activity of Thermus-Aquaticus Dna-Polymerase. Proceedings of the National Academy
of Sciences of the United States of America 88, Nr. 16, 7276–7280
Horzinek, M. (1973): The structure of togaviruses. Prog Med Virol. 16, 109–156
Horzinek, M., E. Reczko und K. Petzoldt (1967): On the morphology of hog cholera
virus. Arch. Gesamte Virusforsch. 21, Nr. 3, 475–478
Huang, C., J. J. Hung, C. Y. Wu und M. S. Chien (2004): Multiplex PCR for
rapid detection of pseudorabies virus, porcine parvovirus and porcine circoviruses. Vet.
Microbiol. 101, Nr. 3, 209–214
Huang, Y. J., M. S. Chien, C. Y. Wu und C. Huang (2005): Mapping of functional
regions conferring nuclear localization and RNA-binding activity of pseudorabies virus
early protein UL54. J Virol Methods 130, Nr. 1-2, 102–107
Hyndman, L., S. Vilcek, J. Conner und P. F. Nettleton (1998): A novel nested
reverse transcription PCR detects bovine viral diarrhoea virus in fluids from aborted
bovine fetuses. J Virol Methods 71, Nr. 1, 69–76
Ilyina, T. V. und E. V. Koonin (1992): Conserved sequence motifs in the initiator
proteins for rolling circle DNA replication encoded by diverse replicons from eubacteria,
eucaryotes and archaebacteria. Nucleic Acids Res 20, Nr. 13, 3279–3285

Literaturverzeichnis 151
Innis, M. A., K. B. Myambo, D. H. Gelfand und M. A. Brow (1988): DNA se-
quencing with Thermus aquaticus DNA polymerase and direct sequencing of polymerase
chain reaction-amplified DNA. Proc Natl Acad Sci U S A 85, Nr. 24, 9436–9440
Jeffreys, A. J., V. Wilson, R. Neumann und J. Keyte (1988): Amplification of
human minisatellites by the polymerase chain reaction: towards DNA fingerprinting of
single cells. Nucleic Acids Res 16, Nr. 23, 10953–10971
Jenkins, C. E. (1992): An enzyme-linked immunosorbent assay for detection of porcine
parvovirus in fetal tissues. J Virol Methods 39, Nr. 1-2, 179–184
Johnson, R. H. und D. F. Collings (1969): Experimental infection of piglets and
pregnant gilts with a parvovirus. Vet Rec 85, Nr. 16, 446–447
Joo, H. S. und R. H. Johnson (1977): Serological responses in pigs vaccinated with
inactivated porcine parvovirus. Aust Vet J 53, Nr. 11, 550–552
Joo, H. S., C. R. Donaldson-Wood, R. H. Johnson und R. S. Campbell (1977):
Pathogenesis of porcine parvovirus infection: pathology and immunofluorescence in the
foetus. J Comp Pathol 87, Nr. 3, 383–391
Kaden, V., H. Steyer, G. Strebelow, E. Lange, P. Hubert und P. Steinhagen
(1999): Detection of low-virulent classical swine fever virus in blood of experimentally
infected animals: comparison of different methods. Acta Virol. 43, Nr. 6, 373–380
Kaerber, G. (1931): Beitrag zur kollektiven Behandlung pharmakologischer Reihenver-
suche. Arch. Exp. Path. Pharm. 162, 480–483
Kainz, P. (2000): The PCR plateau phase - towards an understanding of its limitations.
Biochim. Biophys. Acta 1494, Nr. 1-2, 23–27
Kearns, A. M., A. J. Turner, G. J. Eltringham und R. Freeman (2002): Rapid
detection and quantification of CMV DNA in urine using LightCycler-based real-time
PCR. J Clin Virol 24, Nr. 1-2, 131–134
Kemp, D. J., D. B. Smith, S. J. Foote, N. Samaras und M. G. Peterson (1989):
Colorimetric detection of specific DNA segments amplified by polymerase chain reacti-
ons. Proc Natl Acad Sci U S A 86, Nr. 7, 2423–2427

152 Literaturverzeichnis
Kennedy, S., D. Moffett, F. McNeilly, B. Meehan, J. Ellis, S. Krakowka und
G. M. Allan (2000): Reproduction of lesions of postweaning multisystemic wasting
syndrome by infection of conventional pigs with porcine circovirus type 2 alone or in
combination with porcine parvovirus. J. Comp Pathol. 122, Nr. 1, 9–24
Kim, J. und C. Chae (2001): Differentiation of porcine circovirus 1 and 2 in formalin-
fixed, paraffin-wax-embedded tissues from pigs with postweaning multisystemic wasting
syndrome by in-situ hybridisation. Res. Vet. Sci. 70, Nr. 3, 265–269
Kim, J. und C. Chae (2002): Double in situ hybridization for simultaneous detection
and differentiation of porcine circovirus 1 and 2 in pigs with postweaning multisystemic
wasting syndrome. Vet. J. 164, Nr. 3, 247–253
Kim, J. und C. Chae (2003): Multiplex nested PCR compared with in situ hybridization
for the differentiation of porcine circoviruses and porcine parvovirus from pigs with
postweaning multisystemic wasting syndrome. Can. J. Vet. Res. 67, Nr. 2, 133–137
Kim, J. und C. Chae (2004): A comparison of virus isolation, polymerase chain reaction,
immunohistochemistry, and in situ hybridization for the detection of porcine circovirus
2 and porcine parvovirus in experimentally and naturally coinfected pigs. J. Vet. Diagn.
Invest 16, Nr. 1, 45–50
Kim, J., C. Choi, D. U. Han und C. Chae (2001): Simultaneous detection of porcine
circovirus type 2 and porcine parvovirus in pigs with PMWS by multiplex PCR. Vet
Rec 149, Nr. 10, 304–305
Kim, J., C. Choi und C. Chae (2003a): Pathogenesis of postweaning multisystemic
wasting syndrome reproduced by co-infection with Korean isolates of porcine circovirus
2 and porcine parvovirus. J. Comp Pathol. 128, Nr. 1, 52–59
Kim, J., D. U. Han, C. Choi und C. Chae (2003b): Simultaneous detection and
differentiation between porcine circovirus and porcine parvovirus in boar semen by
multiplex seminested polymerase chain reaction. Journal of Veterinary Medical Science
65, Nr. 6, 741–744
King, D. P., S. M. Reid, G. H. Hutchings, S. S. Grierson, P. J. Wilkinson,
L. K. Dixon, A. D. Bastos und T. W. Drew (2003): Development of a TaqMan

Literaturverzeichnis 153
PCR assay with internal amplification control for the detection of African swine fever
virus. J. Virol. Methods 107, Nr. 1, 53–61
Kleiboeker, S. B. (2002): Swine fever: classical swine fever and African swine fever.
Vet. Clin. North Am. Food Anim Pract. 18, Nr. 3, 431–451
Kleiboeker, S. B., G. F. Kutish, J. G. Neilan, Z. Lu, L. Zsak und D. L. Rock
(1998): A conserved African swine fever virus right variable region gene, l11L, is non-
essential for growth in vitro and virulence in domestic swine. J. Gen. Virol. 79 ( Pt
5), 1189–1195
Kleiboeker, S. B., S. K. Schommer, S. M. Lee, S. Watkins, W. Chittick und
D. Polson (2005): Simultaneous detection of North American and European porci-
ne reproductive and respiratory syndrome virus using real-time quantitative reverse
transcriptase-PCR. J. Vet. Diagn. Invest 17, Nr. 2, 165–170
Klupp, B. G., H. Granzow, W. Fuchs, E. Mundt und T. C. Mettenleiter
(2004a): Pseudorabies virus UL3 gene codes for a nuclear protein which is dispensable
for viral replication. J. Virol. 78, Nr. 1, 464–472
Klupp, B. G., C. J. Hengartner, T. C. Mettenleiter und L. W. Enquist
(2004b): Complete, annotated sequence of the pseudorabies virus genome. J. Virol. 78,
Nr. 1, 424–440
Kono, Y., T. Kanno, M. Shimizu, S. Yamada, S. Ohashi, M. Nakamine und
J. Shirai (1996): Nested PCR for detection and typing of porcine reproductive and
respiratory syndrome (PRRS) virus in pigs. J Vet Med Sci 58, Nr. 10, 941–946
Krafft, A. E., K. L. Russell, A. W. Hawksworth, S. McCall, M. Irvine,
L. T. Daum, J. L. Connoly, A. H. Reid, J. C. Gaydos und J. K. Tauben-
berger (2005): Evaluation of PCR testing of ethanol-fixed nasal swab specimens as
an augmented surveillance strategy for influenza virus and adenovirus identification. J
Clin Microbiol 43, Nr. 4, 1768–1775
Krakowka, S., J. A. Ellis, B. Meehan, S. Kennedy, F. McNeilly und G. Allan
(2000): Viral wasting syndrome of swine: Experimental reproduction of postweaning

154 Literaturverzeichnis
multisystemic wasting syndrome in gnotobiotic swine by coinfection with porcine cir-
covirus 2 and porcine parvovirus. Veterinary Pathology 37, Nr. 3, 254–263
Krakowka, S., J. A. Ellis, F. McNeilly, S. Ringler, D. M. Rings und G. Allan
(2001): Activation of the immune system is the pivotal event in the production of
wasting disease in pigs infected with porcine circovirus-2 (PCV-2). Vet. Pathol. 38,
Nr. 1, 31–42
Kresse, J. I., W. D. Taylor, W. W. Stewart und K. A. Eernisse (1985): Par-
vovirus infection in pigs with necrotic and vesicle-like lesions. Vet Microbiol 10, Nr. 6,
525–531
Kreuzer, K. A., A. Bohn, U. Lass, U. R. Peters und C. A. Schmidt (2000):
Influence of DNA polymerases on quantitative PCR results using TaqMan probe format
in the LightCycler instrument. Mol Cell Probes 14, Nr. 2, 57–60
Krumbholz, A., R. Wurm, O. Scheck, E. Birch-Hirschfeld, R. Egerer,
A. Henke, P. Wutzler und R. Zell (2003): Detection of porcine teschoviruses
and enteroviruses by LightCycler real-time PCR. J. Virol. Methods 113, Nr. 1, 51–63
Kuiper, A. (1985): Epizootologie und Erfahrungen mit der Impfprophylaxe des parvo-
virusbedingten SMEDI-Syndroms. Prakt. Tierarzt 66, 420–422
Laddomada, A. (2000): Incidence and control of CSF in wild boar in europe [In Process
Citation]. Vet. Microbiol. 73, Nr. 2-3, 121–130
Lager, K. M., W. L. Mengeling und S. L. Brockmeier (1997): Duration of ho-
mologous porcine reproductive and respiratory syndrome virus immunity in pregnant
swine. Vet Microbiol 58, Nr. 2-4, 127–133
Landolt, G. A., A. I. Karasin, C. Hofer, J. Mahaney, J. Svaren und C. W.
Olsen (2005): Use of real-time reverse transcriptase polymerase chain reaction assay
and cell culture methods for detection of swine influenza A viruses. Am J Vet Res 66,
Nr. 1, 119–124
Lange, K. D., N. Haddad, M. F. L. Potier, C. Agier, V. M. Le, P. Amar
und B. Toma (2003): Specificity of three ELISA-gE kits for screening pig meat for
antibodies to Aujeszky’s disease. Vet Rec 153, Nr. 20, 621–624

Literaturverzeichnis 155
Larochelle, R. und R. Magar (1997): Evaluation of the presence of porcine repro-
ductive and respiratory syndrome virus in packaged pig meat using virus isolation and
polymerase chain reaction (PCR) method. Vet Microbiol 58, Nr. 1, 1–8
Larochelle, R., M. Antaya, M. Morin und R. Magar (1999): Typing of porcine
circovirus in clinical specimens by multiplex PCR. J. Virol. Methods 80, Nr. 1, 69–75
Liess, B. (1981): Hog cholera. In: E. P. J. Gibbs (Hrsg.) Virus diseases of food animals.
A world geography of epidemiology and control, Vol.II. Academic Press, Institut fur
Virologie der Tierarztlichen Hochschule Hannover, S. 627–650
Liess, B. und V. Moennig (1990): Ruminant pestivirus infection in pigs. Rev. Sci.
Tech. 9, Nr. 1, 151–161
Liu, Q., L. Wang, P. Willson und L. A. Babiuk (2000): Quantitative, competitive
PCR analysis of porcine circovirus DNA in serum from pigs with postweaning multi-
systemic wasting syndrome. J Clin Microbiol 38, Nr. 9, 3474–3477
Livak, K. J. (1999): Allelic discrimination using fluorogenic probes and the 5’ nuclease
assay. Genet. Anal. 14, Nr. 5-6, 143–149
Loan, R. W. und M. M. Storm (1968): Propagation and transmission of hog cholear
virus in nonporcine hosts. Am. J. Vet. Res. 29, Nr. 4, 807–811
Locatelli, G., V. Urso und M. Malnati (2000): Quantitative analysis of GMO food
contaminations using real time PCR. Ital. J Biochem. 49, Nr. 3-4, 61–63
Long, B. C., T. L. Goldberg, S. L. Swenson, G. Erickson und G. Scherba
(2004): Adaptation and limitations of established hemagglutination inhibition assays
for the detection of porcine anti-swine influenza virus H1N2 antibodies. J Vet Diagn
Invest 16, Nr. 4, 264–270
Lowings, P., G. Ibata, J. Needham und D. Paton (1996): Classical swine fever virus
diversity and evolution. J. Gen. Virol. 77 ( Pt 6), 1311–1321
Lukert, P., B. De und J.L.Dale (1995): The Circoviridae. In: F. A. Murphy,
C.M.Faquet und D.H.L.Bishop (Hrsg.) Virus taxonomy. Sixth report of the Inter-

