role of plasmacytoid dendritic cells and other accessory ... · role of plasmacytoid dendritic...
TRANSCRIPT

Role of plasmacytoid dendritic cells and other accessory cells in the
activation of human natural killer cells by herpes simplex virus type 1
Die Rolle plasmazytoider dendritischer Zellen und anderer akzessorischer
Zellen in der Aktivierung humaner natürlicher Killer-Zellen durch
Herpes-Simplex-Virus-1
Der Naturwissenschaftlichen Fakultät
der Friedrich-Alexander-Universität
Erlangen-Nürnberg
zur
Erlangung des Doktorgrades Dr. rer. nat.
vorgelegt von
Karin Petra Vogel
aus Nürnberg

Als Dissertation genehmigt von der Naturwissenschaftlichen Fakultät
der Friedrich-Alexander-Universität Erlangen-Nürnberg
Tag der mündlichen Prüfung 23.01.2015
Vorsitzender des Promotionsorgans Prof. Dr. Jörn Wilms
Gutachter Prof. Dr. Barbara Schmidt
Prof. Dr. Andreas Burkovski

1
Table of contents
1 Summary .......................................................................................................................... 3
1 Zusammenfassung ........................................................................................................... 4
2 Introduction ..................................................................................................................... 5
2.1 Herpes simplex virus type 1 ................................................................................ 5
2.2 Natural killer cells ............................................................................................... 7
2.3 Plasmacytoid dendritic cells .............................................................................. 10
2.4 Mononuclear phagocytes ................................................................................... 12
2.5 Interactions of PDC and NK cells in HSV infection ......................................... 15
3 Rationale ........................................................................................................................ 16
4 Materials and Methods ................................................................................................. 17
4.1 Materials ............................................................................................................ 17
4.1.1 Instruments .......................................................................................... 17
4.1.2 Consumables ........................................................................................ 18
4.1.3 Reagents .............................................................................................. 19
4.1.4 Software .............................................................................................. 20
4.1.5 Commercial Kits .................................................................................. 20
4.1.6 Cell Culture ......................................................................................... 20
4.1.7 Viruses ................................................................................................. 21
4.1.8 Media and Buffers ............................................................................... 21
4.1.9 Antibodies ............................................................................................ 22
4.1.10 Isotype Controls ................................................................................... 24
4.2 Methods ............................................................................................................. 25
4.2.1 Isolation of primary human cells ......................................................... 25
4.2.2 Determination of cell numbers ............................................................ 26
4.2.3 Herpes simplex virus type 1 stocks ..................................................... 27
4.2.4 PDC supernatants ................................................................................ 28
4.2.5 Stimulation and infection of cells with HSV-1 ................................... 29
4.2.6 FACS analysis of cells ......................................................................... 30
4.2.7 Determination of secreted cytokines within supernatants ................... 31
4.2.8 Quantification of HSV-1 DNA ............................................................ 34

2
4.2.9 Virological analysis of hyperproliferative lesions ............................... 34
4.2.10 Statistical analysis ............................................................................... 34
5 Results ............................................................................................................................ 35
5.1 Stimulation of PBMC with HSV-1 leads to NK cell activation ........................ 35
5.2 Only infectious HSV-1 induces NK cell effector functions .............................. 38
5.3 HSV-1 activates NK cells in part via type I IFN induction .............................. 40
5.4 TNF- plays a major role in HSV-1-induced NK cell activation ..................... 43
5.5 Monocytes contribute to HSV-1-induced TNF- production ........................... 47
5.6 Monocytes can be infected by HSV-1 ............................................................... 49
5.7 Monocytes up-regulate MHC-I upon exposure to infectious HSV-1 ............... 51
5.8 HSVd106S affects monocytes similar to HSVGFP ................................................ 56
5.9 Monocytes mediate NK cell effector functions upon HSV-1 infection within the
PBMC context ............................................................................................................. 59
5.10 PDC serve as crucial accessory cell population in NK cell activation by HSV-1-
infected HFF ................................................................................................................ 61
5.11 PDC supernatants inhibit HSV-1 replication in HFF ........................................ 64
5.12 PDC-NK cell interactions are hampered in an HIV-1-infected woman suffering
from persisting genital ulcers ...................................................................................... 67
6 Discussion ....................................................................................................................... 70
7 Abbreviations ................................................................................................................ 85
8 References ...................................................................................................................... 88
9 Publications .................................................................................................................... 99

Summary
3
1 Summary
Herpes simplex virus type 1 (HSV-1), a member of the herpes virus family, is characterized
by a short replication cycle, high cytopathogenicity, and distinct neurotropism. Primary
infection and reactivation may cause severe diseases in immunocompetent and
immunosuppressed individuals. Since studies of human natural killer (NK) cell activation by
HSV-1 are limited, this study investigated mechanisms of NK cell activation by HSV-1 in
vitro, using sucrose gradient-purified UV-inactivated (HSVUV) and infectious (HSVINF)
HSV-1 to stimulate peripheral blood mononuclear cells (PBMC). HSVUV and HSVINF
exhibited distinct stimulatory differences: While both induced IFN- secretion within PBMC
and CD69 up-regulation on NK cells, only HSVINF caused TNF- and IL-1 secretion within
PBMC and NK cell effector functions degranulation and IFN- secretion. IFN- and TNF-
contributed to CD69 up-regulation, and TNF- proved important for IFN- secretion, as
evident from neutralization experiments. Degranulation was independent from IFN-,
TNF-, and IL-1, but dependent on monocytes, as evident from depletion experiments.
Infection experiments evidenced non-productive infection of monocytes by HSV-1,
suggesting recognition of infected monocytes by NK cells as possible cause for degranulation.
MHC-I down-regulation and MICA/MICB expression were excluded as activating signals for
NK cells. Plasmacytoid dendritic cells (PDC), however, proved to suppress HSV-1 replication
within fibroblasts via secreted cytokines. Furthermore, in case of an HIV-1-positive patient
suffering from HSV-2- and HPV-54-induced hyperproliferative lesions, stimulation of PBMC
with HSV-1 resulted in severely reduced IFN- secretion and impaired NK cell activation,
suggesting a role for hampered PDC-NK cell interactions in the patient’s disease. Altogether,
our data suggest a model in which HSV-1-stimulated PDC and monocytes activate NK cells
via IFN- and TNF-, while infection of monocytes induces NK cell effector functions via
TNF--dependent and -independent mechanisms. Furthermore, PDC inhibit HSV-1
replication within susceptible cells and therefore limit viral spread. Thus, PDC and monocytes
appear to have important bystander functions for NK cells to control viral infections.

