formation of fullerene molecules from carbon nanotubes: a quantum chemical molecular dynamics...
TRANSCRIPT

Formation of Fullerene Molecules fromCarbon Nanotubes: A QuantumChemical Molecular Dynamics StudyStephan Irle,† Guishan Zheng,† Marcus Elstner,‡,§ and Keiji Morokuma*,†
Cherry L. Emerson Center for Scientific Computation and Department of Chemistry,Emory UniVersity, Atlanta, Georgia 30322, UniVersitat Paderborn, FachbereichPhysik, 33095 Paderborn, Germany, and Deutsches Krebsforschungszentrum,Abteilung Molekulare Biophysik, Im Neuenheimer Feld 280,69120 Heidelberg, Germany
Received January 13, 2003; Revised Manuscript Received February 14, 2003
ABSTRACT
The first quantum chemical molecular dynamics calculations for the formation of fullerene molecules from carbon nanotubes are presented.The use of quantum chemical potential is shown to be essential to describe important effects of π-conjugation in fullerene chemistry. Trajectoriesfor open-ended carbon nanotubes with various tube lengths were run at 1000−5000 K, using the density functional tight-binding potential. Attemperatures around 3000 K, extremely fast fullerene formation was observed in about 14 ps and shorter.
Although fullerenes have been around for some time, themechanism of their formation still remains unknown andconstitutes one of the most challenging topics in fullerenechemistry. The conditions under which fullerene moleculesare formed are very harsh and involve either vaporizationof graphite by laser1 or combustion of hydrocarbon moleculesin oxygen-rich diffusion flames under low pressure.2,3 In theprocess only reaction products have been identified, andintermediate species with potentially helpful information forunderstanding of the formation mechanism have not beendetected. The abundance of closed carbon cage structuresin the products (up to 40%) clearly indicates that there mustexist some mechanisms of building highly organized fullerenesand nanotubes from small carbon fragments. The mostimportant structural requirement is known to be the introduc-tion of pentagons into the hexagon lattice for graphitestructures to develop curvature.
Over the years, a plethora of fullerene growth mechanismshave appeared in the literature to rationalize how highlystructured, chemically almost inert species such as fullerenemolecules can emerge from a chaotic mixture of small carbonfragments in surprising abundance. Among the most promi-nent are the “pentagon road,”4 the “fullerene road,”5 the“ring-stacking” mechanism,6 and the “ring fusion spiralzipper” mechanism.7,8 While these mechanisms are more orless guesswork and rely on different assumptions, they all
share an underlying principal of order by which fullerenemolecules are either constructed stepwise from well-definedsmaller carbon fragments or spontaneously formed by thecollapse of a highly preorganized structure.
Quantum mechanical (QM) electronic structure calcula-tions have been performed on numerous hypothetical inter-mediate structures,9-12 and even an attempt has been madeto locate transition states connecting intermediate structuresand describing entire pathways for the formation of the C28
fullerene molecule starting from small rings such as C9 andC13.13 It is, however, more than questionable whether anorderly growth process along a single or multiple reactionpathways with well-defined intermediate species can beassumed to take place under the high-temperature nonequi-librium conditions during which fullerene molecules areproduced. The high-temperature conditions of the formationprocesses allow the carbon clusters to climb upward on hillsof the potential energy surface, and let rapidly fluctuatingstructures play a key role in the formation mechanism.Although molecular dynamical (MD) studies for energeticsof intermediate structures along a proposed reaction pathwayhave been performed,14 direct insight into the formationmechanism cannot be gained from such studies. Reactivedynamical approaches allowing bond formation and breakingare required for this purpose.
Monte Carlo15 as well as MD studies on the formationmechanism of fullerene molecules from atomic carbon andsmall carbon clusters16-18 have been reported in the literature.Most of these studies use semiclassical reactive empirical
* Corresponding author: E-mail [email protected].† Emory University.‡ Universitat Paderborn.§ Abteilung Molekulare Biophysik.
NANOLETTERS
2003Vol. 3, No. 4
465-470
10.1021/nl034023y CCC: $25.00 © 2003 American Chemical SocietyPublished on Web 03/13/2003

bond-order (REBO) interatomic carbon-carbon molecularmechanics (MM) force field developed by Brenner for study-ing vapor deposition of diamond.19 Unlike classical MMforce fields, the REBO potential allows for the formationand dissociation of covalent chemical bonds during asimulation by determination of next neighbors and switchingbond functions. Fullerene formation was observed in MM/MD studies17,18on a nanosecond time scale at a temperatureof 3000 K.