156 Literaturverzeichnis
national Committee on Taxonomy of Viruses. Springer Verlag, Wien und New York, S.
166–168
Lyoo, K. S., Y. H. Park und B. K. Park (2001): Prevalence of porcine reproductive
and respiratory syndrome virus, porcine circovirus type 2 and porcine parvovirus from
aborted fetuses and pigs with respiratory problems in Korea. J. Vet. Sci. 2, Nr. 3,
201–207
Mackay, I. M., K. E. Arden und A. Nitsche (2002): Real-time PCR in virology.
Nucleic Acids Res. 30, Nr. 6, 1292–1305
Malmquist, W. A. und D. HAY (1960): Hemadsorption and cytopathic effect produced
by African Swine Fever virus in swine bone marrow and buffy coat cultures. Am J Vet
Res 21, 104–108
Mankertz, A., K. Hattermann, B. Ehlers und D. Soike (2000): Cloning and
sequencing of columbid circovirus (coCV), a new circovirus from pigeons. Arch. Virol.
145, Nr. 12, 2469–2479
Mardassi, H., L. Wilson, S. Mounir und S. Dea (1994): Detection of porcine repro-
ductive and respiratory syndrome virus and efficient differentiation between Canadian
and European strains by reverse transcription and PCR amplification. J. Clin. Micro-
biol. 32, Nr. 9, 2197–2203
Matsuzaki, Y. (2003): [Detection of influenza virus (RT-PCR assay and others)]. Nippon
Rinsho 61, Nr. 11, 1909–1913
Mayr, A., P. A. Bachmann, G. Siegl, H. Mahnel und B. E. Sheffy (1968):
Characterization of a small porcine DNA virus. Arch Gesamte Virusforsch. 25, Nr. 1,
38–51
McCracken, R. M., J. B. McFerran und C. Dow (1973): The neural spread of
pseudorabies virus in calves. J Gen Virol 20, Nr. 1, 17–28
McGoldrick, A., J. P. Lowings, G. Ibata, J. J. Sands, S. Belak und D. J.
Paton (1998): A novel approach to the detection of classical swine fever virus by RT-
PCR with a fluorogenic probe (TaqMan). J. Virol. Methods 72, Nr. 2, 125–135

Literaturverzeichnis 157
McGoldrick, A., E. Bensaude, G. Ibata, G. Sharp und D. J. Paton (1999):
Closed one-tube reverse transcription nested polymerase chain reaction for the detection
of pestiviral RNA with fluorescent probes. J. Virol. Methods 79, Nr. 1, 85–95
McNeilly, F., I. McNair, M. O’Connor, S. Brockbank, D. Gilpin, C. Lasag-
na, G. Boriosi, B. Meehan, J. Ellis, S. Krakowka und G. M. Allan (2002):
Evaluation of a porcine circovirus type 2-specific antigen-capture enzyme-linked immu-
nosorbent assay for the diagnosis of postweaning multisystemic wasting syndrome in
pigs: comparison with virus isolation, immunohistochemistry, and the polymerase chain
reaction. J Vet Diagn Invest 14, Nr. 2, 106–112
Mebus, C. A. (1988): African swine fever. Adv. Virus Res. 35, 251–269
Meehan, B. M., J. L. Creelan, M. S. McNulty und D. Todd (1997): Sequence of
porcine circovirus DNA: affinities with plant circoviruses. J. Gen. Virol. 78 ( Pt 1),
221–227
Meehan, B. M., F. McNeilly, D. Todd, S. Kennedy, V. A. Jewhurst, J. A.
Ellis, L. E. Hassard, E. G. Clark, D. M. Haines und G. M. Allan (1998):
Characterization of novel circovirus DNAs associated with wasting syndromes in pigs.
J. Gen. Virol. 79 ( Pt 9), 2171–2179
Meehan, B. M., F. McNeilly, I. McNair, I. Walker, J. A. Ellis, S. Krakowka
und G. M. Allan (2001): Isolation and characterization of porcine circovirus 2 from
cases of sow abortion and porcine dermatitis and nephropathy syndrome. Arch Virol
146, Nr. 4, 835–842
Meng, Q., C. Wong, A. Rangachari, S. Tamatsukuri, M. Sasaki, E. Fiss,
L. Cheng, T. Ramankutty, D. Clarke, H. Yawata, Y. Sakakura, T. Hirose
und C. Impraim (2001): Automated multiplex assay system for simultaneous detection
of hepatitis B virus DNA, hepatitis C virus RNA, and human immunodeficiency virus
type 1 RNA. J Clin Microbiol 39, Nr. 8, 2937–2945
Meng, X. J. (2000): Heterogeneity of porcine reproductive and respiratory syndrome
virus: implications for current vaccine efficacy and future vaccine development. Veteri-
nary Microbiology 74, Nr. 4, 309–329

158 Literaturverzeichnis
Mengeling, W. L. (1975): Porcine parvovirus: frequency of naturally occurring trans-
placental infection and viral contamination of fetal porcine kidney cell cultures. Am J
Vet Res 36, Nr. 1, 41–44
Mengeling, W. L. (1978): Prevalence of porcine parvovirus-induced reproductive fai-
lure: an abattoir study. J Am Vet Med Assoc 172, Nr. 11, 1291–1294
Mengeling, W. L. und R. C. Cutlip (1976): Reproductive disease experimentally
induced by exposing pregnant gilts to porcine parvovirus. Am J Vet Res 37, Nr. 12,
1393–1400
Mengeling, W. L., P. S. Paul und T. T. Brown (1980): Transplacental infection
and embryonic death following maternal exposure to porcine parvovirus near the time
of conception. Arch Virol 65, Nr. 1, 55–62
Mengeling, W. L., P. S. Paul und K. M. Lager (1993): Virus-induced maternal
reproductive failure of swine. J Am Vet Med Assoc 203, Nr. 9, 1268–1272
Mengeling, W. L., K. M. Lager und A. C. Vorwald (1995): Diagnosis of Porcine
Reproductive and Respiratory Syndrome. Journal of Veterinary Diagnostic Investiga-
tion 7, Nr. 1, 3–16
Mengeling, W. L., A. C. Vorwald, K. M. Lager und S. L. Brockmeier (1996a):
Comparison among strains of porcine reproductive and respiratory syndrome virus for
their ability to cause reproductive failure. American Journal of Veterinary Research
57, Nr. 6, 834–839
Mengeling, W. L., A. C. Vorwald, K. M. Lager und S. L. Brockmeier (1996b):
Diagnosis of porcine reproductive and respiratory syndrome using infected alveolar
macrophages collected from live pigs. Veterinary Microbiology 49, Nr. 1-2, 105–115
Meulenberg, J. J., E. J. de Meijer und R. J. Moormann (1993a): Subgenomic
RNAs of Lelystad virus contain a conserved leader-body junction sequence. J. Gen.
Virol. 74 ( Pt 8), 1697–1701
Meulenberg, J. J., M. M. Hulst, E. J. de Meijer, P. L. Moonen, B. A. den,
E. P. D. Kluyver, G. Wensvoort und R. J. Moormann (1993b): Lelystad virus,

Literaturverzeichnis 159
the causative agent of porcine epidemic abortion and respiratory syndrome (PEARS),
is related to LDV and EAV. Virology 192, Nr. 1, 62–72
Meyer, H., B. Liess, W. Hermanns und G. Trautwein (1980): Experimentelle,
diaplazentare Infektion von Schweinefeten mit dem Virus der Europaischen Schweine-
pest (ESP).Virologische und serologische Untersuchungen in der postnatalen Phase.
Fortschritte der Veterinrrmedizin. Zbl. Vet. Med. 30, Nr. Suppl., 140–144
Meyer, H., B. Liess, H. R. Frey, W. Hermanns und G. Trautwein (1981): Ex-
perimental transplacental transmission of hog cholera virus in pigs. IV. Virological and
serological studies in newborn piglets. Zentralbl. Veterinarmed. B 28, Nr. 8, 659–668
Meyers, G. und H. J. Thiel (1996): Molecular characterization of pestiviruses. Ad-
vances in Virus Research, Vol 47 47, 53–118
Meyers, G., T. Rumenapf und H. J. Thiel (1989): Molecular cloning and nucleotide
sequence of the genome of hog cholera virus. Virology 171, Nr. 2, 555–567
Mulhard, C. (2002): Der Experimentator: Molekularbiologie Genomics. Spektrum Aka-
demischer Verlag, Berlin, 3. Aufl.
Modrow, S., D. Falke und U. Truyen (2003): Molekulare Virologie. Spektrum Aka-
demischer Verlag, Heidelberg - Berlin, 2 Aufl.
Moennig, V. (1992): The hog cholera virus. Comp. Immunol. Microbiol. Infect. Dis. 15,
Nr. 3, 189–201
Moennig, V. (2000): Introduction to classical swine fever: virus, disease and control
policy. Vet. Microbiol. 73, Nr. 2-3, 93–102
Moennig, V. und P. G. Plagemann (1992): The pestiviruses. Adv. Virus Res. 41,
53–98
Moennig, V., I. Greiser-Wilke, H. R. Frey, L. Haas, E. Liebler, J. Pohlenz
und B. Liess (1993): Prolonged persistence of cytopathogenic bovine viral diarrhea
virus (BVDV) in a persistently viremic cattle. Zentralbl. Veterinarmed. B 40, Nr. 5,
371–377

160 Literaturverzeichnis
Moennig, V., G. Floegel-Niesmann und I. Greiser-Wilke (2003): Clinical signs
and epidemiology of classical swine fever: a review of new knowledge. Vet. J. 165, Nr. 1,
11–20
Molitor, T. W., H. S. Joo und M. S. Collett (1983): Porcine parvovirus: virus
purification and structural and antigenic properties of virion polypeptides. J Virol 45,
Nr. 2, 842–854
Molitor, T. W., H. S. Joo und M. S. Collett (1984): Porcine parvovirus DNA:
characterization of the genomic and replicative form DNA of two virus isolates. Virology
137, Nr. 2, 241–254
Molitor, T. W., K. Oraveerakul, Q. Q. Zhang, C. S. Choi und L. R. Ludemann
(1991): Polymerase chain reaction (PCR) amplification for the detection of porcine
parvovirus. J Virol Methods 32, Nr. 2-3, 201–211
Montgomery, R. E. (1921): On a form of swine fever occurring in British East Africa
(Kenya Colony). J. Comp. Pathol. 34, 159–191
Moody, A., S. Sellers und N. Bumstead (2000): Measuring infectious bursal disease
virus RNA in blood by multiplex real-time quantitative RT-PCR. J Virol Methods 85,
Nr. 1-2, 55–64
Moormann, R. J. und M. M. Hulst (1988): Hog cholera virus: identification and
characterization of the viral RNA and the virus-specific RNA synthesized in infected
swine kidney cells. Virus Res. 11, Nr. 4, 281–291
Moormann, R. J., P. A. Warmerdam, M. B. van der, W. M. Schaaper,
G. Wensvoort und M. M. Hulst (1990): Molecular cloning and nucleotide se-
quence of hog cholera virus strain Brescia and mapping of the genomic region encoding
envelope protein E1. Virology 177, Nr. 1, 184–198
Morimoto, T., Y. Fujisaki, Y. Ito und Y. Tanaka (1972): Biological and physico-
chemical properties of porcine parvovirus recovered from stillborn piglets. Natl Inst.
Anim Health Q (Tokyo) 12, Nr. 3, 137–144

Literaturverzeichnis 161
Morozov, I., T. Sirinarumitr, S. D. Sorden, P. G. Halbur, M. K. Morgan,
K. J. Yoon und P. S. Paul (1998): Detection of a novel strain of porcine circovirus in
pigs with postweaning multisystemic wasting syndrome. J. Clin. Microbiol. 36, Nr. 9,
2535–2541
Mullis, K. B. und F. A. Faloona (1987): Specific synthesis of DNA in vitro via a
polymerase-catalyzed chain reaction. Methods Enzymol. 155, 335–350
Murphy, F. A., C. M. Fauquet, D. H. L. Bishop, S. A. Ghabrial, A. W. Jarvis,
G. P. Martinelli, M. A. Mayo und M. D. Summers (1995): Virus taxonomy. Sixth
report of the international committee on taxonomy of viruses. Report
Murphy, F. A., E. P. J. Gibbs, M. A. Horzinek und M. J. Studdert (1999):
Asfarviridae and Iridoviridae. In: F. A. Murphy, E. P. J. Gibbs, M. A. Horzinek
und M. J. Studdert (Hrsg.) Veterinary virology. Academic Press, S. 293–300
Murray, G. und S. McCutcheon (1999): Model framework and principles of emer-
gency management. Rev Sci Tech 18, Nr. 1, 15–20
Murtaugh, M. P., M. R. Elam und L. T. Kakach (1995): Comparison of the struc-
tural protein coding sequences of the VR-2332 and Lelystad virus strains of the PRRS
virus. Arch. Virol. 140, Nr. 8, 1451–1460
Narita, M., S. Inui, Y. Kawakami, K. Kitamura und A. Maeda (1975): Histopa-
thological changes of the brain in swine fetuses naturally infected with procine parvo-
virus. Natl Inst. Anim Health Q (Tokyo) 15, Nr. 1, 24–28
Nawagitgul, P., I. Morozov, S. R. Bolin, P. A. Harms, S. D. Sorden und P. S.
Paul (2000): Open reading frame 2 of porcine circovirus type 2 encodes a major capsid
protein. J Gen Virol 81, Nr. Pt 9, 2281–2287
Nolan, T. (2004): Getting started - The basics of setting up a qPCR assay. In: S. A.
Bustin (Hrsg.) A-Z of Quantitative PCR. International University Line, book chapter
13.2., S. 531–543
Oguzoglu, T. C., G. Floegel-Niesmann, H. R. Frey und V. Moennig (2001):
Differential diagnosis of Classical Swine Fever and Border Disease: Serological investi-

162 Literaturverzeichnis
gations of a pestivirus infection in a mixed farm. Dtsch. Tierarztl. Wochenschr. 108,
210–213
Oleksiewicz, M. B., A. Botner, K. G. Madsen und T. Storgaard (1998): Sen-
sitive detection and typing of porcine reproductive and respiratory syndrome virus by
RT-PCR amplification of whole viral genes. Vet. Microbiol. 64, Nr. 1, 7–22
Olsen, C. W., M. W. McGregor, A. J. Cooley, B. Schantz, B. Hotze und
V. S. Hinshaw (1993): Antigenic and genetic analysis of a recently isolated H1N1
swine influenza virus. Am J Vet Res 54, Nr. 10, 1630–1636
Olvera, A., M. Sibila, M. Calsamiglia, J. Segales und M. Domingo (2004):
Comparison of porcine circovirus type 2 load in serum quantified by a real time PCR in
postweaning multisystemic wasting syndrome and porcine dermatitis and nephropathy
syndrome naturally affected pigs. J Virol Methods 117, Nr. 1, 75–80
Ophuis, R. J., C. J. Morrissy und D. B. Boyle (2006): Detection and quantitative
pathogenesis study of classical swine fever virus using a real time RT-PCR assay. J
Virol Methods 131, Nr. 1, 78–85
Oraveerakul, K., C. S. Choi und T. W. Molitor (1990): Detection of porcine
parvovirus using nonradioactive nucleic acid hybridization. J Vet Diagn Invest 2, Nr. 2,
85–91
Ouardani, M., L. Wilson, R. Jette, C. Montpetit und S. Dea (1999): Multiplex
PCR for detection and typing of porcine circoviruses. J. Clin. Microbiol. 37, Nr. 12,
3917–3924
Pallares, F. J., P. G. Halbur, T. Opriessnig, S. D. Sorden, D. Villar, B. H.
Janke, M. J. Yaeger, D. J. Larson, K. J. Schwartz, K. J. Yoon und L. J.
Hoffman (2002): Porcine circovirus type 2 (PCV-2) coinfections in US field cases of
postweaning multisystemic wasting syndrome (PMWS). J Vet Diagn Invest 14, Nr. 6,
515–519
Parkhurst, K. M. und L. J. Parkhurst (1995a): Donor-acceptor distance distri-
butions in a double-labeled fluorescent oligonucleotide both as a single strand and in
duplexes. Biochemistry 34, Nr. 1, 293–300