Zusammenfassung
4
1 Zusammenfassung
Herpes-simplex-Virus-1 (HSV-1), ein Mitglied der Familie der Herpesviren, zeichnet sich
durch einen kurzen Replikationszyklus, hohe Pathogenität, und starken Neurotropismus aus.
Primärinfektion und Reaktivierung können in immunkompetenten und immunsupprimierten
Individuen schwere Krankheiten verursachen. Da es nur wenige Untersuchungen zur
Aktivierung humaner natürlicher Killer (NK)-Zellen durch HSV-1 gibt, wurden in dieser
Arbeit Mechanismen der NK-Zell-Aktivierung durch HSV-1 in vitro untersucht, wofür
mononukleäre Zellen des peripheren Bluts (PBMCs) mit über Succrosegradient
aufgereinigtem UV-inaktivierten (HSVUV) und infektiösen (HSVINF) HSV-1 stimuliert
wurden. HSVUV und HSVINF wiesen deutliche stimulatorische Unterschiede auf: Während
beide zu IFN--Sekretion in PBMCs und CD69-Hochregulierung auf NK-Zellen führten,
induzierte nur HSVINF TNF-- und IL-1-Sekretion in PBMCs sowie die NK-Zell-
Effektorfunktionen Degranulation und IFN--Sekretion. Neutralisationsversuche wiesen die
Beteiligung von IFN- und TNF- an der CD69-Hochregulierung nach, sowie die
Wichtigkeit von TNF- für die IFN--Sekretion. Die Degranulation war nicht abhängig von
IFN-, TNF- oder IL-1, sondern von Monozyten, wie Depletionsversuche zeigten.
Infektionsversuche bewiesen die nicht-produktive Infektion von Monozyten durch HSV-1,
was auf die Erkennung infizierter Monozyten durch NK-Zellen als mögliche Ursache der
Degranulation hindeutet, wobei MHC-I-Herabregulierung und MICA/MICB-Expression als
aktivierende Signale für NK-Zellen ausgeschlossen wurden. Plasmazytoide dendritische
Zellen (PDCs) unterdrückten dagegen die HSV-1-Replikation in Fibroblasten über Zytokin-
Sekretion. Im Fall einer HIV-1-positiven Patientin mit HSV-2- und HPV-54-induzierten
hyperproliferativen Läsionen resultierte die Stimulation von PBMCs mit HSV-1 in stark
verringerter IFN--Sekretion und NK-Zell-Aktivierung, was eine Rolle von verminderten
PDC-NK-Zell-Interaktionen in der Krankheit der Patientin andeutet. Unsere Daten legen ein
Modell nahe, nach dem HSV-1-stimulierte PDCs und Monozyten NK-Zellen über IFN- und
TNF- aktivieren, während die Infektion von Monozyten NK-Zell-Effektorfunktionen über
TNF--abhängige und -unabhängige Mechanismen induziert. PDCs inhibieren außerdem die
HSV-1-Replikation und dadurch eine Ausbreitung des Virus. Die Anwesenheit von PDCs und
Monozyten erscheint daher wichtig für die Kontrolle viraler Infektionen durch NK-Zellen.

Introduction
5
2 Introduction
2.1 Herpes simplex virus type 1
Herpes simplex virus type 1 (HSV-1) belongs to the family of herpesviruses and is highly
prevalent worldwide (Bernard Roizman et al., 2007b). It possesses a linear DNA genome
encoding more than 90 genes, which is enclosed by a capsid built of diverse viral capsid
proteins. The capsid itself is surrounded by the so called tegument, which consists of various
viral tegument proteins. A host cell-derived membrane containing several viral glycoproteins
envelops the viral particle (FIG. 1) (Bernard Roizman et al., 2007e). The viral DNA exists as
circular episome within the nucleus of the infected cell. Viral gene expression is organized
into three phases during replication: immediate-early or , early or and late or (Bernard
Roizman et al., 2007d).
FIG. 1. Herpes simplex virus type 1 (HSV-1) particle. The linear viral DNA genome is enclosed by the capsid
which itself is surrounded by the tegument. Both capsid and tegument are composed of viral proteins. A host
cell-derived membrane containing several viral glycoproteins (gB - gN) envelops the viral particle.
Together with HSV-2 and varicella zoster virus (VZV), HSV-1 belongs to the subfamily of
-herpesviruses and displays high cytopathogenicity, a short replication cycle and a distinct
neurotropism (Philip E.Pellett and Bernard Roizman, 2007). Primary infection and lytic
replication take place at oral or genital mucocutaneous sites. From there, viral particles are
transported along peripheral sensory nerves to the trigeminal or dorsal root ganglia, where
HSV-1 establishes lifelong latency. After reactivation viral particles are transported back to
the primary infection site, where lytic replication leads to viral shedding and potentially but
not necessarily to disease (Bernard Roizman et al., 2007c). Common symptoms of