However, because fullerenes and carbon nanotubes are allmade from sp2 hybridized atoms whereπ-conjugationaleffects are important, we strongly feel that classical orsemiclassical MM force field methods are inappropriate forreaction dynamics of all-carbon clusters. Concepts such asaromaticity and delocalization stabilization are crucial for arealistic description of conjugated carbon clusters and absentin such force fields. The influence of the electronic structureon relative stabilities and locations of bond formation orbreaking cannot be taken into account by the very nature ofthe atomic force field approach. Therefore, MD studies basedon such MM force fields cannot be even qualitatively correct.
Thus for the study of the mechanism of fullerene forma-tion, it is essential to use QM electronic structure methodsand perform molecular dynamics (QM/MD) calculations.Spontaneous capping on single ends of carbon nanotubeshas been observed in Car-Parinello type QM/MD calcula-tions;20 however, except for a discussion of relative stabilities,no analysis was attempted as to how the closure occurredand what the role of electronic structure is for individualsteps. Here we report for the first time results of QM/MDsimulations of carbon nanotubes leading to completely closedcage fullerene structures and identify key steps during thesedynamics. We believe that, even though using carbonnanotubes as initial structures is certainly somewhat artificialfor the study of fullerene formation, the knowledge of “majorplayers” emerging from our QM/MD simulations will leadto deeper understanding of general growth mechanisms incarbon nanochemistry.
We ran trajectories for a canonical ensemble with avelocity Verlet integrator, using 1.2 fs as for time step unlessotherwise noted. Temperature was kept constant by scalingof atomic velocities at random intervals plus regular intervalsof 12 fs with an overall probability of 20%. (In the case ofa (5,5) tube with 7.5 Å length at 3000 K we tested scalingof temperature at a lower rate (5%) and actually observedfaster closing of both ends, indicating that a scaling prob-ability of 20% does not artificially speed up the dynamicssimulation.) All calculations were carried out using a 30 Åcubic periodic cell. Random velocities are applied at the firststep of each trajectory, making the choice of initial geom-etries irrelevant to the dynamics. To obtain energies andgradients, all-valence electronic structure calculations werecarried out using the computationally inexpensive densityfunctional tight binding (DFTB) method.21,22 DFTB is amethod approximating density functional theory (DFT)utilizing an optimized minimal LCAO basis set in combina-tion with a two-center approximation for the Hamilton matrixelements. All parameters of the method are calculated from
DFT, and no fitting to experimental data is involved. DFTBhas been successfully used in the past to explain relativestabilities of fullerene isomers and aggregates.23-27 Orbitaloccupation numbers were determined for each time step;when energy differences are smaller than 10-4 a.u., orbitalsare considered to be degenerate and electrons are distributedbased on the Hund rule. While total energies are not affectedby the way that electrons are distributed among degenerateorbitals, gradients depend on the spatial distribution of thewave function.
It is well known that neutral carbon clusters adapt differentconfigurations upon cluster growth. Cn clusters withn e 5prefer linear cumulene-type structures, and in the intermedi-ate regime of 6e n < 18, monocyclic isomers are preferred,before fullerenoid structures become the most stable speciesfor n e 18.11,28 In contrast to the semiempirical AM129 andPM330 methods, DFTB is capable of reproducing thesecrucial configuration preferences for carbon clusters and morein line with the full DFT calculations such as B3LYP/6-31G(d).31
We chose to run MD trajectories starting with open-endedcarbon nanotubes of different chirality and lengths at varioustemperatures. In total, we ran more than 70 trajectories, mostof them for at least 12 ps. The low temperature 1000 Kregime was nonreactive and the high-temperature regimeabove 4000 K led to fragmentation. Three different types of(n,m) nanotubes were chosen with about the same diameterd: armchair (5,5),d ) 6.88 Å, chiral (7,3),d ) 7.15 Å, andzigzag (9,0),d ) 7.06 Å. In addition, a (10,5) nanotube witha much larger diameter ofd ) 10.5 Å was studied. Threedifferent tube lengths (7.5, 10, and 20 Å) were adopted foreach species. An important finding is that the diameter ofthe (10,5) nanotube appears to be too large to allow itsopenings to be closed within 12 ps, regardless of the tubelength.