Literaturverzeichnis 163
Parkhurst, K. M. und L. J. Parkhurst (1995b): Kinetic studies by fluorescence
resonance energy transfer employing a double-labeled oligonucleotide: hybridization to
the oligonucleotide complement and to single-stranded DNA. Biochemistry 34, Nr. 1,
285–292
Pastor, M. J., M. Arias und J. M. Escribano (1990): Comparison of two antigens for
use in an enzyme-linked immunosorbent assay to detect African swine fever antibody.
Am J Vet Res 51, Nr. 10, 1540–1543
Paton, D. J. und S. H. Done (1994): Congenital infection of pigs with ruminant-type
pestiviruses. J. Comp. Pathol. 111, Nr. 2, 151–163
Paton, D. J. und I. Greiser-Wilke (2003): Classical swine fever–an update. Res.
Vet. Sci. 75, Nr. 3, 169–178
Paton, D. J., J. P. Lowings und G. C. Ramirez (1994): Stability of the gp53 gene
of a bovine viral diarrhoea virus isolated at different times from a persistently infected
steer. Brit. Vet. J. 150, Nr. 6, 603–607
Paton, D. J., A. McGoldrick, I. Greiser-Wilke, S. Parchariyanon, J. Song,
P. P. Liou, T. Stadejek, J. P. Lowings, H. Bjorklund und S. Belak (2000a):
Genetic typing of classical swine fever virus [In Process Citation]. Vet. Microbiol. 73,
Nr. 2-3, 137–157
Paton, D. J., A. McGoldrick, I. Greiser-Wilke, S. Parchariyanon, J. Y.
Song, P. P. Liou, T. Stadejek, J. P. Lowings, H. Bjorklund und S. Belak
(2000b): Genetic typing of classical swine fever virus. Vet. Microbiol. 73, Nr. 2-3,
137–157
Payungporn, S., S. Chutinimitkul, A. Chaisingh, S. Damrongwantanapokin,
C. Buranathai, A. Amonsin, A. Theamboonlers und Y. Poovorawan (2006):
Single step multiplex real-time RT-PCR for H5N1 influenza A virus detection. J Virol
Methods 131, Nr. 2, 143–147
Pensaert, M., K. Ottis, J. Vandeputte, M. M. Kaplan und P. A. Bachmann
(1981): Evidence for the natural transmission of influenza A virus from wild ducks to
swine and its potential importance for man. Bull World Health Organ 59, Nr. 1, 75–78

164 Literaturverzeichnis
Peters, I. R., C. R. Helps, E. J. Hall und M. J. Day (2004): Real-time RT-PCR:
considerations for efficient and sensitive assay design. J. Immunol. Methods 286, Nr.
1-2, 203–217
Pfaffl, M. W. (2004): Quantification Strategies in Real-Time PCR. In: S. A. Bustin
(Hrsg.) A-Z of Quantitative PCR. International University Line, book chapter 3, S.
87–120
Plonait, H. (2001): Fortpflanzungsphysiologie und Gynakologie der Sau. In: K.-H.
Waldmann und M. Wendt (Hrsg.) Lehrbuch der Schweinekrankheiten. 3., durchge-
sehene auflage Aufl., Parey Buchverlag, book chapter 15, S. 399–470
Plowright, W., C. T. Perry und A. Greig (1974): Sexual transmission of African
swine fever virus in the tick, Ornithodoros moubata porcinus, Walton. Res Vet Sci 17,
Nr. 1, 106–113
Prieto, C. und J. M. Castro (2005): Porcine reproductive and respiratory syndrome
virus infection in the boar: a review. Theriogenology 63, Nr. 1, 1–16
Prieto, C., C. Garcıa, I. Simarro und J. M. Castro (2003): Temporal localiza-
tion of porcine reproductive and respiratory syndrome virus in reproductive tissues of
experimentally infected boars. Theriogenology 60, Nr. 8, 1505–1514
Prikhod’ko, G. G., H. Reyes, I. Vasilyeva und T. F. Busby (2003): Establishment
of a porcine parvovirus (PPV) DNA standard and evaluation of a new lightcycler nested-
PCR assay for detection of PPV. J Virol Methods 111, Nr. 1, 13–19
Pringle, C. R. (1999): Virus taxonomy - 1999 - The Universal System of Virus Taxo-
nomy, updated to include the new proposals ratified by the International Committee
on Taxonomy of Viruses during 1998. Archives of Virology 144, Nr. 2, 421–429
Rasmussen, T. B., A. Uttenthal, S. K. de, S. Belak und T. Storgaard (2003):
Development of a novel quantitative real-time RT-PCR assay for the simultaneous
detection of all serotypes of foot-and-mouth disease virus. Arch Virol 148, Nr. 10,
2005–2021

Literaturverzeichnis 165
Rasmussen, T. B., A. Uttenthal, J. Fernandez und T. Storgaard (2005): Quan-
titative multiplex assay for simultaneous detection and identification of Indiana and
New Jersey serotypes of vesicular stomatitis virus. J Clin Microbiol 43, Nr. 1, 356–362
Reeth, K. V., I. Brown, S. Essen und M. Pensaert (2004): Genetic relationships,
serological cross-reaction and cross-protection between H1N2 and other influenza A
virus subtypes endemic in European pigs. Virus Res 103, Nr. 1-2, 115–124
Revilla-Fernandez, S., B. Wallner, K. Truschner, A. Benczak, G. Brem,
F. Schmoll, M. Mueller und R. Steinborn (2005): The use of endogenous and
exogenous reference RNAs for qualitative and quantitative detection of PRRSV in por-
cine semen. J. Virol. Methods 126, Nr. 1-2, 21–30
Rice, C. M., E. M. Lenches, S. R. Eddy, S. J. Shin, R. L. Sheets und J. H.
Strauss (1985): Nucleotide sequence of yellow fever virus: implications for flavivirus
gene expression and evolution. Science 229, Nr. 4715, 726–733
Richt, J. A., K. M. Lager, D. F. Clouser, E. Spackman, D. L. Suarez und
K. J. Yoon (2004): Real-time reverse transcription-polymerase chain reaction assays
for the detection and differentiation of North American swine influenza viruses. J Vet
Diagn Invest 16, Nr. 5, 367–373
Ridpath, J. F. und S. R. Bolin (1997): Comparison of the complete genomic sequence
of the border disease virus, BD31, to other pestiviruses. Virus Res. 50, Nr. 2, 237–243
Ririe, K. M., R. P. Rasmussen und C. T. Wittwer (1997): Product differentia-
tion by analysis of DNA melting curves during the polymerase chain reaction. Anal.
Biochem. 245, Nr. 2, 154–160
Risatti, G., L. Holinka, Z. Lu, G. Kutish, J. D. Callahan, W. M. Nelson,
T. E. Brea und M. V. Borca (2005): Diagnostic evaluation of a real-time reverse
transcriptase PCR assay for detection of classical swine fever virus. J. Clin. Microbiol.
43, Nr. 1, 468–471
Risatti, G. R., J. D. Callahan, W. M. Nelson und M. V. Borca (2003): Rapid
detection of classical swine fever virus by a portable real-time reverse transcriptase
PCR assay. J. Clin. Microbiol. 41, Nr. 1, 500–505

166 Literaturverzeichnis
Ritchie, B. W., F. D. Niagro, P. D. Lukert, I. W. L. Steffens und K. S.
Latimer (1989): Characterization of a new virus from cockatoos with psittacine beak
and feather disease. Virology 171, Nr. 1, 83–88
Rumenapf, T., G. Meyers, R. Stark und H. J. Thiel (1989): Hog cholera virus–
characterization of specific antiserum and identification of cDNA clones. Virology 171,
Nr. 1, 18–27
Rumenapf, T., G. Unger, J. H. Strauss und H. J. Thiel (1993): Processing of the
envelope glycoproteins of pestiviruses. J. Virol. 67, Nr. 6, 3288–3294
Rodeffer, H. E., A. D. Leman, H. W. Dunne, M. Cropper und D. J. Sprecher
(1975): Reproductive failure in swine associated with maternal seroconversion for por-
cine parvovirus. J Am Vet Med Assoc 166, Nr. 10, 991–992
Rose, N., P. Blanchard, G. Larour, G. L. Diguerher, E. Eveno, J. P. Jol-
ly, A. Oger, M. L. Dimna, A. Jestin und F. Madec (2002): PMWS in France:
Serological Profiles of Affected Versus Non-affected Herds and Preliminary Analytical
Epidemiology. Pig Journal 50, 124–134
Rosell, C., J. Segales, J. Plana-Duran, M. Balasch, G. M. Rodrıguez-
Arrioja, S. Kennedy, G. M. Allan, F. McNeilly, K. S. Latimer und
M. Domingo (1999): Pathological, immunohistochemical, and in-situ hybridization
studies of natural cases of postweaning multisystemic wasting syndrome (PMWS) in
pigs. J. Comp Pathol. 120, Nr. 1, 59–78
Rosell, C., J. Segales, J. A. Ramos-Vara, J. M. Folch, G. M. Rodrıguez-
Arrioja, C. O. Duran, M. Balasch, J. Plana-Duran und M. Domingo (2000):
Identification of porcine circovirus in tissues of pigs with porcine dermatitis and nephro-
pathy syndrome. Vet Rec 146, Nr. 2, 40–43
Rossow, K. D. (1998): Porcine reproductive and respiratory syndrome. Vet Pathol 35,
Nr. 1, 1–20
Rossow, K. D., E. M. Bautista, S. M. Goyal, T. W. Molitor, M. P. Murtaugh,
R. B. Morrison, D. A. Benfield und J. E. Collins (1994): Experimental porcine

Literaturverzeichnis 167
reproductive and respiratory syndrome virus infection in one-, four-, and 10-week-old
pigs. J Vet Diagn Invest 6, Nr. 1, 3–12
Rovira, A., M. Balasch, J. Segales, L. Garcıa, J. Plana-Duran, C. Rosell,
H. Ellerbrok, A. Mankertz und M. Domingo (2002): Experimental inoculation
of conventional pigs with porcine reproductive and respiratory syndrome virus and
porcine circovirus 2. J. Virol. 76, Nr. 7, 3232–3239
Saiki, R. K., S. Scharf, F. Faloona, K. B. Mullis, G. T. Horn, H. A. Erlich
und N. Arnheim (1985): Enzymatic amplification of beta-globin genomic sequences
and restriction site analysis for diagnosis of sickle cell anemia. Science 230, Nr. 4732,
1350–1354
Sambrook, J., E. F. Fritsch und T. Maniatis (1989): Molecular cloning. A labora-
tory manual. Cold Spring Harbor Laboratory Press, Cold Spring Habor, NewYork, 2.
Aufl.
Sandvik, T., D. J. Paton und P. J. Lowings (1997): Detection and identification
of ruminant and porcine pestiviruses by nested amplification of 5’ untranslated cDNA
regions. J. Virol. Methods 64, Nr. 1, 43–56
Sandvik, T., H. Crooke, T. W. Drew, S. Blome, I. Greiser-Wilke,
V. Moennig, T. A. Gous, S. Gers, J. A. Kitching, G. Buhrmann und G. K.
Bruckner (2005): Classical swine fever in South Africa after 87 years’ absence. Vet
Rec 157, Nr. 9, 267
SantaLucia, J. J. (1998): A unified view of polymer, dumbbell, and oligonucleotide
DNA nearest-neighbor thermodynamics. Proc Natl Acad Sci U S A 95, Nr. 4, 1460–
1465
Schang, L. M. und F. A. Osorio (1993): A quantitative technique for the study of
the latency of Aujeszky virus. Rev. Sci. Tech. 12, Nr. 2, 505–521
Scheibner, H. (2000): Methoden zur Isolierung von Ribonukleinsaure (RNA) zum Vi-
rusgenomnachweis mittels Polymerasekettenreaktion nach Reverser Transkription (RT-
PCR). Dissertation, Institut fur Virologie, Tierarztliche Hochschule Hannover

168 Literaturverzeichnis
Schmidt, S. P., W. A. Hagemoser, J. P. Kluge und H. T. Hill (1987): Patho-
genesis of ovine pseudorabies (Aujeszky’s disease) following intratracheal inoculation.
Can J Vet Res 51, Nr. 3, 326–333
Schmittgen, T. D. und B. A. Zakrajsek (2000): Effect of experimental treatment
on housekeeping gene expression: validation by real-time, quantitative RT-PCR. J
Biochem. Biophys. Methods 46, Nr. 1-2, 69–81
Segales, J. und M. Domingo (2002): Postweaning multisystemic wasting syndrome
(PMWS) in pigs. A review. Vet Q 24, Nr. 3, 109–124
Segales, J., C. Rosell und M. Domingo (2004): Pathological findings associated
with naturally acquired porcine circovirus type 2 associated disease. Vet. Microbiol.
98, Nr. 2, 137–149
Shope, R. E. (1931a): An experimental study of”mad itch“ with especial reference to
its relationship to pseudorabies. The Journal of Experimental Medicine 54, 233–248
Shope, R. E. (1931b): Swine influenza. Filtration experiments and etiology. The Journal
of Experimental Medicine 54, 373–385
Smith, I. L., K. Halpin, D. Warrilow und G. A. Smith (2001): Development of a
fluorogenic RT-PCR assay (TaqMan) for the detection of Hendra virus. J Virol Methods
98, Nr. 1, 33–40
Smith, W. J., J. R. Thomson und S. Done (1993): Dermatitis/nephropathy syndrome
of pigs. Vet Rec 132, Nr. 2, 47
Snowdon, W. A. und E. L. French (1968): The bovine mucosal disease-swine fever
virus complex in pigs. Aust. Vet. J. 44, Nr. 4, 179–184
Soares, R. M., E. L. Durigon, J. G. Bersano und L. J. Richtzenhain (1999):
Detection of porcine parvovirus DNA by the polymerase chain reaction assay using
primers to the highly conserved nonstructural protein gene, NS-1. J Virol Methods 78,
Nr. 1-2, 191–198