Introduction
6
reactivation are cold sores and genital herpes. In rare cases, however, reactivation of HSV-1
as well as primary infection can cause severe diseases in immunocompetent individuals, like
acute retinal necrosis (ARN) or encephalitis, while in immunosuppressed individuals it can
lead to disseminated, systemic infections (Bernard Roizman et al., 2007a).
HSV-1 infections are tightly controlled by the immune system, including a wide variety of
immune cells (Cunningham et al., 2006). Cells of both innate and adaptive immunity
participate in the suppression of HSV-1 replication, and interactions between different cell
types take place within and across the innate-adaptive barrier (Schuster et al., 2011). Innate
immunity is crucial for the early, fast response to primary HSV-1 infection (Ashkar and
Rosenthal, 2003), and also appears to play a role in reactivation (Donaghy et al., 2009; Kittan
et al., 2007). Type I interferons (IFN), mainly produced by plasmacytoid dendritic cells
(PDC) (Siegal et al., 1999; Cella et al., 1999), are key factors in the anti-herpesviral response
(Zhang et al., 2007). They lead to an antiviral state of HSV-1 infected and susceptible cells on
the one hand (Härle et al., 2001), and they activate cells of the innate as well as the adaptive
immune system and thus trigger the immune response on the other hand (Gill et al., 2011;
Tough et al., 1996). Natural killer (NK) cells mediate recognition and killing of infected cells
as well as early production of IFN- (Lodoen and Lanier, 2006). Adaptive immunity appears
to contribute to maintenance of latency and limiting of viral spread. While the role of humoral
immunity is not clear, contributions of cell-mediated immunity against HSV-1, especially the
role of cluster of differentiation (CD)4+ T cells and CD8
+ T cells, have been well described
(Johnson et al., 2008; Koelle et al., 1998; Ghiasi et al., 1999).

Introduction
7
2.2 Natural killer cells
NK cells are a large granular lymphocyte subset distinct from B and T cells, which aroused
the interest of researchers due to its ability to lyse tumor cells as well as virus-infected cells
without prior sensitization and without restriction by major histocompatibility (MHC)
antigens (Trinchieri, 1989). Human NK cells, which are defined as CD3-CD56
+ cells, divide
into two phenotypic subsets, according to their expression of CD56 and CD16 (Cooper et al.,
2001a). CD56 was found to be identical with neural cell adhesion molecule (NCAM) (Lanier
et al., 1989), which belongs to the immunoglobulin (Ig) superfamily and mediates homotypic
adhesion between cells. It is expressed in nervous tissues of many vertebrates and plays a
major role in the embryonic development of the nervous system (Rutishauser and Jessell,
1988). CD16 is part of the low affinity fragment, cristallizable receptor IIIA (FcRIIIA),
which recognizes and binds the Fc part of antibodies bound to cell-associated antigens,
thereby inducing antibody-dependent cellular cytotoxicity (ADCC) towards opsonized target
cells (Leibson, 1997). CD56bright
CD16dim/-
cells account for about 10%, CD56dim
CD16bright
cells for about 90% of circulating NK cells. Besides their distinct phenotype researchers
observed functional differences between those two subtypes (FIG. 2). CD56bright
CD16dim/-
cells, which constitutively express the high affinity interleukin 2 (IL-2) receptor (IL-2R),
proliferate in response to low amounts of IL-2 and primarily account for the secretion of
cytokines such as IFN- or tumor necrosis factor (TNF)-, while CD56dim
CD16bright
cells
exhibit high cytotoxicity, mediated either through binding of activating NK cell receptors to
their ligands or through binding of CD16 to opsonized target cells (Cooper et al., 2001a).

Introduction
8
FIG. 2. Natural killer (NK) cell subsets. CD56bright
CD16dim/-
cells show high expression of CD56 and low or no
expression of CD16, they constitutively express the high affinity interleukin (IL)-2 receptor (IL-2R) and
primarily account for the secretion of cytokines such as interferon (IFN)- or tumor necrosis factor (TNF)-.
CD56dim
CD16bright
cells show low expression of CD56 and high expression of CD16 and exhibit high
cytotoxicity.
In contrast to B and T cells, NK cells recognize their target cells independently of antigen-
specific receptors. The activation status of NK cells is determined by a balance of signals
resulting from binding of inhibitory and activating NK cell receptors to their respective
ligands (Lanier, 2005). Inhibitory receptors recognize MHC class I (MHC-I) molecules,
activating receptors recognize stress-induced or virus-derived molecules on a target cell
(Kärre et al., 1986; Bauer et al., 1999). Cytokines secreted by other immune cells further
influence NK cell activation and functions (Nguyen et al., 2002). NK cells play a crucial role
in the immune defense against various pathogens such as viruses, bacteria and parasites. They
contribute to the control of infection by secretion of IFN- and by killing of infected cells
(Lodoen and Lanier, 2006). NK cells kill target cells via two main mechanisms: via granule-
dependent cytotoxicity, where cytotoxic granules containing perforin and granzymes are
released towards the target cell (Kägi et al., 1994; Metkar et al., 2002), and via stimulation of
death receptors on the target cell by TNF-related apoptosis-inducing ligand (TRAIL) (Zamai
et al., 1998), Fas ligand (FasL) (Arase et al., 1995) or TNF- (Paya et al., 1988). In addition
to their effector functions, NK cells exhibit regulatory functions and engagement in reciprocal