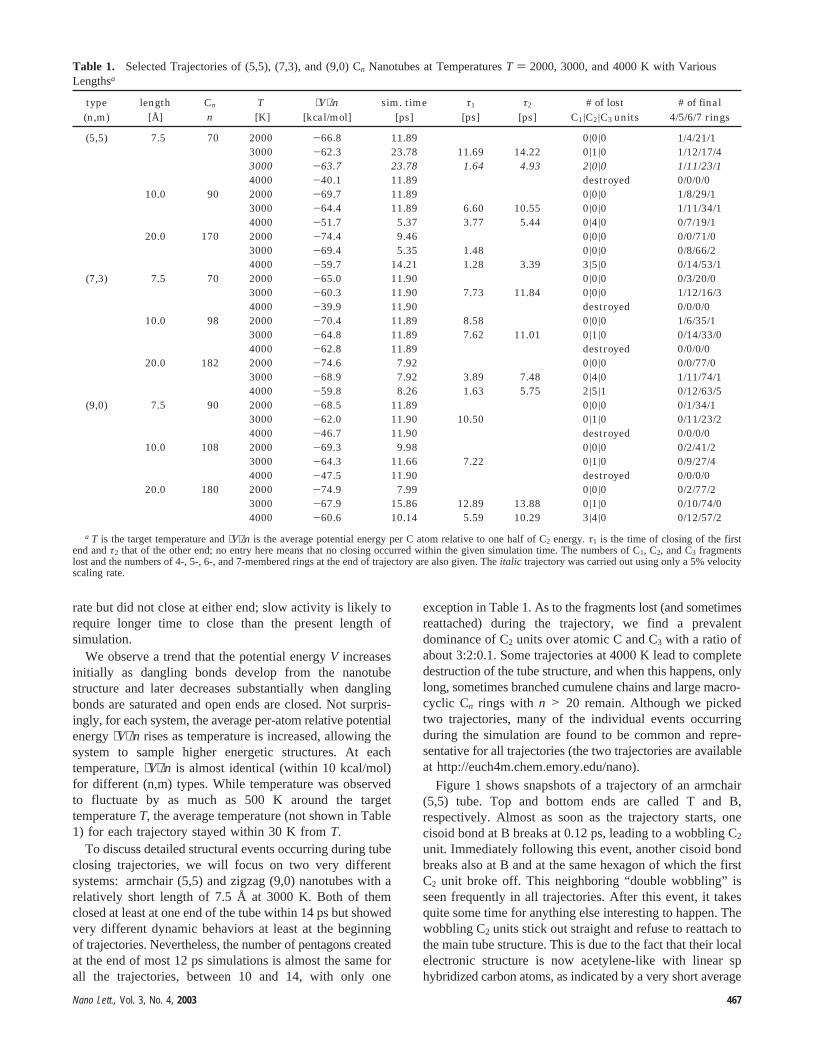
Table 1 gives an overview of trajectories at 2000, 3000,and 4000 K (omitting our 2500 and 3500 K simulations forbrevity) for (5,5), (7,3), and (9,0) open nanotubes. We callthe end of a tube “closed” when the largest rings present inthe opening involve not more than 8 carbon atoms. Ameasure of 1.8 Å was used as a threshold for bondedinteraction. Since only one or a few trajectories were runfor each case, we make no claims on the statistical validityof the findings. However, we want to extract importantdynamic events common to many of these trajectories. Themost striking finding of the present study is that manytrajectories at 3000 and 4000 K closed both ends and formedfullerene structures within 14 ps. The time required forclosure does not seem to depend on the length of the tube.As will be described in detail later, the key dynamics is aninterplay between wobbling C2 fragments, which are formedat the ends of the tube and occasionally catch neighboringhexagons to form pentagons, and creation, migration, andisomerization of pentagons. Trajectories at 4000 K, comparedto 3000 K, show higher C2 formation activities and tend toclose faster, accompanied with loss of C1 to C3 fragments.Trajectories at 2000 K also formed wobbling bonds at lower
466 Nano Lett., Vol. 3, No. 4, 2003
rate but did not close at either end; slow activity is likely torequire longer time to close than the present length ofsimulation.