Literaturverzeichnis 169
Solorzano, R. F., J. E. Thigpen, D. M. Bedell und W. L. Schwartz (1966):
The diagnosis of hog cholera by a fluorescent antibody test. J. Am. Vet. Med. Assoc.
149, Nr. 1, 31–34
Spagnuolo-Weaver, M., I. W. Walker, S. T. Campbell, F. McNeilly, B. M.
Adair und G. M. Allan (2000): Rapid detection of porcine reproductive and respira-
tory syndrome viral nucleic acid in blood using a fluorimeter based PCR method. Vet.
Microbiol. 76, Nr. 1, 15–23
Stadejek, T., A. Stankevicius, T. Storgaard, M. B. Oleksiewicz, S. Belak,
T. W. Drew und Z. Pejsak (2002): Identification of radically different variants of
porcine reproductive and respiratory syndrome virus in Eastern Europe: towards a
common ancestor for European and American viruses. Journal of General Virology 83,
1861–1873
Steiger, Y., M. Ackermann, C. Mettraux und U. Kihm (1992): Rapid and biolo-
gically safe diagnosis of African swine fever virus infection by using polymerase chain
reaction. J Clin Microbiol 30, Nr. 1, 1–8
Stone, B., J. Burrows, S. Schepetiuk, G. Higgins, A. Hampson, R. Shaw und
T. W. Kok (2004): Rapid detection and simultaneous subtype differentiation of influ-
enza A viruses by real time PCR. Journal of Virological Methods 117, Nr. 2, 103–112
Studdert, M. J. (1993): Circoviridae - New Viruses of Pigs, Parrots and Chickens.
Australian Veterinary Journal 70, Nr. 4, 121–122
Teifke, J. P., E. Lange, R. Klopfleisch und V. Kaden (2005): Nictitating mem-
brane as a potentially useful postmortem diagnostic specimen for classical swine fever.
J Vet Diagn Invest 17, Nr. 4, 341–345
Temin, H. M. und S. Mizutani (1970): RNA-dependent DNA polymerase in virions of
Rous sarcoma virus. Nature 226, Nr. 5252, 1211–1213
Templeton, K. E., S. A. Scheltinga, M. F. Beersma, A. C. Kroes und E. C.
Claas (2004): Rapid and sensitive method using multiplex real-time PCR for diagnosis
of infections by influenza a and influenza B viruses, respiratory syncytial virus, and
parainfluenza viruses 1, 2, 3, and 4. J. Clin. Microbiol. 42, Nr. 4, 1564–1569

170 Literaturverzeichnis
Terpstra, C. und G. Wensvoort (1988): Natural infections of pigs with bovine viral
diarrhoea virus associated with signs resembling swine fever. Res. Vet. Sci. 45, Nr. 2,
137–142
Terpstra, C., G. Wensvoort und J. M. A. Pol (1991): Experimental Reproduction
of Porcine Epidemic Abortion and Respiratory Syndrome (Mystery Swine Disease) by
Infection with Lelystad Virus - Koch Postulates Fulfilled. Veterinary Quarterly 13,
Nr. 3, 131–136
Terry, C. F., D. J. Shanahan, L. D. Ballam, N. Harris, D. G. McDowell und
H. C. Parkes (2002): Real-time detection of genetically modified soya using Lightcy-
cler and ABI 7700 platforms with TaqMan, Scorpion, and SYBR Green I chemistries.
J AOAC Int 85, Nr. 4, 938–944
Teuffert, J., T. Muller, M. Ziller und T. Selhorst (2005): [Proposal for changing
the spot checking examination regarding the control of the maintenance of freedom from
Aujeszky’s disease (AD)]. Dtsch Tierarztl Wochenschr 112, Nr. 8, 286–1
Thacker, E. L., B. J. Thacker, T. F. Young und P. G. Halbur (2000): Effect of
vaccination on the potentiation of porcine reproductive and respiratory syndrome virus
(PRRSV)-induced pneumonia by Mycoplasma hyopneumoniae. Vaccine 18, Nr. 13,
1244–1252
Thellin, O., W. Zorzi, B. Lakaye, B. B. De, B. Coumans, G. Hennen,
T. Grisar, A. Igout und E. Heinen (1999): Housekeeping genes as internal stan-
dards: use and limits. J Biotechnol. 75, Nr. 2-3, 291–295
Thompson, J. D., D. G. Higgins und T. J. Gibson (1994): CLUSTAL W: improving
the sensitivity of progressive multiple sequence alignment through sequence weighting,
position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22, Nr. 22,
4673–4680
Thur, B. und M. A. Hofmann (1998): Comparative detection of classical swine fe-
ver virus in striated muscle from experimentally infected pigs by reverse transcription
polymerase chain reaction, cell culture isolation and immunohistochemistry. J. Virol.
Methods 74, Nr. 1, 47–56

Literaturverzeichnis 171
Tischer, I., R. Rasch und G. Tochtermann (1974): Characterization of papovavirus-
and picornavirus-like particles in permanent pig kidney cell lines. Zentralbl. Bakteriol.
[Orig. A] 226, Nr. 2, 153–167
Tischer, I., H. Gelderblom, W. Vettermann und M. A. Koch (1982): A very
small porcine virus with circular single-stranded DNA. Nature 295, Nr. 5844, 64–66
Tischer, I., W. Mields, D. Wolff, M. Vagt und W. Griem (1986): Studies on
Epidemiology and Pathogenicity of Porcine Circovirus. Archives of Virology 91, Nr.
3-4, 271–276
Tischer, I., D. Peters, R. Rasch und S. Pociuli (1987): Replication of porcine
circovirus: induction by glucosamine and cell cycle dependence. Arch. Virol. 96, Nr.
1-2, 39–57
Todd, D., J. L. Creelan, D. P. Mackie, F. Rixon und M. S. McNulty (1990):
Purification and biochemical characterization of chicken anaemia agent. J. Gen. Virol.
71 ( Pt 4), 819–823
Todd, D., J. Weston, N. W. Ball, B. J. Borghmans, J. A. Smyth, L. Gelmini
und A. Lavazza (2001a): Nucleotide sequence-based identification of a novel circovirus
of canaries. Avian Pathology 30, Nr. 4, 321–325
Todd, D., J. H. Weston, D. Soike und J. A. Smyth (2001b): Genome sequence
determinations and analyses of novel circoviruses from goose and pigeon. Virology 286,
Nr. 2, 354–362
Trautwein, G. (1988): Pathology and pathogenesis of the disease. In: B. Liess (Hrsg.)
Classical Swine Fever and Related Viral Infections. Martinus Nijhoff Publishing, S.
27–54
Truyen, U. (2003): Schweineinfluenza. In: Virusinfektionen bei Haus- und Nutztieren.
2nd Aufl., Schlutersche GmbH & Co. KG, book chapter 4.10, S. 88–90
Turner, L. W., L. N. Brown, E. A. Carbrey, W. L. Mengeling, D. H. Perella
und R. F. Solorzano (1968): Recommended minimum standards for the isolation and

172 Literaturverzeichnis
identification of hog cholera by the fluorescent antibody-cell culture technique. Proc.
Ann. Meet. US. Anim. Health Assoc. 72, 444–447
Uttenthal, A., T. Storgaard, M. B. Oleksiewicz und S. K. de (2003): Experi-
mental infection with the Paderborn isolate of classical swine fever virus in 10-week-old
pigs: determination of viral replication kinetics by quantitative RT-PCR, virus isolation
and antigen ELISA. Vet. Microbiol. 92, Nr. 3, 197–212
van der Molen, E. J. und J. T. van Oirschot (1981): Congenital persistent swi-
ne fever (hog cholera). I. Pathomorphological lesions in lymphoid tissues, kidney and
adrenal. Zentralbl. Veterinarmed. B. 28, Nr. 2, 89–101
van Elden, L. J., M. Nijhuis, P. Schipper, R. Schuurman und A. M. van Loon
(2001): Simultaneous detection of influenza viruses A and B using real-time quantitative
PCR. J Clin Microbiol 39, Nr. 1, 196–200
van Oirschot, J. T. (1999a): Diva vaccines that reduce virus transmission. J. Biotech-
nol. 73, Nr. 2-3, 195–205
van Oirschot, J. T. (1999b): Hog cholera. In: B. E. Straw, S. D’Allaire, W. L.
Mengeling und D. J. Taylor (Hrsg.) Diseases of swine. 8th Aufl., Iowa State Uni-
versity Press, book chapter 14, S. 159–172
van Oirschot, J. T. und C. A. Terpstra (1977): A congenital persistent swine fever
infection. I. Clincal and virological observations. II. Immune response to swine fever
and unrelated antigens. Vet. Microbiol. 2, 121–142
van Oirschot, J. T., D. J. Houwers, H. J. Rziha und P. J. Moonen (1988): Deve-
lopment of an ELISA for detection of antibodies to glycoprotein I of Aujeszky’s disease
virus: a method for the serological differentiation between infected and vaccinated pigs.
J. Virol. Methods 22, Nr. 2-3, 191–206
van Rijn, P. A., G. J. Wellenberg, H. R. H. van der, L. Jacobs, P. L. Moonen
und H. Feitsma (2004): Detection of economically important viruses in boar semen
by quantitative RealTime PCR technology. J. Virol. Methods 120, Nr. 2, 151–160

Literaturverzeichnis 173
Vandesompele, J., K. D. Preter, F. Pattyn, B. Poppe, N. van Roy, A. D.
Paepe und F. Speleman (2002): Accurate normalization of real-time quantitative
RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol.
3, Nr. 7
Vilcek, S., P. F. Nettleton, J. A. Herring und A. J. Herring (1994): Rapid
detection of bovine herpesvirus 1 (BHV 1) using the polymerase chain reaction. Vet.
Microbiol. 42, Nr. 1, 53–64
Vilcek, S., T. Stadejek, I. Takacsova, L. Strojny und M. Mojzis (1997): Genetic
analysis of classical swine fever virus isolates from a small geographic area. Dtsch.
Tierarztl. Wochenschr. 104, Nr. 1, 9–12
Ward, C. L., M. H. Dempsey, C. J. Ring, R. E. Kempson, L. Zhang, D. Gor,
B. W. Snowden und M. Tisdale (2004): Design and performance testing of quan-
titative real time PCR assays for influenza A and B viral load measurement. J. Clin.
Virol. 29, Nr. 3, 179–188
Wardley, R. C., E. M. bu Elzein, J. R. Crowther und P. J. Wilkinson (1979):
A solid-phase enzyme linked immunosorbent assay for the detection of African swine
fever virus antigen and antibody. J Hyg (Lond) 83, Nr. 2, 363–369
Wasilk, A., J. D. Callahan, J. Christopher-Hennings, T. A. Gay, Y. Fang,
M. Dammen, M. E. Reos, M. Torremorell, D. Polson, M. Mellencamp,
E. Nelson und W. M. Nelson (2004): Detection of U.S., Lelystad, and European-
like porcine reproductive and respiratory syndrome viruses and relative quantitation
in boar semen and serum samples by real-time PCR. J. Clin. Microbiol. 42, Nr. 10,
4453–4461
Watzinger, F., M. Suda, S. Preuner, R. Baumgartinger, K. Ebner, L. Basko-
va, H. G. Niesters, A. Lawitschka und T. Lion (2004): Real-time quantitative
PCR assays for detection and monitoring of pathogenic human viruses in immunosup-
pressed pediatric patients. J Clin Microbiol 42, Nr. 11, 5189–5198
Wengler, G. (1991): Family Flaviviridae. Report

174 Literaturverzeichnis
Wengler, G., D. W. Bradley, M. S. Collett, F. X. Heinz, R. W. Schlesinger
und J. H. Strauss (1995): Family Flaviviridae. Report
Wensvoort, G., C. Terpstra, J. M. Pol, E. A. ter Laak, M. Bloemraad,
E. P. D. Kluyver, C. Kragten, B. L. van, B. A. den, F. Wagenaar und .
(1991): Mystery swine disease in The Netherlands: the isolation of Lelystad virus. Vet
Q 13, Nr. 3, 121–130
Wensvoort, G., E. P. D. Kluyver, J. M. Pol, F. Wagenaar, R. J. Moormann,
M. M. Hulst, R. Bloemraad, B. A. den, T. Zetstra und C. Terpstra (1992):
Lelystad virus, the cause of porcine epidemic abortion and respiratory syndrome: a
review of mystery swine disease research at Lelystad. Vet Microbiol 33, Nr. 1-4, 185–
193
Westaway, E. G., M. A. Brinton, S. Y. Gaidamovich, M. C. Horzinek,
A. Igarashi, L. Kaariainen, D. K. Lvov, J. S. Porterfield, P. K. Russell
und D. W. Trent (1985): Flaviviridae. Intervirology 24, Nr. 4, 183–192
Wills, R. W., J. J. Zimmerman, K. J. Yoon, S. L. Swenson, L. J. Hoffman,
M. J. McGinley, H. T. Hill und K. B. Platt (1997): Porcine reproductive and
respiratory syndrome virus: routes of excretion. Vet Microbiol 57, Nr. 1, 69–81
Wilson, G. M., H. K. Jindal, D. E. Yeung, W. Chen und C. R. Astell (1991):
Expression of minute virus of mice major nonstructural protein in insect cells: purifi-
cation and identification of ATPase and helicase activities. Virology 185, Nr. 1, 90–98
Wrathall, A. E. und W. L. Mengeling (1979): Effect of inseminating seropositive
gilts with semen containing porcine parvovirus. Br Vet J 135, Nr. 5, 420–425
Yoon, H. A., S. K. Eo, A. G. Aleyas, S. O. Park, J. H. Lee, J. S. Chae, J. G.
Cho und H. J. Song (2005): Molecular survey of latent pseudorabies virus infection
in nervous tissues of slaughtered pigs by nested and real-time PCR. J Microbiol 43,
Nr. 5, 430–436
Yoon, I. J., H. S. Joo, W. T. Christianson, R. B. Morrison und G. D. Dial
(1993): Persistent and contact infection in nursery pigs experimentally infected with
porcine reproductive and respiratory syndrome virus. Swine Health Prod. 1, 5–8

Literaturverzeichnis 175
Yu, M., L. F. Wang, B. J. Shiell, C. J. Morrissy und H. A. Westbury (1996):
Fine mapping of a C-terminal linear epitope highly conserved among the major envelope
glycoprotein E2 (gp51 to gp54) of different pestiviruses. Virology 222, Nr. 1, 289–292
Zhang, F. Q., Z. H. Li und N. Z. Zhang (2005): [Identification and comparison of
neutralizing epitopes of glycoprotein E(rns) of classical swine fever virus]. Wei Sheng
Wu Xue. Bao. 45, Nr. 1, 66–71
Zhang, X., J. Shin, T. W. Molitor, L. B. Schook und M. S. Rutherford (1999):
Molecular responses of macrophages to porcine reproductive and respiratory syndrome
virus infection. Virology 262, Nr. 1, 152–162
Zimmermann, W. und H. Plonait (2001): Erkrankungen des Atmungsapparates. In:
K.-H. Waldmann und M. Wendt (Hrsg.) Lehrbuch der Schweinekrankheiten. 3.,
durchgesehene auflage Aufl., Parey Buchverlag, book chapter 7, S. 111–150
Zsak, L., M. V. Borca, G. R. Risatti, A. Zsak, R. A. French, Z. Lu, G. F.
Kutish, J. G. Neilan, J. D. Callahan, W. M. Nelson und D. L. Rock (2005):
Preclinical diagnosis of African swine fever in contact-exposed swine by a real-time
PCR assay. J. Clin. Microbiol. 43, Nr. 1, 112–119
Zuker, M. (2003): Mfold web server for nucleic acid folding and hybridization prediction.
Nucleic Acids Res 31, Nr. 13, 3406–3415