Introduction
9
interactions with various cell types, amongst others T cells, macrophages and dendritic cells
(Vivier et al., 2008).
Studies with NK cell-depleted mice could demonstrate the in vivo contribution of NK cells
particularly in the initial control of HSV infections (Habu et al., 1984; Tanigawa et al., 2000),
but there is also evidence for an accessory role of NK cells in adaptive immunity
(Nandakumar et al., 2008), and even HSV-induced NK cell memory has recently been
described (Abdul-Careem et al., 2012). Humans with NK cell deficiencies show increased
susceptibility to herpesviral infection, indicating an important role for NK cells in human
HSV immunity (Jawahar et al., 1996; Dalloul et al., 2004; Orange, 2002). In vitro studies
showed the ability of NK cells to recognize HSV-1-infected cells, leading to secretion of
IFN- and lysis of infected cells. NK cell activation occurred early enough in infection to
reduce spread of virus progeny and therefore limit viral replication in tissue culture
(Fitzgerald et al., 1985; Leibson et al., 1986). A role in NK cell recognition of HSV-infected
cells has been described for MHC-I molecules, which are known to be down-regulated by the
HSV-1 protein infected cell polypeptide (ICP)47 (Hill et al., 1995; Früh et al., 1995): HeLa
cells infected with HSV-1 or transfected with ICP47 down-regulated human leukocyte antigen
(HLA)-C molecules, which was sufficient to mediate NK cell cytotoxicity by NK cell clones
expressing an inhibitory killer cell immunoglobulin-like receptor (KIR) that recognizes
HLA-C (Huard and Früh, 2000). Other groups have shown that expression of HSV-1
immediate early proteins, particularly ICP0, is necessary and sufficient for NK cell
recognition of HSV-1-infected cells (Fitzgerald-Bocarsly et al., 1991; Chisholm et al., 2007).
In addition, cytokines influence NK cell activity, for example IL-15, which seems to be of
importance in the activation of NK cells in the context of peripheral blood mononuclear cells
(PBMC) (Ahmad et al., 2000), and type I IFN, which seem to be involved in activating NK
cells to lyse HSV-1-infected fibroblasts (Feldman et al., 1992). Also, mouse models suggest
roles for the IFN-/ receptor and hence type I IFN (Gill et al., 2011), IL-18 (Reading et al.,
2007), and dendritic cells as important accessory cells (Kassim et al., 2009; Frank et al.,
2012).

Introduction
10
2.3 Plasmacytoid dendritic cells
In 1999 PDC were identified as major producers of type I IFN (Siegal et al., 1999; Cella et al.,
1999). While they are negative for the expression of lineage markers, they express the specific
receptors blood dendritic cell antigen 2 (BDCA-2) and BDCA-4 (FIG. 3) (Dzionek et al.,
2001; Dzionek et al., 2002). PDC play a crucial role in the immune response to viral
infections. Their contributions to the control of viral infection appear to be not only of a direct
manner, but also of an indirect manner by interacting with various immune cells and thus
linking innate and adaptive immunity (Colonna et al., 2004). Stimulation of PDC with HSV-1
leads to secretion of high amounts of IFN-. This stimulation does not require infectivity,
since ultraviolet light (UV)-inactivated HSV-1 is able to stimulate PDC, and HSV-1 does not
replicate in PDC (Schuster et al., 2010; Donaghy et al., 2009). PDC stimulation occurs via
recognition of the viral genome by toll-like receptor 9 (TLR-9) within endosomes (Krug et al.,
2004), which has also been shown for HSV-2 (Lund et al., 2003).
FIG. 3. Plasmacytoid dendritic cell (PDC). PDC are identified by their expression of blood dendritic cell
antigen 2 (BDCA-2) and BDCA-4 and by lacking expression of lineage markers. They recognize HSV-1 via toll-
like receptor 9 (TLR-9) which is expressed in endosomes. Stimulation by HSV-1 leads to secretion of high
amounts of IFN-.
Coordinated regulation of surface receptors upon stimulation indicates various aspects of PDC
function, like attraction to inflamed tissue, antigen recognition and subsequent migration into
secondary lymphatic tissue (Schuster et al., 2010). In fact, upon vaginal HSV-2 infection,
PDC are recruited to the infected tissue and suppress viral replication (Lund et al., 2006).

Introduction
11
Furthermore, virally stimulated PDC are able to induce migration (Megjugorac et al., 2004)
and also activation of cells of the innate and adaptive immune system (Feldman et al., 1992;
Kadowaki et al., 2000). The capacity of PDC to engulf antigen and present it to T cells is
controversially discussed (Villadangos and Young, 2008). The significance of PDC in HSV
infections has been demonstrated in different mouse models. In this respect, Lund et al.
observed an increase in pathogenesis of genital HSV-2 infections after antibody-dependent
PDC depletion (Lund et al., 2006), while Swiecki et al. found that PDC depletion in
CLEC4-DTR mice diminished type I IFN as well as pro-inflammatory cytokine production,
NK cell activation and CD8+ T cell responses during systemic HSV-1 and HSV-2 infections
(Swiecki et al., 2013).

Introduction
12
2.4 Mononuclear phagocytes
Mononuclear phagocytes constitute an important and early component of the immune system.
Monocytes are normally circulating in the blood, while macrophages and dendritic cells,
which represent differentiated stages of monocytes, reside in lymphoid and non-lymphoid
tissues. Macrophages serve as first line defense against invading pathogens as well as
initiators of inflammation, and dendritic cells are particularly important in initiating and
supporting adaptive immune responses. Upon pathogen invasion and inflammation, blood
monocytes support resident macrophages and dendritic cells by infiltrating the tissue and
differentiating into one or the other, depending on the cytokine milieu (Michael Ehrenstein et
al., 2008b; van and Cohn, 1968; Randolph et al., 1999). Mononuclear phagocytes are
equipped with receptors that recognize a wide range of ligands, like pathogen-derived
molecules, so called opsonizing molecules of the humoral immune system that are bound to
pathogens, and chemokines as well as cytokines, which enables them to recognize pathogens
directly and indirectly, and to communicate with other cells of the immune system. Two of
those receptors are predominantly expressed by monocytes and macrophages: CD14 binds
bacterial lipopolysaccharide (LPS), thereby leading to its recognition by TLR-4, and CD64,
also known as FcRI, binds the Fc part of antibodies and thereby recognizes opsonized
pathogens (FIG. 4) (Michael Ehrenstein et al., 2008c; Michael Ehrenstein et al., 2008d). Their
importance in pathogen defense is due to their secretion of various inflammatory cytokines,
like TNF- and IL-1, their secretion of chemokines and their ability to phagocytose invading
pathogens. Macrophages, which have exceptionally high phagocytic properties, destroy
ingested pathogens by digesting them. For that purpose, phagosomes fuse with lysosomes,
which have a low pH and are filled with enzymes, nitric oxide (NO) and reactive oxygen
species (ROS) (Michael Ehrenstein et al., 2008b; Michael Ehrenstein et al., 2008a; Dale et al.,
2008). Dendritic cells promote adaptive immune responses by presenting antigens derived
from ingested pathogens to T cells and thereby activating them (Leon et al., 2007).