We observe a trend that the potential energyV increasesinitially as dangling bonds develop from the nanotubestructure and later decreases substantially when danglingbonds are saturated and open ends are closed. Not surpris-ingly, for each system, the average per-atom relative potentialenergy⟨V⟩/n rises as temperature is increased, allowing thesystem to sample higher energetic structures. At eachtemperature,⟨V⟩/n is almost identical (within 10 kcal/mol)for different (n,m) types. While temperature was observedto fluctuate by as much as 500 K around the targettemperatureT, the average temperature (not shown in Table1) for each trajectory stayed within 30 K fromT.
To discuss detailed structural events occurring during tubeclosing trajectories, we will focus on two very differentsystems: armchair (5,5) and zigzag (9,0) nanotubes with arelatively short length of 7.5 Å at 3000 K. Both of themclosed at least at one end of the tube within 14 ps but showedvery different dynamic behaviors at least at the beginningof trajectories. Nevertheless, the number of pentagons createdat the end of most 12 ps simulations is almost the same forall the trajectories, between 10 and 14, with only one
exception in Table 1. As to the fragments lost (and sometimesreattached) during the trajectory, we find a prevalentdominance of C2 units over atomic C and C3 with a ratio ofabout 3:2:0.1. Some trajectories at 4000 K lead to completedestruction of the tube structure, and when this happens, onlylong, sometimes branched cumulene chains and large macro-cyclic Cn rings with n > 20 remain. Although we pickedtwo trajectories, many of the individual events occurringduring the simulation are found to be common and repre-sentative for all trajectories (the two trajectories are availableat http://euch4m.chem.emory.edu/nano).
Figure 1 shows snapshots of a trajectory of an armchair(5,5) tube. Top and bottom ends are called T and B,respectively. Almost as soon as the trajectory starts, onecisoid bond at B breaks at 0.12 ps, leading to a wobbling C2
unit. Immediately following this event, another cisoid bondbreaks also at B and at the same hexagon of which the firstC2 unit broke off. This neighboring “double wobbling” isseen frequently in all trajectories. After this event, it takesquite some time for anything else interesting to happen. Thewobbling C2 units stick out straight and refuse to reattach tothe main tube structure. This is due to the fact that their localelectronic structure is now acetylene-like with linear sphybridized carbon atoms, as indicated by a very short average
Table 1. Selected Trajectories of (5,5), (7,3), and (9,0) Cn Nanotubes at TemperaturesT ) 2000, 3000, and 4000 K with VariousLengthsa
type(n,m)
length[Å]
Cn
nT[K]
⟨V⟩/n[kcal/mol]
sim. time[ps]
τ1
[ps]τ2
[ps]# of lost
C1|C2|C3 units# of final
4/5/6/7 rings
(5,5) 7.5 70 2000 -66.8 11.89 0|0|0 1/4/21/13000 -62.3 23.78 11.69 14.22 0|1|0 1/12/17/43000 -63.7 23.78 1.64 4.93 2|0|0 1/11/23/14000 -40.1 11.89 destroyed 0/0/0/0
10.0 90 2000 -69.7 11.89 0|0|0 1/8/29/13000 -64.4 11.89 6.60 10.55 0|0|0 1/11/34/14000 -51.7 5.37 3.77 5.44 0|4|0 0/7/19/1
20.0 170 2000 -74.4 9.46 0|0|0 0/0/71/03000 -69.4 5.35 1.48 0|0|0 0/8/66/24000 -59.7 14.21 1.28 3.39 3|5|0 0/14/53/1
(7,3) 7.5 70 2000 -65.0 11.90 0|0|0 0/3/20/03000 -60.3 11.90 7.73 11.84 0|0|0 1/12/16/34000 -39.9 11.90 destroyed 0/0/0/0
10.0 98 2000 -70.4 11.89 8.58 0|0|0 1/6/35/13000 -64.8 11.89 7.62 11.01 0|1|0 0/14/33/04000 -62.8 11.89 destroyed 0/0/0/0
20.0 182 2000 -74.6 7.92 0|0|0 0/0/77/03000 -68.9 7.92 3.89 7.48 0|4|0 1/11/74/14000 -59.8 8.26 1.63 5.75 2|5|1 0/12/63/5
(9,0) 7.5 90 2000 -68.5 11.89 0|0|0 0/1/34/13000 -62.0 11.90 10.50 0|1|0 0/11/23/24000 -46.7 11.90 destroyed 0/0/0/0
10.0 108 2000 -69.3 9.98 0|0|0 0/2/41/23000 -64.3 11.66 7.22 0|1|0 0/9/27/44000 -47.5 11.90 destroyed 0/0/0/0
20.0 180 2000 -74.9 7.99 0|0|0 0/2/77/23000 -67.9 15.86 12.89 13.88 0|1|0 0/10/74/04000 -60.6 10.14 5.59 10.29 3|4|0 0/12/57/2
a T is the target temperature and⟨V⟩/n is the average potential energy per C atom relative to one half of C2 energy.τ1 is the time of closing of the firstend andτ2 that of the other end; no entry here means that no closing occurred within the given simulation time. The numbers of C1, C2, and C3 fragmentslost and the numbers of 4-, 5-, 6-, and 7-membered rings at the end of trajectory are also given. Theitalic trajectory was carried out using only a 5% velocityscaling rate.