A. Anhang
A.1 Verwendete Virusisolate, Organ- und Blutpro-
ben, Antikorper
Die verwendeten Virusisolate sowie Organ- und Blutproben sind in Kapitel 3.1.2.1
aufgefuhrt.
PorcilisR© PRRS
Fa. Intervet, Unterschleißheim
Basiert auf europaischem PRRSV-Stamm DV
2 ml Dosis enthalt mind. 104 KID50 des PRRSV DV
IngelvacR© PRRS MLV
Fa. Boehringer Ingelheim Vetmedica, Ingelheim
20 ml, 10 Impfdosen, MLV (modified live virus)
Porcine Reproductive and Respiratory Syndrome Virus MoAb
Fa. VMRD, USA
Kat. Nr. 2D6
BVD/C16 Antikorper, Peroxidase gekoppelt
177

178 A. Anhang
Institut fur Virologie
TiHo Hannover
A.2 Enzyme und kommerzielle Kits
QuantiTectR© Multiplex PCR Kit
25 ml PCR Kit (1000) QuantiTectR© Multiplex PCR Master Mix (mit ROX)
20 ml RNase-freies Wasser
Kat. Nr.: 204545, Fa. Qiagen, Hilden
BrilliantR© SYBR R©-Green QPCR Master Mix
2 × Brilliant SYBR R©-Green QPCR Master Mix
Referenzfarbstoff ROX (1 mM)
Kat. Nr.: 600548, Fa. Stratagene, Amsterdam, Niederlande
Abgene Thermo-Start Mastermix
20 × 1,8 ml
Kat. Nr.: AB-0938/15/B, Fa. ABgene, Hamburg
High Pure Viral RNA Kit
Kat. Nr.: 1858882, Fa. Roche Diagnostics GmbH, Mannheim
RNeasy Lipid Tissue Mini Kit
Kat. Nr.: 74804, Fa. Qiagen GmbH, Hilden
QIAamp DNA Mini Kit
Kat. Nr.: 51304, Fa. Qiagen GmbH, Hilden

A.2. Enzyme und kommerzielle Kits 179
DNeasy Tissue Kit
Kat. Nr.: 69504, Fa. Qiagen GmbH, Hilden
P-GEM R©T-easy Kit
Kat.Nr.: A1380, Fa. Promega, Mannheim
TOPO TA Cloning R© Kit
Kat. Nr.: K4500-40 Fa. Invitrogen, Carlsbad, Kalifornien, USA
SuperScriptTMIII Reverse Transkriptase
Kat. Nr.: 18080-044, Fa. Invitrogen, Karlsruhe
Alkalische Phosphatase mit Puffer
Kat. Nr.: 2392376, Boehringer, Mannheim
SpeI mit Puffer Tango
Kat. Nr.: #ER1251, Fa. Fermentas, St.Leon-Rot
EcoRV mit Puffer REact
Kat. Nr.: 15425-010, Fa. Invitrogen, Karlsruhe
XmnI mit Puffer NEB2
Kat. Nr.: #ER1531, Fa. Fermentas, St.Leon-Rot

180 A. Anhang
A.3 Medien, Puffer, Losungen, Antibiotika
A.3.1 Zellkulturmedien
EMEM (Eagle´s minimum essential medium)
Pulvermedium 9,60 g Fa. Invitrogen, Karlsruhe
NaHCO 2,20 g Fa. Merck, Darmstadt
Phenolrot 0,016 g Fa. Merck, Darmstadt
A. bidest. ad 1000,00 ml
Der pH-Wert wurde durch CO2-Begasung auf 6,9 eingestellt.
EMEM wurde als Erhaltungsmedium fur die PK(15)A-Zelllinie unter Zugabe von 50 ml
fetalem Kalberserum (Fa. Biochrom, Hamburg) eingesetzt.
Fur zellkulturbasierte Nachweissysteme in Mikro- und Makrotiterplatten wurden dem
EMEM folgende Inhaltsstoffe zugefugt:
Penicillin G 50 mg Fa. Sigma, Deisenhofen
Streptomycinsulfat 60 mg Fa. Sigma, Deisenhofen
fetales Kalberserum 100 ml Fa. Biochrom, Hamburg
EMEM/MEM-Hanks
MEM-Earle-Fertigpulver 4,76 g Fa. Invitrogen, Karlsruhe
MEM-Hanks-Fertigpulver 5,32 g Fa. Invitrogen, Karlsruhe
NaHCO3 1,52 g Fa. Merck, Darmstadt
Nichtessentielle Aminosauren 10 ml Fa. Biochrom, Hamburg
Natrium-Pyruvat 0,6 g Fa. Riedel-de Haen, Seelze
A. bidest. ad 1000 ml
Fetales Kalberserum 100 ml Fa. Biochrom, Hamburg

A.3. Medien, Puffer, Losungen, Antibiotika 181
Der pH-Wert wurde durch CO2-Begasung auf 7,2 eingestellt.
EDulb (EMEM, modifiziert nach Dulbecco und Freeman)
EDulb-Pulvermedium 13,53 g Fa. Serva, Heidelberg
NaHCO2 2,20 g Fa. Merck, Darmstadt
Penicilli G 60 mg Fa. Sigma, Deisenhofen
Streptomycin Sulfat 50 mg Fa. Sigma, Deisenhofen
Fetales Kalberserum 100 ml Fa. Biochrom, Hamburg
A. bidest. ad 1000 ml
Der pH-Wert wurde durch CO2-Begasung auf 6,9 eingestellt.
Einfriermedium
EMEM 700 ml
DMSO 100 ml Fa. Sigma, Deisenhofen
Fetales Kalberserum 200 ml Fa. Biochrom, Hamburg
ATV (adjusted trypsin-versen)-Losung
NaCl 8,00 g Fa. Merck, Darmstadt
KCl 0,40 g Fa. Merck, Darmstadt
NaHCO3 0,58 g Fa. Merck, Darmstadt
Dextrose 1,00 g Fa. Sigma, Deisenhofen
EDTA (Versen) 0,20 g Fa. Merck, Darmstadt
Trypsin(3U/mg) 0,50 g Fa. Serva, Heidelberg
A. bidest. ad 1000 ml
Versen-Trypsin-Losung (0,125%)
NaCl 8,0 g Fa. Merck, Darmstadt

182 A. Anhang
KCl 0,2 g Fa. Merck, Darmstadt
KH2PO4 0,2 g Fa. Merck, Darmstadt
Na2HPO4 × 12 H2O 2,37 g Fa. Merck, Darmstadt
CaCl2 × 2 H2O 0,132 g Fa. Merck, Darmstadt
Trypsin (3 U/mg) 1,25 g Fa. Serva, Heidelberg
EDTA (Versen) 1,25 g Fa. Merck, Darmstadt
Penicillin G 50 mg Fa. Sigma, Deisenhofen
Streptomycinsulfat 60 mg Fa. Sigma, Deisenhofen
MgCl2 × 6 H2O 0,1 g Fa. Sigma, Deisenhofen
A. bidest. ad 1000 ml
Einstellung des pH-Wertes auf 7,0 mit 1 N NaOH
AEC-Losung
3-Amino-9-Ethylcarbazol (AEC) 20,00 mg Fa. Sigma, Deisenhofen
Dimethylformamid 3,00 ml Fa. Merck, Darmstadt
Natriumazetat-Puffer
(0,05 M, pH 5,0) ad 50,00 ml Fa. Merck, Darmstadt
H2O2 (3 %) 0,40 ml Fa. Merck, Darmstadt
PBS (phosphatgepufferte Kochsalzlosung)
NaCl 8,00 g Fa. Merck, Darmstadt
KCl 0,20 g Fa. Merck, Darmstadt
Na2HPO4 × 12 H2O 2,37 g Fa. Merck, Darmstadt
KH2PO4 0,20 g Fa. Merck, Darmstadt
CaCl2 x 2 H2O 0,132 g Fa. Merck, Darmstadt
MgCl2 × 6 H2O 0,10 g Fa. Sigma, Deisenhofen
A. bidest. ad 1000 ml

A.3. Medien, Puffer, Losungen, Antibiotika 183
PBSM (phosphatgepufferte Kochsalzlosung ohne Kalzium und Magnesium)
NaCl 8,00 g Fa. Merck, Darmstadt
KCl 0,20 g Fa. Merck, Darmstadt
Na2HPO4 × 12 H2O 2,37 g Fa. Merck, Darmstadt
KH2PO4 0,20 g Fa. Merck, Darmstadt
A. bidest. ad 1000 ml
PBS/Tween
PBSM 1000 ml
Tween R© 20 100µl Fa. Serva, Heidelberg
10 × TBE-Puffer
TRIS 108 g Fa. USB, Cleveland, Ohio, USA
Borsaure 53,4 g Fa. Riedel-de-Haen, Seelze
EDTA 7,4 g
A. bidest. ad 1000 ml
5 × Probenpuffer zum Beladen von Agarose-Gelen
Glyzerin 25 % Fa. Merck, Darmstadt
EDTA 0,5 mM Fa. Merck, Darmstadt
Bromphenolblau 0,2 % Fa. Serva, Darmstadt
A. bidest. ad 50 ml
A.3.2 Puffer und Losungen
DEPC-Wasser
Kat.-Nr. 2420-204, Fa. BIO 101, Vista, USA

184 A. Anhang
A.4 Sonstige Reagenzien
peqGold 100 bp DNA-Leiter Plus
5 × 50µg
Kat. Nr.: 25-2021, Fa. Peqlab - Biotechnologie GmbH, Erlangen
A.5 Primer- und Sondensequenzen
Primer- und Sondensequenzen sind in Tabelle 4.3 auf Seite 83 dargestellt.
A.6 Gerate und Verbrauchsmittel
A.6.1 Gerate
Thermocycler
Mx 4000TMMultiplex Fa. Stratagene, Amsterdam, Niederlande
Quantitative PCR System Kat.-Nr.:401260
T personal Cycler Fa. Biometra, Gottingen
T3 Thermocycler Fa. Biometra, Gottingen
Agarosegelelektrophoresesystem
Mini Sub Cell Fa. Bio-Rad, Munchen
Wide Mini Sub Cell Fa. Bio-Rad, Munchen
Spannungsquelle Power Supply Fa. Bio-Rad, Munchen

A.6. Gerate und Verbrauchsmittel 185
Mikrowellengerat Fa. Privileg
UV-Transilluminator Fa. UVP, USA
Bildverarbeitungssystem Fa. MWG-Biotech AG, Ebersberg
Inkubatoren
CO2-Inkubator Typ B 5060 EK/CO2 Fa. Heraeus, Hanau
pH-Meter
Calimatic 766 Fa. Jurgens, Hannover
Zentrifugen
Biofuge pico Fa. Heraeus, Hanau
Kuhlzentrifuge 5417 R Fa. Eppendorf, Hamburg
Labofuge 400R Fa. Heraeus, Hanau
Vortex
K-550-GE Bender Fa. Hobein, Zurich, Schweiz
REAX 2000 Fa. Heidolph Elektro GmbH, Kelheim
Waagen
1213 MP Fa. Sartorius, Gottingen
EW Kern Fa. Landgraf, Langenhagen
Analysenwaage 1712 MP 8 Fa. Sartorius, Gottingen

186 A. Anhang
A.6.2 Verbrauchsmittel
Reaktionsgefaße
Volumen 0,5 ml Fa. Eppendorf, Hamburg
Volumen 1,5 ml Fa. Eppendorf, Hamburg
Volumen 2 ml Fa. Eppendorf, Hamburg
Glasgefaße 25 ml bis 3 l Fa. Jurgens, Hannover
Thermo-Strips and Ultra Clear Caps
Kat. Nr.: AB-1183, Fa. ABgene, Hamburg
Strip Tube, 8 × 0,2 ml Format
Kat. Nr.: 410022
Optical Cap, 8 × Strip
Kat. Nr.: 410024 Fa. Stratagene, Amsterdam, Niederlande
Gewebekulturflasche 50 ml / 25 cm2 Fa. Greiner, Frickenhausen
Gewebekulturflasche 250 ml / 75 cm2 Fa. Greiner, Nurtingen
96-Well-Gewebekulturplatten Fa. Costar, Bodenheim
24-Well-Gewebekulturplatten Fa. Greiner, Frickenhausen
Kryo-Vials 2 ml Fa. Greiner, Frickenhausen
Thomakammer neu Fa. Jurgens, Hannover
A.7 Software
MicrosoftR© Office Excel 2003 (11.6355.6408) SP1
Bestandteil von Microsoft Office Professional Edition 2003
Copyright c© 1985-2003 Microsoft Corporation.
Unterschleißheim

A.8. Tabellen und Abbildungen 187
Clustal W (1.81) Multiple Sequence Alignments
Toby Gibson, Des Higgins (now at the EBI, Hinxton, Great Britain), Julie Thompson
EMBL, Heidelberg
Thompson et al. (1994)
BioEdit Version 7.0.1.1
Copyright c© 1997-2004, Tom Hall
Isis Pharmaceuticals, Inc., Carlsbad, USA
Beacon Designer 2.13 Build 31904
Premier Biosoft International
Palo Alto, Kalifornien, USA
Copyright c© 2003 Premier Biosoft International
Mx4000 v4.20 Build 221, Schema 60
Fa. Stratagene, Kalifornien, USA
Copyright c© 2004 Stratagene
Kat.-Nr.:401260
Restriktionsmusteranalyse
The restriction enzyme database. New England Biolabs
http://rebase.neb.com/rebase/rebase.html
A.8 Tabellen und Abbildungen