Introduction
13
FIG. 4. Mononuclear phagocyte. Mononuclear phagocytes express the Fc receptor CD64, as well as CD14 and
TLR-4 which bind and recognize bacterial lipopolysaccharide (LPS). They secrete pro-inflammatory cytokines
like TNF- and IL-1 and contain lysosomes which serve for digestion of pathogens.
In mouse models researchers have investigated the role of macrophages in viral infections and
subsequently demonstrated the importance of macrophages in innate resistance to viruses
(Mogensen, 1979). In the case of HSV infection macrophages are among the first immune
cells to be activated and to exert antiviral activity (Ellermann-Eriksen, 2005). They can be
infected by HSV, but are non- or barely permissive for viral replication, depending on their
state of differentiation (Bruun et al., 1998; Daniels et al., 1978). Macrophages of HSV-
infected mice have been demonstrated to exert extrinsic antiviral activity in vitro, thereby
limiting viral replication in cell culture, independently of the virus or the host cell species
used (Morahan et al., 1980). The observed extrinsic antiviral activity is due to various
cytokines and anti-microbial molecules secreted by HSV-activated macrophages, like
IFN-/, ROS and NO, which directly inhibit HSV replication. Other cytokines like TNF-
and IL-12 activate other immune cells like NK cells (Voth et al., 1988; Wolf et al., 1991). In
HSV-infected mice mononuclear phagocytes are among the first cell populations recruited to
the infection site (Frank et al., 2012). They limit viral replication via TNF- secretion and NO
production (Fields et al., 2006; Kodukula et al., 1999) and are also required for the
development of an adaptive immune response (Cheng et al., 2000). Monocytes and
macrophages furthermore serve as important accessory cells in NK cell activation not only by
HSV-1 but by diverse viral, bacterial, and also protozoan pathogens. They activate NK cells

Introduction
14
via secretion of various cytokines, like IL-12, IL-15 and IL-18, as well as direct cell contact
through different receptor-ligand interactions, like natural killer group 2, member D
(NKG2D)-MHC class I polypeptide-related sequence (MIC) A or NKG2D-UL-16-binding
proteins (ULBP), natural cytotoxicity triggering receptor 1 (NKp46)-DNAX accessory
molecule-1 (DNAM1), and 2B4-CD48, depending on the respective pathogen (Michel et al.,
2012).

Introduction
15
2.5 Interactions of PDC and NK cells in HSV infection
Interactions between PDC and NK cells in HSV infection have been investigated in vivo and
in vitro. Barr et al. described PDC-NK cell interactions via IL-18 as important for NK cell
IFN- secretion after HSV-1 infection in mice. However, PDC were not the only cell
population activating NK cells, and CD69 expression as well as cytotoxicity of NK cells was
independent of IL-18 (Barr et al., 2007). Feldman et al. showed a role for accessory cells
(AC) in human NK cell-mediated lysis of HSV-1-infected fibroblasts: NK cell cytotoxicity
against infected fibroblasts was only accomplished in the presence of AC, which were, at least
in part, so called interferon producing cells (IPC), later identified as PDC. Participation of AC
was described to be IFN--dependent as well as IFN--independent, and probably cell
contact-dependent (Feldman et al., 1992). Another study demonstrated the in vitro ability of
HSV-1-stimulated PDC to induce migration of NK cells via secretion of chemokine (C-C
motif) ligand (CCL)4 and chemokine (C-X-C motif) ligand (CXCL)10 (Megjugorac et al.,
2004). A mouse study conducted by Persson et al. showed that HSV-1-stimulated PDC
recruited and activated NK cells in vivo (Persson and Chambers, 2010), and Swiecki et al.
demonstrated a critical role for PDC in NK cell activation in systemic HSV-1 and HSV-2
infections in mice (Swiecki et al., 2013). In a study of recurrent human HSV-2 infection, PDC
and NK cells co-localized in recurrent genital herpes lesions (Donaghy et al., 2009).
Several studies of human PDC-NK cell interaction after stimulation with CpG
oligodeoxynucleotides (CpG-ODN) indicated a major role for cytokines, particularly IFN-,
and a minor role for direct cell contact. In all studies CD69 expression on NK cells was cell
contact-independent but dependent on IFN- and other soluble factors like TNF-(Gerosa et
al., 2005; Benlahrech et al., 2009; Romagnani et al., 2005; Marshall et al., 2006). Cytotoxicity
was described to be induced by either soluble factors alone (Gerosa et al., 2005; Romagnani et
al., 2005) or demanded direct cell contact (Benlahrech et al., 2009), while IFN- secretion
induced by PDC was reported to be cytokine-mediated (Benlahrech et al., 2009; Romagnani
et al., 2005; Marshall et al., 2006).

Rationale
16
3 Rationale
Altogether, studies of NK cell activation in human HSV-1 infections, and particularly NK cell
interactions with potential accessory cells, like PDC, are limited, and the so far existing data
are controversial and insufficient. There are only few in vitro studies concerning human NK
cell activation by HSV-1, and in vivo studies which were mostly conducted in mice. In most
in vitro studies investigating NK cell-PDC interaction CpG-ODN were used as surrogate for
DNA viruses like HSV-1, but these studies only cover stimulatory effects of viral DNA, not
of other components of the HSV-1 particle nor the possible impact of viral replication.
Therefore, the goal of this study was to analyze the potential of HSV-1 to activate human NK
cells in vitro within the PBMC context, and to decipher mechanisms leading to HSV-1-
induced NK cell activation, in particular PDC-NK cell interactions, to identify further
accessory cell populations interacting with NK cells, and to determine cytokines involved in
the cellular crosstalk between NK cells and accessory cells. For these purposes, sucrose
gradient-purified UV-inactivated (HSVUV) as well as infectious (HSVINF) HSV-1 were used to
stimulate primary human PBMC.