Nano Lett., Vol. 3, No. 4, 2003 467

C-C bond distance of only about 1.22 Å. Their linearconfiguration prevents them from coming close to the rimsof the tube opening, and therefore they wobble relativelyindependent with a long lifetime. At 0.49 ps another cisoidbond breaks at the opposite T end of the tube, and at 0.94ps the very same pattern of neighboring bond breaking occurshere as was observed previously at the B end. At 1.11 ps, awobbling C2 unit at T bridges to a next neighbored hexagon,giving rise to a pentagon structure with one carbon atomattached to its top. However, this kind of five-membered ringis relatively short-lived for just 0.36 ps before it breaks againand the C2 unit reappears. Creation and destruction of thispentagon in this way occurs repeatedly afterward, and at 2.52ps we notice pentagon formation with one carbon atomattached at the top at the B end. Both hexagons adjacent tothis pentagon have wobbling C2 units attached to them(“double wobbling”), making the formation of a heptagonpossible with the atom on top of the pentagon in the center.At 2.64 ps, one of them catches the carbon atom at the topof a pentagon, and a fused 5- and 7-membered rings(hereafter called a 5/7 fused ring) is formed which provesto be relatively stable. The 5/7 ring combination producedin this way continues to occur frequently, and one of theircharacteristics is that the 5/7 fused bond separating pentagonand heptagon is long and weak, giving this structure a largerflexibility than a regular hexagon lattice. Reisomerizationof the 5/7 fused ring system to two hexagons (often calledStones-Wales32 isomerization) is observed especially attemperatures 3000 K or below. At 5.35 ps, the T end hasfour wobbling C2 units, while the B end is completely healedback to that of a regular armchair tube. Shortly after, at 5.98ps, two independent T-C2 units reattach to the opening rim,and again a pentagon with a single wobbling carbon atomat its top is formed. This time, this carbon atom bends overthe next-neighbored four carbon atoms to its left, and two
adjacent pentagons are formed in a very short time at 6.11ps. Almost immediately, the pentagon on the right side ofthe figure separates itself from its pentagon neighbor bymigration through a hexagon, as shown in the structure at6.23 ps. Three 5/7 combinations appear at 6.88 ps, one at Tand two at B, with two of them displaying a stretchedcarbon-carbon bond at their 5/7 junctions, making themmore look like a 10-membered ring. The tube has beenrotated in the figure to indicate how far the 5/7, 10-memberedcyclic structure reached inside the tube opening, withwobbling C2 units around it. At 9.48 ps, a C2 unit attachesto a nearby 7-membered ring, leading to a new heptagonand the first 7/7 junction of this simulation. This effectivelyreduces the number of members in the macrocyclic opening,which is consequently reduced in size to a 12-membered ring.The structure at 11.39 ps shows a system which only containsthree 12-membered rings as openings, and two fused 12-membered rings are visible in the front of the picture whichwere created shortly before by a wobbling C2 unit acting asa “bridge”. Another C2 unit seemingly acts as a bystanderwhile being attached to one of the 12-membered rings. Thesnapshot taken at 11.42 shows how simultaneous collapseof a 12-membered ring into a 5/6/5 combination occurs bya [2+4] cycloaddition-like formation of twoσ bonds. Thisring collapse causes the adjacent 12-membered ring to adapt,and very shortly afterward at 11.60 ps, two carbon atomson opposite sides of the macrocycle come close to each other,leading to the formation of a pentagon and heptagon withthe C2 unit now wobbling outside the cage structure. Thisconstitutes end closure, which occurred within 0.2 ps fromthe simultaneous collapse of two fused 12-membered rings.Subsequently, it takes about 1.4 ps before the wobbling C2
unit is lost. The other end closes at 13.74 ps, when the 12-membered ring structure reemerges from a 14-macrocycleplus wobbling C2 unit and repeating the [2+4] cycloadditionstyle collapse into a 5/6/5 combination of the other end,leaving an almost perfect C68 fullerene molecule with 12pentagons, 4 heptagons, and 1 four-membered ring.