188 A. Anhang
Tabelle A.1: Ergebnisse der ASPV-Primertitrationen mit SYBRTM-Green real-time
PCR. Die Tests wurden in Doppelansatzen mit in Standardvektoren klonierten PCR-
Produkten durchgefuhrt. Die jeweiligen Primerpaare sind unter Name aufgefuhrt; F steht
fur Forward, R fur Reverse-Primer. Als ∆Rn ist die finale Fluoreszenz angegeben. Ct, Ct
mittel und Ct SA bezeichnen die Ct-Werte, deren Mittel und die Standardabweichung. Die
Schmelztemperatur des PCR-Produktes befindet sich in der letzten Spalte.
Name ∆Rn Ct Ct mittel Ct SA Tm Produkt
50F50R 0,210 22,13 22,13 0,10 75,5
50F100R 0,217 20,83 20,83 0,04 75,5
50F300R 0,222 20,09 20,09 0,13 75,5
50F600F 0,231 19,93 19,93 0,04 75,5
50F900F 0,224 19,76 19,76 0,11 75,5
100F50R 0,224 22,79 22,79 0,16 74,5
100F100R 0,282 20,08 20,06 0,37 75,5
100F300R 0,307 19,35 19,36 0,22 75,5
100F600R 0,268 19,58 19,58 0,02 75,5
100F900F 0,245 19,49 19,53 0,31 75,5
300F50R 0,206 22,48 22,48 0,04 75,5
300F100R 0,279 20,03 20,03 0,01 75,5
300F300R 0,299 19,59 19,59 0,02 75,5
300F600R 0,328 19,33 19,33 0,04 75,5
300F900F 0,330 19,28 19,28 0,03 75,5
600F50R 0,230 21,51 21,51 0,13 75,5
600F100R 0,284 19,94 19,94 0,18 75,5
600F300R 0,304 19,51 19,51 0,14 75,5
600F600R 0,323 19,43 19,43 0,03 75,5
600F900F 0,329 19,31 19,32 0,13 75,5
900F50R 0,204 22,54 22,67 0,95 75,5
900F100R 0,269 20,12 20,13 0,23 75,5
900F300R 0,305 19,58 19,58 0,08 75,5
900F600R 0,224 19,01 18,99 0,24 75,5
900F900F 0,231 19,07 19,07 0,10 75,5
1µM-NTC 0,014 No Ct No Ct No Ct 68,5
5µM-NTC 0,245 34,09 34,07 0,49 72,5

A.8. Tabellen und Abbildungen 189
Tabelle A.2: Ergebnisse der SHV-1-Primertitrationen mit SYBRTM-Green real-time
PCR. Die Tests wurden in Doppelansatzen mit in Standardvektoren klonierten PCR-
Produkten durchgefuhrt. Die jeweiligen Primerpaare sind unter Name aufgefuhrt; F steht
fur Forward, R fur Reverse-Primer. Als ∆Rn ist die finale Fluoreszenz angegeben. Ct, Ct
mittel und Ct SA bezeichnen die Ct-Werte, deren Mittel und die Standardabweichung. Die
Schmelztemperatur des PCR-Produktes befindet sich in der letzten Spalte.
Name ∆Rn Ct Ct mittel Ct SA Tm Produkt
50F50R 0,214 19,83 19,83 0,12 82,5
50F100R 0,205 20,03 20,03 0,12 82,5
50F300R 0,192 19,91 19,92 0,18 82,5
50F600F 0,203 19,72 19,73 0,25 82,5
50F900F 0,212 19,56 19,56 0,10 81,5
100F50R 0,229 19,22 19,22 0,02 81,5
100F100R 0,250 19,15 19,15 0,09 81,5
100F300R 0,281 19,08 19,08 0,06 82,5
100F600R 0,263 19,11 19,11 0,02 82,5
100F900F 0,203 19,35 19,35 0,04 82,5
300F50R 0,200 19,33 19,33 0,04 82,5
300F100R 0,246 19,12 19,13 0,17 82,5
300F300R 0,276 19,11 19,11 0,06 82,5
300F600R 0,284 19,08 19,08 0,19 82,5
300F900F 0,308 19,02 19,01 0,10 82,5
600F50R 0,206 19,33 19,33 0,04 81,5
600F100R 0,265 19,06 19,06 0,09 82,5
600F300R 0,273 19,24 19,24 0,03 82,5
600F600R 0,296 19,26 19,26 0,09 82,5
600F900F 0,304 19,26 19,26 0,17 82,5
900F50R 0,200 19,47 19,47 0,12 82,5
900F100R 0,232 19,29 19,29 0,16 82,5
900F300R 0,243 19,64 19,67 0,36 82,5
900F600R 0,316 19,22 19,22 0,14 82,5
900F900F 0,316 19,31 19,31 0,07 82,5
1µM-NTC 0,003 No Ct No Ct No Ct 73,5
5µM-NTC 0,197 35,06 35,05 0,31 79,5

190 A. Anhang
Tabelle A.3: Ergebnisse der PCV-2-Primertitrationen mit SYBRTM-Green real-time
PCR. Die Tests wurden in Doppelansatzen mit in Standardvektoren klonierten PCR-
Produkten durchgefuhrt. Die jeweiligen Primerpaare sind unter Name aufgefuhrt; F steht
fur Forward, R fur Reverse-Primer. Als ∆Rn ist die finale Fluoreszenz angegeben. Ct, Ct
mittel und Ct SA bezeichnen die Ct-Werte, deren Mittel und die Standardabweichung. Die
Schmelztemperatur des PCR-Produktes befindet sich in der letzten Spalte.
Name ∆Rn Ct Ct mittel Ct SA Tm Produkt
50F50R 0,286 19,60 19,60 0,10 80,5
50F100R 0,320 18,18 18,19 0,19 80,5
50F300R 0,322 17,60 17,60 0,06 80,5
50F600F 0,331 17,38 17,38 0,13 80,5
50F900F 0,313 17,46 17,47 0,13 80,5
100F50R 0,307 19,54 19,54 0,01 80,5
100F100R 0,378 17,82 17,82 0,19 80,5
100F300R 0,397 17,33 17,33 0,10 80,5
100F600R 0,408 17,19 17,19 0,14 80,5
100F900F 0,386 17,16 17,16 0,07 80,5
300F50R 0,297 19,31 19,31 0,04 80,5
300F100R 0,365 17,62 17,64 0,27 80,5
300F300R 0,417 17,11 17,12 0,11 80,5
300F600R 0,412 17,13 17,13 0,05 80,5
300F900F 0,417 17,08 17,08 0,11 80,5
600F50R 0,313 19,01 19,00 0,09 80,5
600F100R 0,379 17,69 17,69 0,06 80,5
600F300R 0,406 17,27 17,27 0,00 80,5
600F600R 0,410 17,12 17,12 0,05 80,5
600F900F 0,425 17,02 17,00 0,11 80,5
900F50R 0,278 19,49 19,50 0,13 80,5
900F100R 0,339 17,88 17,88 0,21 80,5
900F300R 0,348 17,60 17,63 0,39 80,5
900F600R 0,399 17,32 17,33 0,11 80,5
900F900F 0,422 17,19 17,19 0,06 80,5
1µM-NTC 0,001 No Ct No Ct No Ct 68,5
5µM-NTC 0,205 36,33 36,34 0,16 69,5
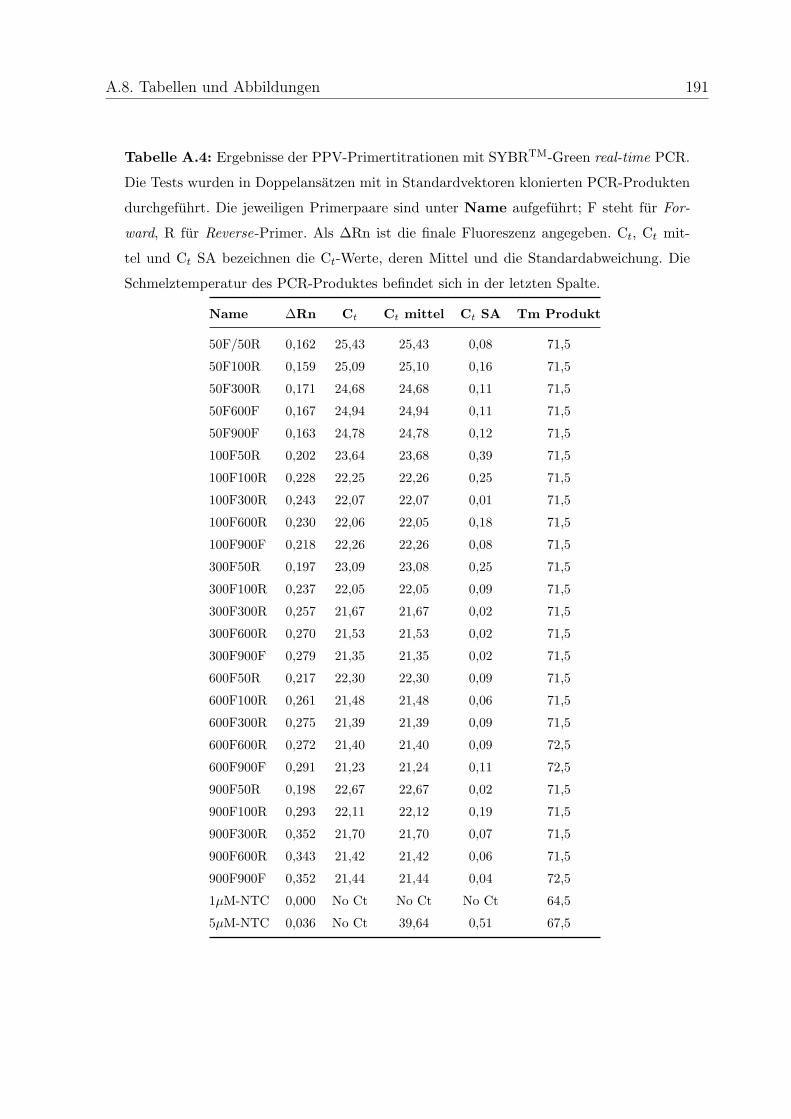
A.8. Tabellen und Abbildungen 191
Tabelle A.4: Ergebnisse der PPV-Primertitrationen mit SYBRTM-Green real-time PCR.
Die Tests wurden in Doppelansatzen mit in Standardvektoren klonierten PCR-Produkten
durchgefuhrt. Die jeweiligen Primerpaare sind unter Name aufgefuhrt; F steht fur For-
ward, R fur Reverse-Primer. Als ∆Rn ist die finale Fluoreszenz angegeben. Ct, Ct mit-
tel und Ct SA bezeichnen die Ct-Werte, deren Mittel und die Standardabweichung. Die
Schmelztemperatur des PCR-Produktes befindet sich in der letzten Spalte.
Name ∆Rn Ct Ct mittel Ct SA Tm Produkt
50F/50R 0,162 25,43 25,43 0,08 71,5
50F100R 0,159 25,09 25,10 0,16 71,5
50F300R 0,171 24,68 24,68 0,11 71,5
50F600F 0,167 24,94 24,94 0,11 71,5
50F900F 0,163 24,78 24,78 0,12 71,5
100F50R 0,202 23,64 23,68 0,39 71,5
100F100R 0,228 22,25 22,26 0,25 71,5
100F300R 0,243 22,07 22,07 0,01 71,5
100F600R 0,230 22,06 22,05 0,18 71,5
100F900F 0,218 22,26 22,26 0,08 71,5
300F50R 0,197 23,09 23,08 0,25 71,5
300F100R 0,237 22,05 22,05 0,09 71,5
300F300R 0,257 21,67 21,67 0,02 71,5
300F600R 0,270 21,53 21,53 0,02 71,5
300F900F 0,279 21,35 21,35 0,02 71,5
600F50R 0,217 22,30 22,30 0,09 71,5
600F100R 0,261 21,48 21,48 0,06 71,5
600F300R 0,275 21,39 21,39 0,09 71,5
600F600R 0,272 21,40 21,40 0,09 72,5
600F900F 0,291 21,23 21,24 0,11 72,5
900F50R 0,198 22,67 22,67 0,02 71,5
900F100R 0,293 22,11 22,12 0,19 71,5
900F300R 0,352 21,70 21,70 0,07 71,5
900F600R 0,343 21,42 21,42 0,06 71,5
900F900F 0,352 21,44 21,44 0,04 72,5
1µM-NTC 0,000 No Ct No Ct No Ct 64,5
5µM-NTC 0,036 No Ct 39,64 0,51 67,5