Materials and Methods
17
4 Materials and Methods
4.1 Materials
4.1.1 Instruments
Instrument Manufacturer
BD LSRII BD Biosciences (Heidelberg, DE)
Biogard hood The Baker Company (Sanford, ME, US)
Bio-Link 254 UV crosslinker Vilber Lourmat (Eberhardzell, DE)
Eclipse TS 100 inverted microscope Nikon (Düsseldorf, DE)
ELx800 Absorbance Microplate Reader BioTek (Bad Friedrichshall, DE)
Finnpipette 300µl multi channel pipet Thermo scientific (Langenselbold, DE)
Heraeus Labofuge M Thermo scientific (Langenselbold, DE)
L7-55 ultracentrifuge Beckman Coulter (Krefeld, DE)
Micro 200R centrifuge Hettich lab technology (Tuttlingen, DE)
Neubauer Chamber hemocytometer Marienfeld Superior (Lauda-Königshofen, DE)
Pipetman 20µl pipet Gilson (Middleton, WI, US)
Pipetman 200µl pipet Gilson (Middleton, WI, US)
Pipetman 1000µl pipet Gilson (Middleton, WI, US)
pipetus electrical pipette filler Hirschmann (Eberstadt, DE)
Reax top vortexer Heidolph (Schwabach, DE)
Research plus 20µl pipet Eppendorf (Wesseling-Berzdorf, DE)
Research plus 200µl pipet Eppendorf (Wesseling-Berzdorf, DE)
Research plus 1000µl pipet Eppendorf (Wesseling-Berzdorf, DE)
Research 100µl multi channel pipet Eppendorf (Wesseling-Berzdorf, DE)
Rotina 380R centrifuge Hettich lab technology (Tuttlingen, DE)
Rotilabo mini centrifuge Roth (Karlsruhe, DE)
Stericult 200 incubator Labotect (Göttingen, DE)
SW 32Ti rotor Beckman Coulter (Krefeld, DE)
Thermomixer comfort 2ml Eppendorf (Wesseling-Berzdorf, DE)

Materials and Methods
18
4.1.2 Consumables
Consumable Manufacturer
0.5ml Micro-tubes Roth (Karlsruhe, DE)
1.5ml Micro-tubes Brand (Wertheim, DE)
1.5ml Screw cap micro tubes Sarstedt (Nümbrecht, DE)
2ml Micro-tubes Sarstedt (Nümbrecht, DE)
5ml FACS tubes Sarstedt (Nümbrecht, DE)
15ml centrifuge tubes Sarstedt (Nümbrecht, DE)
38.5ml polyallomer tubes Beckman Coulter (Krefeld, DE)
38.5ml ultra clear tubes Beckman Coulter (Krefeld, DE)
50ml centrifuge tubes Sarstedt (Nümbrecht, DE)
caps for FACS tubes Sarstedt (Nümbrecht, DE)
Cellstar filtertop cell culture flasks 650ml Greiner bio-one (Solingen, DE)
Cellstar filtertop cell culture flasks 250ml Greiner bio-one (Solingen, DE)
Cellstar filtertop cell culture flasks 50ml Greiner bio-one (Solingen, DE)
Cellstar 24 well plates Greiner bio-one (Solingen, DE)
Cellstar 96 well plates Greiner bio-one (Solingen, DE)
Cellstar cell culture plates 100mm Greiner bio-one (Solingen, DE)
Cellstar cell culture plates 60mm Greiner bio-one (Solingen, DE)
Costar 5mL Stripette, Polystyrene Corning (Wiesbaden, DE)
Costar 10mL Stripette, Polystyrene Corning (Wiesbaden, DE)
Costar 25mL Stripette, Polystyrene Corning (Wiesbaden, DE)
filter, 0.22µm BD Biosciences (Heidelberg, DE)
microscope cover slips Menzel-Gläser (Braunschweig, DE)
Nunc MaxiSorp 96 well plates Thermo scientific (Langenselbold, DE)
paper towels Tork (Mannheim, DE)
Pipet tips 1000µl Ratiolab (Dreieich, DE)
Pipet tips 200µl Sarstedt (Nümbrecht, DE)
SafeGuard filter tips 1250µl Peqlab (Erlangen, DE)
Safety Multifly needle Sarstedt (Nümbrecht, DE)
Silver Nitrile gloves S Kimberly-Clark (Koblenz, DE)
S-Monovette EDTA K2 gel Sarstedt (Nümbrecht, DE)
Stericup Filter Unit, 0.22µm, 150ml Merck Millipore (Darmstadt, DE)
Stericup Filter Unit, 0.22µm, 250ml Merck Millipore (Darmstadt, DE)
Stericup Filter Unit, 0.22µm, 500ml Merck Millipore (Darmstadt, DE)
syringe, 10ml BD Biosciences (Heidelberg, DE)

Materials and Methods
19
4.1.3 Reagents
Reagent Manufacturer
acetic acid (C2H4O2) Merck Millipore (Darmstadt, DE)
Biocoll 1.077g/ml Biochrom (Tutzing, DE)
bovine serum albumin (BSA) Sigma-Aldrich (München, DE)
CpG-A 6016 (5´-T*C-G-A-C-G-T-C-G-T-G-
G*G*G*G-3´)
* stands for phosphorothioate
- stands for phosphodiester bonds
Coley Pharmaceutical (Düsseldorf, DE)
disodium phosphate (Na2HPO4) Merck Millipore (Darmstadt, DE)
Dulbecco`s Modified Eagle Medium (DMEM) Invitrogen (Darmstadt, DE)
ethylenediaminetetraacetic acid (EDTA) Sigma-Aldrich (München, DE)
fetal calf serum (FCS) Sigma-Aldrich (München, DE)
glucose Merck Millipore (Darmstadt, DE)
glutamine Invitrogen (Darmstadt, DE)
recombinant human interferon-2b (rhIFN-) Miltenyi Biotec (Bergisch Gladbach, DE)
recombinant human interleukin 2 (rhIL-2) Roche-Pharma (Grenzach-Wyhlen, DE)
recombinant human interleukin 3 (rhIL-3) R&D Systems (Wiesbaden-Nordenstadt, DE)
hydrogen chloride (HCl) Merck Millipore (Darmstadt, DE)
monopotassium phosphate (KH2PO4) Merck Millipore (Darmstadt, DE)
paraformaldehyde (PFA) Sigma-Aldrich (München, DE)
penicillin Invitrogen (Darmstadt, DE)
phenol red Merck Millipore (Darmstadt, DE)
potassium chloride (KCl) Merck Millipore (Darmstadt, DE)
Roswell Park Memorial Institute (RPMI) 1640
Medium
Invitrogen (Darmstadt, DE)
sodium chloride (NaCl) Merck Millipore (Darmstadt, DE)
streptomycin Invitrogen (Darmstadt, DE)
sulfuric acid (H2SO4) Merck Millipore (Darmstadt, DE)
tris(hydroxymethyl)aminomethane (Tris) Roth (Karlsruhe, DE)
trypan blue Sigma-Aldrich (München, DE)
Tween 20 Roth (Karlsruhe, DE)