Figure 2 illustrates a trajectory of a 7.5 Å long (9,0) zigzagtube also at 3000 K. In contrast to the armchair case wherethe first C2 unit was created within 0.12 ps, it takes 1.70 psfor the zigzag tube to develop a wobbling C2 unit. Stone-Wales type 5/7 pair formation and reisomerization is thedominant pattern generally observed for the first 2 ps ofzigzag tubes at 3000 K, indicating that the polyacetylenicrim of the zigzag tube openings is much more stable againstbond cleavage than the armchair tube openings. The wob-bling C2 unit is created by bond cleavage of a 5/7 heptagonat the T end. Once the rim of a zigzag tube has been brokenopen at a heptagon, next-neighbored hexagons become morelikely to break because of lack ofπ-conjugational stabiliza-tion. The structure at 2.04 ps depicts such a situation.Occasionally it is observed that wobbling C2 units break off,here after 2.66 ps. Upon loss of C2, a next-neighboredwobbling C2 unit attacks at the break-off point, giving riseto a hexagon and a pentagon at 2.93 ps. Two fused pentagonsare created at 3.72 ps at B as a result of heptagonisomerization toward a pentagon with wobbling C2 formation.
Figure 1. Twelve representative snapshots of a trajectory for a(5,5) tube with 7.5 Å at 3000 K. Carbon-carbon bonds are drawnwith a threshold of 1.8 Å.
468 Nano Lett., Vol. 3, No. 4, 2003

In this case, they isomerize and reisomerize into a 5/7combination by incorporation of the attached C2 unit repeat-edly as shown for 6.05 ps. At this time, no hexagon survivedat either tube opening, which is now completely dominatedby 5/7 ring combinations. Eventually, at 8.04 ps, three fusedpentagons with a heptagon attached are developed at thesame end, now rotated in the figure for better clarity. Here,the 5/7 combination develops into a 10-membered cyclicstructure reaching into the opening similar as in the armchairtube case. The three fused pentagons are responsible for agreater curvature in the opening, and at 8.95 ps a heptagonis created in the opening by attaching a C2 unit across thestrongly curved polypentagon system. Eventually restructur-ing and separation of the pentagons by hexagons occur, andtwo pentagons with attached hexagons enforce a curvaturestrong enough to form a bond over the opening. This leadsfinally to a 6-membered ring with an 8-membered ringattached to it, closing this end at 10.50 ps.
The DFTB MD simulations described above give anexcellent insight into key elements involved in converting athree-dimensionally curved hexagon cluster of carbon atomsto closed cage structures with a high pentagon-to-hexagonratio at the open ends. To summarize, the key dynamic stepsare:
1. Armchair-type openings show a high tendency towardbreaking of cisoid bonds, forming many wobbling C2 units,whereas C2 units are formed an order of magnitude slowerfor zigzag type openings as a result of bond breaking inheptagons, preceded by a large number of 6/6 to 5/7 ringisomerizations.
2. Wobbling C2 units have a high lifetime and have atendency to be created pairwise at same hexagons (“doublewobbling”). They show acetylene-type electronic structuresand stand out upright from the rim of the opening, withoccasional approach of next neighbored rings to formpentagons or other rings.