192 A. Anhang
Tabelle A.5: Ergebnisse der Pestivirenspezifischen-Primertitrationen mit SYBRTM-
Green real-time PCR. Die Tests wurden in Doppelansatzen mit in Standardvektoren
klonierten PCR-Produkten durchgefuhrt. Die jeweiligen Primerpaare sind unter Name
aufgefuhrt; F steht fur Forward, R fur Reverse-Primer. Als ∆Rn ist die finale Fluoreszenz
angegeben. Ct, Ct mittel und Ct SA bezeichnen die Ct-Werte, deren Mittel und die Stan-
dardabweichung. Die Schmelztemperatur des PCR-Produktes befindet sich in der letzten
Spalte.
Name ∆Rn Ct Ct mittel Ct SA Tm Produkt
50F50R 0,300 21,81 21,81 0,10 82,5
50F100R 0,308 21,99 21,98 0,11 82,5
50F300R 0,298 22,15 22,15 0,14 82,5
50F600F 0,282 22,27 22,27 0,12 82,5
50F900F 0,291 22,69 22,69 0,10 82,5
100F50R 0,349 20,56 20,56 0,29 82,5
100F100R 0,406 20,26 20,27 0,22 82,5
100F300R 0,421 20,16 20,16 0,29 82,5
100F600R 0,366 20,51 20,51 0,11 82,5
100F900F 0,346 21,11 21,11 0,07 82,5
300F50R 0,320 20,14 20,15 0,31 82,5
300F100R 0,362 20,06 20,06 0,04 82,5
300F300R 0,394 20,33 20,34 0,04 82,5
300F600R 0,438 20,31 20,31 0,01 82,5
300F900F 0,420 20,44 20,44 0,13 82,5
600F50R 0,323 19,63 19,63 0,17 82,5
600F100R 0,419 19,35 19,35 0,10 82,5
600F300R 0,426 19,92 19,92 0,05 82,5
600F600R 0,418 20,27 20,28 0,22 82,5
600F900F 0,456 20,20 20,20 0,06 82,5
900F50R 0,298 20,04 20,03 0,27 82,5
900F100R 0,329 20,07 20,07 0,09 82,5
900F300R 0,399 20,13 20,13 0,01 82,5
900F600R 0,445 19,89 19,89 0,04 82,5
900F900F 0,485 19,86 19,86 0,20 82,5
1µM-NTC 0,155 38,22 38,29 1,43 81,5
5µM-NTC 0,296 34,76 34,78 0,60 82,5
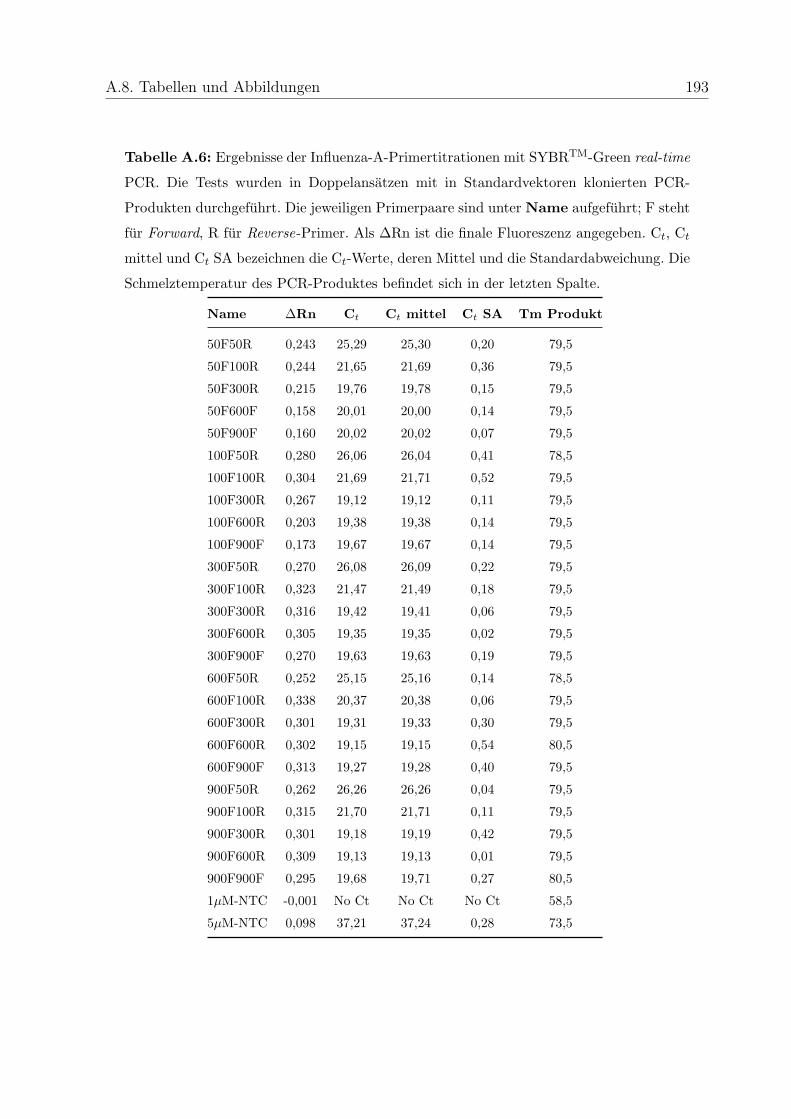
A.8. Tabellen und Abbildungen 193
Tabelle A.6: Ergebnisse der Influenza-A-Primertitrationen mit SYBRTM-Green real-time
PCR. Die Tests wurden in Doppelansatzen mit in Standardvektoren klonierten PCR-
Produkten durchgefuhrt. Die jeweiligen Primerpaare sind unter Name aufgefuhrt; F steht
fur Forward, R fur Reverse-Primer. Als ∆Rn ist die finale Fluoreszenz angegeben. Ct, Ct
mittel und Ct SA bezeichnen die Ct-Werte, deren Mittel und die Standardabweichung. Die
Schmelztemperatur des PCR-Produktes befindet sich in der letzten Spalte.
Name ∆Rn Ct Ct mittel Ct SA Tm Produkt
50F50R 0,243 25,29 25,30 0,20 79,5
50F100R 0,244 21,65 21,69 0,36 79,5
50F300R 0,215 19,76 19,78 0,15 79,5
50F600F 0,158 20,01 20,00 0,14 79,5
50F900F 0,160 20,02 20,02 0,07 79,5
100F50R 0,280 26,06 26,04 0,41 78,5
100F100R 0,304 21,69 21,71 0,52 79,5
100F300R 0,267 19,12 19,12 0,11 79,5
100F600R 0,203 19,38 19,38 0,14 79,5
100F900F 0,173 19,67 19,67 0,14 79,5
300F50R 0,270 26,08 26,09 0,22 79,5
300F100R 0,323 21,47 21,49 0,18 79,5
300F300R 0,316 19,42 19,41 0,06 79,5
300F600R 0,305 19,35 19,35 0,02 79,5
300F900F 0,270 19,63 19,63 0,19 79,5
600F50R 0,252 25,15 25,16 0,14 78,5
600F100R 0,338 20,37 20,38 0,06 79,5
600F300R 0,301 19,31 19,33 0,30 79,5
600F600R 0,302 19,15 19,15 0,54 80,5
600F900F 0,313 19,27 19,28 0,40 79,5
900F50R 0,262 26,26 26,26 0,04 79,5
900F100R 0,315 21,70 21,71 0,11 79,5
900F300R 0,301 19,18 19,19 0,42 79,5
900F600R 0,309 19,13 19,13 0,01 79,5
900F900F 0,295 19,68 19,71 0,27 80,5
1µM-NTC -0,001 No Ct No Ct No Ct 58,5
5µM-NTC 0,098 37,21 37,24 0,28 73,5

194 A. Anhang
Tabelle A.7: Ergebnisse der pestivirusspezifischen PCR. Eigesetzt wurden in Tabelle 3.3
aufgefuhrten Virussuspensionen und Organmaterialien.
Name Farbstoff Ct Ergebnis
137/14 FAM 36,42 +
CSF-573 FAM 23,37 +
CSF-277 FAM 20,31 +
V 487 FAM 18,87 +
217 Milz FAM 24,27 +
209 / CSF0905 FAM 27,16 +
208 / CSF0905 FAM 25,87 +
BVDV NADL FAM 17,10 +
BDV Gifhorn FAM 17,97 +
240-1 / 0695 FAM 25,14 +
CSF0104 FAM 20,66 +
CSF0902 FAM 17,34 +
Moredun FAM 31,65 +
CSF0309 FAM 32,68 +
240-2 FAM 22,07 +
Arnsberg FAM 19,93 +
CSF-573 / 1 FAM 22,86 +
0081/2 FAM 20,18 +
217 Niere FAM 20,84 +
BDV Frijters FAM 32,95 +
219 BDV FAM 38,52 +
CSF-273 FAM 24,39 +
BDV 10574 FAM 20,90 +
277 Milz FAM 22,16 +
-RT FAM No Ct −
NTC FAM No Ct −
NTC FAM No Ct −
NTC FAM No Ct −

A.8. Tabellen und Abbildungen 195
Tabelle A.8: Ergebnisse der Influenza-A-spezifischen PCR. Eingesetzt wurden Influenza-
A-Virus-Isolate wie in Tabelle 3.2 angegeben. Die Auswertung erfolgte softwaregestutzt.
Name Farbstoff Ct Ergebnis
Infl.-A Iso.:1 HEX 25,3 +
Infl.-A Iso.:1 HEX 24,9 +
Infl.-A Iso.:2 HEX 23,8 +
Infl.-A Iso.:2 HEX 23,6 +
Infl.-A Iso.:3 HEX 24,8 +
Infl.-A Iso.:3 HEX 25,3 +
Infl.-A Iso.:4 HEX 22,5 +
Infl.-A Iso.:4 HEX 22,4 +
Infl.-A Iso.:5 HEX 21,2 +
Infl.-A Iso.:5 HEX 21,6 +
Infl.-A Iso.:6 HEX 23,3 +
Infl.-A Iso.:6 HEX 23,4 +
Infl.-A Iso.:7 HEX 21,8 +
Infl.-A Iso.:7 HEX 21,8 +
Infl.-A Iso.:8 HEX 24,5 +
Infl.-A Iso.:8 HEX 24,7 +
Infl.-A Iso.:9 HEX 24,2 +
Infl.-A Iso.:9 HEX 24,3 +
Infl.-A Iso.:10 HEX 28,6 +
Infl.-A Iso.:10 HEX 28,5 +
Infl.-A Iso.:11 HEX 23,0 +
Infl.-A Iso.:11 HEX 23,0 +
Infl.-A Iso.:12 HEX 20,3 +
Infl.-A Iso.:12 HEX 20,4 +
— HEX No Ct −
— HEX No Ct −

196 A. Anhang
Tabelle A.9: Ergebnisse der SHV-1-spezifischen PCR. Eingesetzt wurden SHV-1-Isolate
wie in Tabelle 3.1 angegeben. Die Auswertung erfolgte softwaregestutzt.
Name Farbstoff Ct Ergebnis
SHV-1 I1 HEX 22,36 +
SHV-1 I231 HEX 18,12 +
SHV-1 I427 HEX 20,18 +
SHV-1 I517 HEX 18,81 +
SHV-1 I527 HEX 29,44 +
SHV-1 I450 HEX 19,74 +
SHV-1 I57 HEX 18,52 +
SHV-1 I537 HEX 17,12 +
SHV-1 I311 HEX 17,83 +
SHV-1 I425 HEX 13,62 +
SHV-1 I321 HEX 16,81 +
SHV-1 I612 HEX 21,20 +
SHV-1 I397 HEX 23,83 +
SHV-1 I410 HEX 23,21 +
SHV-1 I351 HEX 19,56 +
SHV-1 I386 HEX 16,37 +
SHV-1 I549 HEX 18,38 +
SHV-1 I37 HEX 19,08 +
SHV-1 I387 HEX 26,54 +
SHV-1 I423 HEX 20,89 +
SHV-1 I18 HEX 18,30 +
SHV-1 I550 HEX 22,80 +
SHV-1 I611 HEX 20,98 +
NTC HEX No Ct −
NTC HEX No Ct −

A.8. Tabellen und Abbildungen 197
Tabelle A.10: Ergebnisse der ASPV- und SHV-1-spezifischen PCRs. Die PCRs wurden
durchgefuhrt mit in Standardvektoren klonierten PCR-Produkten. Es konnte eine Absen-
kung der PCR-Effizienz in multiplex-Ansatzen festgestellt werden.
Virus Farbstoff Ct Kopienzahl/ Ansatz Ergebnis R2 Effizienz (%)
ASPV FAM 16,87 1 × 106 + 0,998 91,0
ASPV FAM 20,12 1 × 105 + 0,998 91,0
ASPV FAM 23,32 1 × 104 + 0,998 91,0
ASPV FAM 27,75 1 × 103 + 0,998 91,0
ASPV FAM 31,09 1 × 102 + 0,998 91,0
ASPV FAM 34,30 1 × 101 + 0,998 91,0
SHV-1 HEX 17,70 1 × 106 + 1,000 96,7
SHV-1 HEX 21,15 1 × 105 + 1,000 96,7
SHV-1 HEX 24,62 1 × 104 + 1,000 96,7
SHV-1 HEX 28,12 1 × 103 + 1,000 96,7
SHV-1 HEX 31,30 1 × 102 + 1,000 96,7
SHV-1 HEX 34,73 1 × 101 + 1,000 96,7
ASPV+SHV-1 FAM 17,71 1 × 106 + 0,998 82,4
ASPV+SHV-1 FAM 21,08 1 × 105 + 0,998 82,4
ASPV+SHV-1 FAM 24,49 1 × 104 + 0,998 82,4
ASPV+SHV-1 FAM 29,05 1 × 103 + 0,998 82,4
ASPV+SHV-1 FAM 32,62 1 × 102 + 0,998 82,4
ASPV+SHV-1 FAM 36,70 1 × 101 + 0,998 82,4
ASPV+SHV-1 HEX 20,20 1 × 106 + 0,996 80,4
ASPV+SHV-1 HEX 23,82 1 × 105 + 0,996 80,4
ASPV+SHV-1 HEX 26,84 1 × 104 + 0,996 80,4
ASPV+SHV-1 HEX 32,19 1 × 103 + 0,996 80,4
ASPV+SHV-1 HEX 35,55 1 × 102 + 0,996 80,4
ASPV+SHV-1 HEX 39,42 1 × 101 + 0,996 80,4

198 A. Anhang
Tabelle A.11: Ergebnisse der PCV-2- und PPV-spezifischen PCRs. Die PCRs wurden
durchgefuhrt mit in Standardvektoren klonierten PCR-Produkten. Es konnte eine Absen-
kung der PCR-Effizienz in multiplex-Ansatzen festgestellt werden.
Virus Farbstoff Ct Kopienzahl/ Ansatz Ergebnis R2 Effizienz (%)
PCV-2 FAM 19,19 1 × 106 + 0,970 98,8
PCV-2 FAM 22,49 1 × 105 + 0,970 98,8
PCV-2 FAM 26,88 1 × 104 + 0,970 98,8
PCV-2 FAM 30,65 1 × 103 + 0,970 98,8
PCV-2 FAM 34,37 1 × 102 + 0,970 98,8
PCV-2 FAM 34,77 1 × 101 + 0,970 98,8
PPV HEX 19,59 1 × 106 + 0,983 99,5
PPV HEX 23,14 1 × 105 + 0,983 99,5
PPV HEX 27,06 1 × 104 + 0,983 99,5
PPV HEX 30,58 1 × 103 + 0,983 99,5
PPV HEX 34,42 1 × 102 + 0,983 99,5
PPV HEX 35,45 1 × 101 + 0,983 99,5
PCV-2+PPV FAM 16,40 1 × 107 + 0,982 85,6
PCV-2+PPV FAM 22,03 1 × 106 + 0,982 85,6
PCV-2+PPV FAM 25,58 1 × 105 + 0,982 85,6
PCV-2+PPV FAM 29,81 1 × 104 + 0,982 85,6
PCV-2+PPV FAM 33,47 1 × 103 + 0,982 85,6
PCV-2+PPV FAM 37,18 1 × 102 + 0,982 85,6
PCV-2+PPV FAM 38,42 1 × 101 + 0,982 85,6
PCV-2+PPV HEX 17,78 1 × 107 + 0,982 90,4
PCV-2+PPV HEX 23,21 1 × 106 + 0,982 90,4
PCV-2+PPV HEX 26,64 1 × 105 + 0,982 90,4
PCV-2+PPV HEX 30,66 1 × 104 + 0,982 90,4
PCV-2+PPV HEX 34,43 1 × 103 + 0,982 90,4
PCV-2+PPV HEX 37,55 1 × 102 + 0,982 90,4
PCV-2+PPV HEX 39,01 1 × 101 + 0,982 90,4