Materials and Methods
20
4.1.4 Software
Software Source
FACSDiva Software BD Biosciences (Heidelberg, DE)
FCS Express 3 Software De Novo Software (Los Angeles, CA, US)
FlowCytomixPro software Affymetrix eBioscience (Frankfurt, DE)
Gen5 Data Analysis Software BioTek (Bad Friedrichshall, DE)
VassarStats Statistical Computation Website http://www.vassarstats.net/
4.1.5 Commercial Kits
Kit Manufacturer
Human CD304 MicroBead Kit Miltenyi Biotec (Bergisch Gladbach, DE)
Human CD14 MicroBeads Miltenyi Biotec (Bergisch Gladbach, DE)
Human IFN- Matched Antibody Pairs Affymetrix eBioscience (Frankfurt, DE)
Human sCD40L Matched Antibody Pairs Affymetrix eBioscience (Frankfurt, DE)
Human IFN- Secretion Assay Detection Kit Miltenyi Biotec (Bergisch Gladbach, DE)
Human NK Cell Isolation Kit Miltenyi Biotec (Bergisch Gladbach, DE)
Human Th1/Th2 11plex RTU FlowCytomix
Multiplex
Affymetrix eBioscience (Frankfurt, DE)
Human TNF- Secretion Assay Detection Kit Miltenyi Biotec (Bergisch Gladbach, DE)
4.1.6 Cell Culture
Cultured cells Cell type Origin
Human foreskin fibroblasts
(HFF)
neonatal foreskin Human
Primary blood cells peripheral blood mononuclear cells
(PBMC)
Human
Primary blood cells plasmacytoid dendritic cells (PDC) Human
Primary blood cells natural killer (NK) cells Human
Primary blood cells monocytes Human
Vero cells deficient for IFN-
and IFN-1 genes (Diaz et al.,
1988)
kidney epithelial cells African green monkey

Materials and Methods
21
4.1.7 Viruses
Virus Clone Source
Herpes simplex virus type 1
(HSV-1), expressing a green
fluorescent protein (GFP)-tagged
VP22
166v Gillian Elliott, Peter O’Hare (Elliott and
O'Hare, 1999)
Herpes simplex virus type 1
(HSV-1), ICP4, ICP22, ICP27,
ICP47 deletion mutant,
expressing GFP under a HCMV
promoter
d106S David M. Knipe (Liu et al., 2009)
Herpes simplex virus type 1
(HSV-1), wild type
primary isolate diagnostic services, Institute of Clinical
and Molecular Virology, Friedrich-
Alexander-University Erlangen-Nürnberg
(Kittan et al., 2007)
4.1.8 Media and Buffers
Medium / Buffer Composition
assay buffer (Matched Antibody Pairs) 0.5% BSA
0.05% Tween 20
in DPBS (Matched Antibody Pairs)
cytokine buffer (cytokine secretion assay) 0.5% BSA
2mM EDTA
in DPBS
coating solution (IFN- Matched Antibody Pairs) 1µg/ml antibody
in DPBS (Matched Antibody Pairs)
coating solution (sCD40L Matched Antibody Pairs) 5µg/ml antibody
in DPBS (Matched Antibody Pairs)
DPBS 138mM NaCl
2.7mM KCl
6.5mM Na2HPO4
1.5mM KH2PO4
DPBS (Matched Antibody Pairs) 8g NaCl
0.2g KCl
2.85g Na2HPO4 x12 H2O
0.2g KH2PO4
ad 1l H2O
FACS buffer 1% FCS
1mM EDTA
in DPBS
MACS buffer 1% FCS
2mM EDTA
in DPBS

Materials and Methods
22
Medium / Buffer Composition
stop solution (Matched Antibody Pairs) 4N H2SO4
supplemented RPMI 1640 0.3mg/ml glutamine
200U/ml penicillin
90U/ml streptomycin
10% FCS
supplemented DMEM 0.3mg/ml glutamine
200U/ml penicillin
90U/ml streptomycin
10% FCS
Trypsin EDTA 140mM NaCl
5mM KCl
0.65mM Na2HPO4
5mM glucose
25mM Tris/HCl
0.01% EDTA
0.1% phenole red
virus standard buffer (VSB) 0.05M Tris
0.012M KCl
0.005M EDTA
pH 7.8
VSB 15% sucrose solution 15% sucrose
0.1% BSA
in VSB
VSB 30% sucrose solution 30% sucrose
0.1% BSA
in VSB
washing buffer (Matched Antibody Pairs) 0.05% Tween 20
in DPBS
4.1.9 Antibodies
Epitope Flurophore Clone Isotype Manufacturer
CD1c FITC L161 mouse IgG1 Biolegend (London, GB)
CD3 FITC UCHT1 mouse IgG1 Biolegend (London, GB)
CD3 PE UCHT1 mouse IgG1 Biolegend (London, GB)
CD3 PE-Cy5 UCHT1 mouse IgG1 AbD Serotec (Düsseldorf, DE)
CD3 AlexaFluor700 UCHT1 mouse IgG1 Biolegend (London, GB)
CD3 PacificBlue UCHT1 mouse IgG1 BD Biosciences (Heidelberg, DE)
CD4 PE-Cy7 RPA-T4 mouse IgG1 Biolegend (London, GB)
CD8 APC MEM-31 mouse IgG2a ImmunoTools (Friesoythe, DE)