3. Once a structural defect such as C2 creation occurs,π-delocalization in that region of the opening is decreased,
and more bonds are likely to break, giving rise to more C2
units and subsequent reorganization steps.4. The 5/7 fused 10-membered ring systems show a high
flexibility; especially the 5/7 fused bond is fragile and oftenbreaks in high-temperature conditions. This gives rise to largebridge-like structures, reaching into the openings and provid-ing a chance for attack of C2 units and other defects fromthe opposite side of the opening.
5. [2+4] cycloaddition “zipper” type reactions occur infinal stages of closing when the opening size has beenreduced and only consists of 12-membered rings. Thecycloaddition reaction in this case leads spontaneously tothe creation of 2 pentagons and 1 hexagon.
6. Pentagons are often formed by 5/7 ring conversions into5/5 + C2. When multiple fused pentagons are created, astrongly deformed region is created at the opening andstabilizes itself by bond formation across the two furthermostends, which gives rise to a hexagon in the opening that canthen attach itself to the opposite side of the opening, leadingto closing.
The MD calculations described above represent simula-tions in the context of fullerene formation in which for thefirst time quantum mechanical potential is used. Open-endednanotubes were successfully converted to fullerene moleculeswithin 10 to 14 ps. We started with (5,5), (7,3), (9,0), and(10,5) open-ended nanotubes of different lengths. Temper-ature is the most important factor. Above 4000 K, the systemsfall apart into fragments. At around 3000 K, the hexagonsof the nanotube ends open and create acetylenic C2 unitsthat wobble at the end of the openings. Such wobbling C2
units have never been observed in MM simulations (see, e.g.,refs 17 and 18) but are the most essential building tool inrestructuring of the tube opening. Some of them recombineand form pentagons which cause the open ends to curl inside.At the curled end, a wobbling C2 chain suddenly binds tothe other side of curl and forms a new ring, narrowing theopening. This occurs repeatedly and preferably at 5/7 ringcombinations with broken fused inter-ring bonds, forming abridge-like structure in the opening. Eventually the openingswill close by systematically reducing the opening size bybridging with C2 units, leaving octagons as the largest cyclicstructures observable at the ends. When this happens at bothends of the nanotubes, the tube is fully closed and a fullerenemolecule is formed. Between 2000 and 3000 K we observedsome, however, less frequent C2 wobbling chain formation,and the tube opening cannot be closed within the givensimulation time. Presumably it takes much longer to closethe nanotubes at this temperature. Around 1000 K, C2
wobbling chain formation and 5/7 isomerization at the openends are very rare, and the nanotube seems to be stableagainst deformations of the hexagonal lattice in the timeframe of several picoseconds. Under experimental conditions,however, fullerene formation occurs typically between 1000and 1500 K, indicating that a simulation time of severalpicoseconds is not sufficient to follow rearrangement pro-cesses in this temperature region.
In conclusion, we expect our findings to be applicable forgeneral carbon clusters with a high degree of curvature,
Figure 2. Twelve representative snapshots of a trajectory for a(9,0) tube with 7.5 Å at 3000 K. Only one end closed. Carbon-carbon bonds are drawn with a threshold of 1.8 Å.
Nano Lett., Vol. 3, No. 4, 2003 469

which is a prerequisite for the final stages of fullereneformation. How such preorganized systems are created isstill an open question, which we are confident to demystifyby future DFTB MD simulations.
Acknowledgment. This work was partially supported bya grant from the Mitsubishi Chemical Corporation and fromthe Petroleum Research Fund, the American ChemicalSociety. Acknowledgment is made to the Cherry L. EmersonCenter of Emory University for the use of its resources,which is in part supported by a National Science Foundationgrant (CHE-0079627) and an IBM Shared University Re-search Award. We also thank the National Center forSupercomputing Applications (NCSA) for valuable computertime.
References
(1) Kratschmer, W.; Lamb, L. D.; Fostiropoulos, K.; Huffman, D. R.Nature1990, 347, 354-358.
(2) Gerhardt, P.; Lo¨ffler, S.; Homann, K.-H.Chem. Phys. Lett.1987,137, 306-310.
(3) Howard, J. B.; McKinnon, J. T.; Makarovsky, Y.; Lafleur, A. L.;Johnson, M. E.Nature1991, 352, 139-141.