A.8. Tabellen und Abbildungen 199
Tabelle A.12: Ergebnisse der Pestiviren- und Influenza-A-spezifischen PCRs. Die PCRs
wurden durchgefuhrt mit in Standardvektoren klonierten Es konnte eine Absenkung der
PCR-Effizienz in multiplex-Ansatzen festgestellt werden. Sie macht sich besonders be-
merkbar im Falle der Influenza-A-PCR.
Virus Farbstoff Ct Kopienzahl/ Ansatz Ergebnis R2 Effizienz (%)
Pestiviren FAM 20,48 1 × 106 + 1,000 92,9
Pestiviren FAM 24,30 1 × 105 + 1,000 92,9
Pestiviren FAM 27,64 1 × 104 + 1,000 92,9
Pestiviren FAM 31,26 1 × 103 + 1,000 92,9
Pestiviren FAM 34,59 1 × 102 + 1,000 92,9
Pestiviren FAM 38,12 1 × 101 + 1,000 92,9
Influenza-A-Virus HEX 21,31 1 × 106 + 0,991 98,8
Infl.-A-Virus HEX 24,50 1 × 105 + 0,991 98,8
Influenza-A-Virus HEX 26,77 1 × 104 + 0,991 98,8
Influenza-A-Virus HEX 30,76 1 × 103 + 0,991 98,8
Influenza-A-Virus HEX 33,75 1 × 102 + 0,991 98,8
Influenza-A-Virus HEX 38,42 1 × 101 + 0,991 98,8
Pesti.+Infl.-A-Virus FAM 22,52 1 × 106 + 0,974 90,8
Pesti.+Infl.-A-Virus FAM 26,38 1 × 105 + 0,974 90,8
Pesti.+Infl.-A-Virus FAM 31,18 1 × 104 + 0,974 90,8
Pesti.+Infl.-A-Virus FAM 35,44 1 × 103 + 0,974 90,8
Pesti.+Infl.-A-Virus FAM 38,01 1 × 102 + 0,974 90,8
Pesti.+Infl.-A-Virus FAM 39,63 1 × 101 + 0,974 90,8
Pesti.+Infl.-A-Virus HEX 20,08 1 × 106 + 0,998 69,9
Pesti.+Infl.-A-Virus HEX 23,77 1 × 105 + 0,998 69,9
Pesti.+Infl.-A-Virus HEX 28,45 1 × 104 + 0,998 69,9
Pesti.+Infl.-A-Virus HEX 32,57 1 × 103 + 0,998 69,9
Pesti.+Infl.-A-Virus HEX 37,41 1 × 102 + 0,998 69,9
Pesti.+Infl.-A-Virus HEX NoCt 1 × 101 − 0,998 69,9

200 A. Anhang
Tabelle A.13: Ergebnisse der PRRSV-EU- und PRRSV-NA-spezifischen PCRs. Die
PCRs wurden durchgefuhrt mit in Standardvektoren klonierten PCR-Produkten.
Virus Farbstoff Ct Kopienzahl/ Ansatz Ergebnis R2 Effizienz (%)
PRRSV-EU 106 kopien FAM 24,29 + 1,00×106 0,982 99,7
PRRSV-EU 105 kopien FAM 27,5 + 1,00×105 0,982 99,7
PRRSV-EU 104 kopien FAM 30,08 + 1,00×104 0,982 99,7
PRRSV-EU 103 kopien FAM 34,56 + 1,00×103 0,982 99,7
PRRSV-EU 102 kopien FAM 38,85 + 1,00×102 0,982 99,7
PRRSV-EU 101 kopien FAM 39,87 + 1,00×101 0,982 99,7
PRRSV-NA-106 kopien HEX 21,69 + 1,00×106 0,999 95,1
PRRSV-NA-105 kopien HEX 25,36 + 1,00×105 0,999 95,1
PRRSV-NA-104 kopien HEX 28,99 + 1,00×104 0,999 95,1
PRRSV-NA-103 kopien HEX 31,96 + 1,00×103 0,999 95,1
PRRSV-NA-102 kopien HEX 35,69 + 1,00×102 0,999 95,1
PRRSV-NA-101 kopien HEX 39,01 + 1,00×101 0,999 95,1

A.8.
Tab
ellenund
Abbild
ungen
201
Tabelle A.14: Ergebnisse der Bestimmung von PCR-Nachweisgrenzen im Vergleich zur Virustitration (Virusisolierung). Die Be-
stimmung erfolgte anhand einer logarithmischen Verdunnungsreihe einer Virussuspension (Stufen bezeichnet als V1 bis V8). Die
Verdunnungsstufen sowie die Originalsuspension wurden in einer Virustitration getestet und die KID50/0,1 ml (VT – Virustiter in
der Virustitration [logKID50/0,1 ml]; OVT – Virustiter der Originalsuspension [KID50/0,1 ml]) ermittelt. Parallel wurden in den
Stufen V1-V8 Genomaquivalente mittels real-time PCR ermittelt (GA – Genomaquivalente pro PCR-Ansatz [logGenomaquivalen-
te/6 µl] um sie mit KID50 (VTK – Virustiter auf PCR-Volumen korrigiert [logKID50/6 µl]) in Beziehung zu setzen. Beispielsweise
betragt beim PRRSV-NA in der Stufe V2 der Virustiter (VT) 1 logKID50/0,1 ml=10 KID50/0,1 ml. Dies entspricht 3,76 logGe-
nomaquivalente/6µl (GA) = 5754 Genomaquivalente/6µl, oder -0,22 logKID50/6 µl (VTK) = 0,6 KID50/6 µl.
V1 V2 V3 V4
OVT VT GA VTK VT GA VTK VT GA VTK VT GA VTK
SHV-1 107,25 6,25 3,84 5,02 5 3,09 3,78 4 2,25 2,78 3 1,37 1,78
PPV 107,00 5,75 5,07 4,53 4,5 4,71 3,28 4 3,95 2,78 2,75 2,8 1,53
Pestiviren 107,25 6,25 5,85 5,03 5 4,89 3,78 3,75 3,42 2,53 3,25 2,64 2,03
Influenza-A-Virus 105,25 4 5,8 2,78 3 4,53 1,78 0,25 3,47 -0,97 0 2,28 -1,22
PRRSV-EU 104,50 3,25 5,93 2,23 2 4,47 0,78 2 2,77 0,78 1 1,42 -0,22
PRRSV-NA 103,00 1,75 4,50 0,53 1 3,76 -0,22 0 2,57 -1,22 − 1,64 −
V5 V6 V7 V8
VT GA VTK VT GA VTK VT GA VTK VT GA VTK
SHV-1 2,25 − 1,03 1 0,96 -0,22 − − -1,22 0,75 − -1,35
PPV 1,75 1,77 0,53 0,75 0,53 -0,47 0,75 0,07 -0,13 − − −
Pestiviren 2,75 1,39 1,53 1 0,86 -0,22 0 − -1,22 − − −
Influenza-A-Virus − 1,29 − − 0,32 − − 0,09 − − − −
PRRSV-EU − 0,03 − − − − − − − − − −
PRRSV-NA − − − − − − − − − − − −


Abbildungsverzeichnis
2.1 Schematische Darstellung des Virus der Klassischen Schweinepest . . . . . 5
2.2 Schematische Darstellung des Pestivirusgenoms . . . . . . . . . . . . . . . 5
2.3 Genetische Diversitat der KSPV-Isolate . . . . . . . . . . . . . . . . . . . . 7
2.4 Schematische Darstellung des ASP-Virus . . . . . . . . . . . . . . . . . . . 13
2.5 Schematische Darstellung eines Herpesvirus . . . . . . . . . . . . . . . . . 16
2.6 Schematische Darstellung des PRRSV-Genoms . . . . . . . . . . . . . . . . 19
2.7 Influenza-A Virus. Schematische Darstellung viraler Proteine. Nach
Modrow et al. (2003). . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 22
2.8 Schematische Darstellung des Genoms des Porzinen Parvovirus. . . . . . . 24
2.9 Schematische Darstellung eines PCV-2 . . . . . . . . . . . . . . . . . . . . 28
2.10 Schematische Darstellung einer Amplifikationsgrafik . . . . . . . . . . . . . 33
2.11 Schematische Darstellung eines TaqMan Assays . . . . . . . . . . . . . . . 34
2.12 Prinzip des PriProET -Systems . . . . . . . . . . . . . . . . . . . . . . . . 36
2.13 Emissonsspektren verschiedener Fluoreszenzfarbstoffe . . . . . . . . . . . . 40
2.14 Das OIE-Validierungsschema . . . . . . . . . . . . . . . . . . . . . . . . . . 45
3.1 Primeroptimierungsmatrix . . . . . . . . . . . . . . . . . . . . . . . . . . . 62
4.1 Ergebnisse der gelbasierten β-Aktin PCR. . . . . . . . . . . . . . . . . . . 80
203

204 Abbildungsverzeichnis
4.2 Ermittlung von DNA-Sekundarstrukturen am Beispiel von PCV-2. . . . . . 81
4.3 Ermittlung von DNA-Sekundarstrukturen des Pestivirusgenoms. . . . . . . 82
4.4 Primertitration. Amplifikationsgraphen. . . . . . . . . . . . . . . . . . . . . 86
4.5 Primertitration. Schmelzkurven. . . . . . . . . . . . . . . . . . . . . . . . . 87
4.6 Ergebnisse der Primertitration am Beispiel der ASPV. Fluoreszenzmessung 88
4.7 Ergebnisse der Primertitration am Beispiel der ASPV. Ermittelte Ct-Werte 89
4.8 β-Aktin Primer- und Sondenlokalisation. . . . . . . . . . . . . . . . . . . . 91
4.9 β-Aktin RNA-DNA Diskriminierung. . . . . . . . . . . . . . . . . . . . . . 92
4.10 PPV PCR / Virustitration . . . . . . . . . . . . . . . . . . . . . . . . . . . 96
4.11 Influenza-A PCR / Virustitration . . . . . . . . . . . . . . . . . . . . . . . 97

Tabellenverzeichnis
3.1 SHV-1 Isolate. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 49
3.2 Influenza-A – Isolate. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 50
3.3 Pestiviren-Isolate . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 51
3.4 Reverse Transkription, thermisches Profil . . . . . . . . . . . . . . . . . . . 58
3.5 Einstellungen und Parameter der Primer- und Sondensuche im Beacon De-
signer 2.13 . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 60
3.6 Einstellungen und Parameter fur mFold . . . . . . . . . . . . . . . . . . . . 60
3.7 Abgene HotStart PCR. Thermisches Profil . . . . . . . . . . . . . . . . . . 61
3.8 SYBRTM-Green PCR. Thermisches Profil . . . . . . . . . . . . . . . . . . . 63
3.9 TaqMan real-time PCR. Zusammensetzung der PCR-Ansatze . . . . . . . 64
3.10 Kolonie-PCR. Zusammensetzung des Mastermixes . . . . . . . . . . . . . . 67
3.11 Kolonie-PCR. Thermische Profil . . . . . . . . . . . . . . . . . . . . . . . . 68
3.12 Restriktionsenzymverdau . . . . . . . . . . . . . . . . . . . . . . . . . . . . 70
4.1 Ergebnisse der Literaturrecherche . . . . . . . . . . . . . . . . . . . . . . . 74
4.2 TaqMan-PCR. Thermisches Profil. . . . . . . . . . . . . . . . . . . . . . . 78
4.3 Ubersicht der eingesetzten Genomregionen und Sequenzen . . . . . . . . . 83
205

206 Tabellenverzeichnis
4.4 Die experimentell ermittelten optimalen Primer- und Sondenkonzentratio-
nen. Die Primer- und Sondenkonzentrationen fur die PRRSV-PCR wurden
aus dem Protokoll von Kleiboeker et al. (2005) ubernommen. . . . . . . 90
4.5 Zusammensetzung einzelner real-time PCR-Ansatze im Gesamtkonzept . . 93
4.6 Ergebnisse der Untersuchung KSPV-positiver sowie -negativer Proben. . . 101
4.7 Ergebnisse der Untersuchung klinischer Proben. . . . . . . . . . . . . . . . 102
4.7 (Fortsetzung) . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 103
A.1 Ergebnisse der ASPV-PCR-Primertitrationen . . . . . . . . . . . . . . . . 188
A.2 Ergebnisse der SHV-1-PCR-Primertitrationen . . . . . . . . . . . . . . . . 189
A.3 Ergebnisse der PCV-2-PCR-Primertitrationen . . . . . . . . . . . . . . . . 190
A.4 Ergebnisse der PPV-PCR-Primertitrationen . . . . . . . . . . . . . . . . . 191
A.5 Ergebnisse der Pestivirenspezifischen-PCR-Primertitrationen . . . . . . . . 192
A.6 Ergebnisse der Influenza-A-PCR-Primertitrationen . . . . . . . . . . . . . 193
A.7 Ergebnisse der pestivirusspezifischen PCR . . . . . . . . . . . . . . . . . . 194
A.8 Ergebnisse der Influenza-A-spezifischen PCR . . . . . . . . . . . . . . . . . 195
A.9 Ergebnisse der SHV-1-spezifischen PCR . . . . . . . . . . . . . . . . . . . . 196
A.10 Ergebnisse der ASPV- und SHV-1-spezifischen PCRs . . . . . . . . . . . . 197
A.11 Ergebnisse der PCV-2- und PPV-spezifischen PCRs . . . . . . . . . . . . . 198
A.12 Ergebnisse der Pestiviren- und Influenza-A-spezifischen PCRs . . . . . . . 199
A.13 Ergebnisse der PRRSV-EU- und PRRSV-NA-spezifischen PCRs . . . . . . 200
A.14 Ergebnisse der Bestimmung von PCR-Nachweisgrenzen im Vergleich zu
Virustitration (Virusisolierung). . . . . . . . . . . . . . . . . . . . . . . . . 201

Veroffentlichungen und Tagungsbeitrage
Teile der vorliegenden Arbeit wurden auf folgenden Tagungen prasentiert:
Gavrilenko A., Meindl-Bohmer A., Greiser-Wilke I., Moennig V.
Development of a real-time multiplex PCR assay for differential diagnosis of classical
swine fever (CSF)
In: 6th Pestivirus Symposium, Thun, Schweiz, September 13–16, 2005
Gavrilenko A.
Workshop on classical swine fever, Hannover, 10–13 October 2005
Kurzvortrag: Differential diagnosis of CSF – laboratory diagnostic approaches

Danksagung
Mein besonderer Dank gilt Herrn Prof. Dr. V. Moennig fur die freundliche Aufnahme im
Institut fur Virologie, die Uberlassung des Themas, die wissenschaftliche Betreuung der
Arbeit sowie die stete Unterstutzung und Gesprachsbereitschaft.
Frau Apl. Prof. Dr. I. Greiser-Wilke fur ihre konstruktiven Vorschlage, stetige wissen-
schaftliche Forderung und die jederzeit gewahrte Unterstutzung.
Dem Institut fur Parasitologie, insbesondere Herrn Prof. Dr. G. von Samson und Frau S.
Buschbaum danke ich fur die Bereitstellung des real-time PCR Gerates und die Einarbei-
tung in die Technik.
Frau Dr. A. Meindl-Bohmer mochte ich fur die fachlichen Diskussionen und vielfaltigen
Hilfestellungen danken.
Frau Prof. Dr. B. Grummer mochte ich fur die fachlichen Diskussionen und vielfaltigen
Anregungen danken.
Sehr herzlich danke ich allen Mitarbeitern des Instituts fur Virologie, insbesondere Frau
I. Betsch, Frau M. Giesecke, Frau G. Muller fur ihre freundliche Aufnahme und Hilfsbe-
reitschaft.
Allen meinen Mitdoktoranden, insbesondere Sandra Blome danke ich fur ihre Diskussi-
onsbereitschaft, die Korrektur der schriftlichen Arbeit und die moralische Unterstutzung.