(4) Haufler, R. E.; Chai, Y.; Chibante, L. P. F.; Conceicao, J.; Jin, C.;Wang, L.-S.; Maruyama, S.; Smalley, R. E.Mater. Res. Soc. Symp.Proc. 1991, 206, 627-637.
(5) Heath, J. R.ACS Symp. Ser.1991, 481, 1-23.(6) Wakabayashi, T.; Shiromaru, H.; Kikuchi, K.; Achiba, Y.Chem. Phys.
Lett. 1993, 201, 470-474.(7) Helden, G.Nature1993, 363, 60-63.(8) Hunter, X.Science1993, 260, 784-786.(9) Strout, D. L.; Scuseria, G. E.J. Phys. Chem.1996, 100, 6492-
6498.(10) Bates, K. R.; Scuseria, G. E.J. Phys. Chem. A1997, 101, 3038-
3042.(11) Jones, R. O.J. Chem. Phys.1999, 110, 5189-5200.
(12) Portmann, S.; Galbraith, J. M.; Schaefer, H. F.; Scuseria, G. E.; Lu¨thi,H. P. Chem. Phys. Lett.1999, 301, 98-104.
(13) Mishra, R. K.; Lin, Y.-T.; Lee, S.-L.J. Chem. Phys.2000, 112,6355-6364.
(14) Hua, X.; Cagin, T.; Che, J.; Goddard, W. A., III.Nanotechnology2000, 11, 85-88.
(15) Schweigert, V. A.; Alexandrov, A. L.; Morokov, Y. N.; Bedanov,V. M. Chem. Phys. Lett.1995, 235, 221-229.
(16) Chelikowsky, J. R.Phys. ReV. B 1992, 45, 12062-12070.(17) Yamaguchi, Y.; Maruyama, S.Chem. Phys. Lett.1998, 286,
336-342.(18) Maruyama, S.; Yamaguchi, Y.Chem. Phys. Lett.1998, 286,
343-349.(19) Brenner, D. W.Phys. ReV. B 1990, 42, 9458-9471.(20) Charlier, J.-C.; De Vita, A.; Blase, X.; Car, R.Science1997, 275,
646-649.(21) Porezag, D.; Frauenheim, T.; Ko¨hler, T.; Seifert, G.; Kaschner, R.
Phys. ReV. B 1995, 51, 12947-12957.(22) Seifert, G.; Porezag, D.; Frauenheim, T.Int. J. Quantum Chem.1996,
58, 185-192.(23) Fowler, P. W.; Heine, T.; Mitchell, D.; Orlandi, G.; Schmidt, R.;
Seifert, G.; Zerbetto, F.J. Phys. Chem.1996, 100, 6984-6991.(24) Fowler, P. W.; Heine, T.; Mitchell, D.; Orlandi, G.; Schmidt, R.;
Seifert, G.; Zerbetto, F.J. Chem. Soc., Faraday Trans.1996, 92,2203-2210.
(25) Jungnickel, G.; Porezag, D.; Ko¨hler, T.; Frauenheim, T.; Pedersen,M. R. In Fullerenes and Fullerene Nanostructures; Kuzmany, H.,Ed.; World Scientific: Singapore: Kirchberg, Austria, 1996; pp305-318.
(26) Domene, M. C.; Fowler, P. W.; Mitchell, D.; Seifert, G.; Zerbetto,F. J. Phys. Chem. A1997, 101, 8339-8344.
(27) Fowler, P. W.; Heine, T.; Rogers, K. M.; Sandall, J. P. B.; Seifert,G.; Zerbetto, F.Chem. Phys. Lett.1999, 300, 369-378.
(28) Martins, J. L.; Reuse, F. A.Condens. Matter Theor.1998, 13,355-362.
(29) Dewar, M. J. S.; Zoebisch, E. G.; Healy, E. F.; Stewart, J. J. P.J.Am. Chem. Soc.1985, 107, 3902-3909.
(30) Stewart, J. J. P.J. Comput. Chem.1989, 10, 209-220.(31) Zheng, G.; Elstner, M.; Irle, S.; Morokuma, K., to be published.(32) Stone, A. J.; Wales, D. J.Chem. Phys. Lett.1986, 128, 501-503.
NL034023Y
470 Nano Lett., Vol. 3, No. 4, 2003