Materials and Methods
23
Epitope Flurophore Clone Isotype Manufacturer
CD8 APC-eFluor780 RPA-T8 mouse IgG1 Affymetrix eBioscience
(Frankfurt, DE)
CD14 PE-Cy5 61D3 mouse IgG1 AbD Serotec (Düsseldorf, DE)
CD16 PE-Cy7 3G8 mouse IgG1 Biolegend (London, GB)
CD19 APC HIB19 mouse IgG1 Biolegend (London, GB)
CD33 PE WM53 mouse IgG1 Biolegend (London, GB)
CD56 PE HCD56 mouse IgG1 Biolegend (London, GB)
CD56 PE-Cy7 HCD56 mouse IgG1 Biolegend (London, GB)
CD64 APC 10.1 mouse IgG1 Biolegend (London, GB)
CD69 FITC FN50 mouse IgG1 Miltenyi Biotec (Bergisch
Gladbach, DE)
CD69 AlexaFluor700 FN50 mouse IgG1 Biolegend (London, GB)
CD107a AlexaFluor488 eBioH4A3 mouse IgG1 Affymetrix eBioscience
(Frankfurt, DE)
CD123 PE AC145 mouse IgG2a Miltenyi Biotec (Bergisch
Gladbach, DE)
CD303 FITC AC144 mouse IgG1 Miltenyi Biotec (Bergisch
Gladbach, DE)
CD304 APC AD5-17F6 mouse IgG1 Miltenyi Biotec (Bergisch
Gladbach, DE)
HLA-ABC PE W6/32 mouse IgG2a Biolegend (London, GB)
HLA-E PE 3D12 mouse IgG1 Biolegend (London, GB)
IFN-/R none MMHAR-2 mouse IgG2a Acris (Herford, DE)
IFN- APC 45-15 mouse IgG1 Miltenyi Biotec (Bergisch
Gladbach, DE)
IL-1 none 8516 mouse IgG1 R&D Systems (Wiesbaden-
Nordenstadt, DE)
MICA /
MICB
APC 6D4 mouse IgG2a Biolegend (London, GB)
TNF- PE cA2 human IgG1 Miltenyi Biotec (Bergisch
Gladbach, DE)
TNF- none 28401 mouse IgG1 R&D Systems (Wiesbaden-
Nordenstadt, DE)

Materials and Methods
24
4.1.10 Isotype Controls
Flurophore Clone Isotype Manufacturer
AlexaFluor700 MOPC-21 mouse IgG1 Biolegend (London, GB)
APC MOPC-21 mouse IgG1 Biolegend (London, GB)
APC PPV-04 mouse IgG2a ImmunoTools (Friesoythe, DE)
APC-eFluor780 MOPC-21 mouse IgG1 Biolegend (London, GB)
FITC MOPC-21 mouse IgG1 Biolegend (London, GB)
none 11711 mouse IgG1 R&D Systems (Wiesbaden-
Nordenstadt, DE)
none PPV-04 mouse IgG2a Acris (Herford, DE)
PacificBlue MOPC-21 mouse IgG1 BD Biosciences (Heidelberg, DE)
PE MOPC-21 mouse IgG1 Biolegend (London, GB)
PE MOPC-173 mouse IgG2a Biolegend (London, GB)
PE-Cy5 MCA928C mouse IgG1 AbD Serotec (Düsseldorf, DE)
PE-Cy7 MOPC-21 mouse IgG1 Biolegend (London, GB)

Materials and Methods
25
4.2 Methods
4.2.1 Isolation of primary human cells
Peripheral blood mononuclear cells
PBMC were isolated from EDTA-anti-coagulated blood of healthy donors using Biocoll
density centrifugation (1.077g/ml). These studies were approved by the Ethical Committee of
the Medical Faculty, Friedrich-Alexander-Universität Erlangen-Nürnberg (Ref. no. 3299).
EDTA blood was centrifuged at 200x g for 10min and plasma was removed. Cells of four
vials were then transferred into a 50ml tube, filled up to 35ml with RPMI 1640 and layered
onto 15ml Biocoll. Separation of PBMC from erythrocytes and granulocytes was achieved by
centrifugation at 440x g for 25min with the brake inactivated. The interphase containing
lymphocytes and monocytes, visible as a ring between the upper cell-free layer and the
Biocoll layer, was transferred into a 50ml tube, filled up to 50ml with RPMI 1640 and
centrifuged at 440x g for 5min. This washing step was repeated, and cells were then re-
suspended in supplemented RPMI 1640. Cell numbers were determined using a Neubauer
chamber.
Plasmacytoid dendritic cells
PDC were isolated from PBMC via magnetic-activated cell sorting (MACS) using the CD304
MicroBead Kit (Miltenyi Biotec). PBMC were centrifuged at 440x g for 5min. Supernatant
was discarded and cells were washed by re-suspension in MACS buffer and centrifugation at
440x g for 5min. Cells were then re-suspended in MACS buffer and incubated with FcR
blocking reagent and CD304 MicroBeads at 4°C for 15min. Per one million cells, 1.5µl
MACS buffer, 0.5µl FcR blocking reagent and 0.5µl CD304 MicroBeads were used. Cells
were then washed, re-suspended in 1ml MACS buffer and applied to a LS MACS column that
had been placed in a MACS separator and equilibrated with 3ml MACS buffer. Magnetically
labeled PDC were retained within the column, while unlabeled cells could flow through. After
three washing steps with 3ml MACS buffer, the column was removed from the separator and
PDC were eluted using 10ml MACS buffer. Flow through was used as PDC-depleted PBMC
for depletion experiments. After centrifugation a second round of isolation followed using a
MS MACS column and volumes of 500µl for equilibration, re-suspension and washing and
4ml for elution. Numbers of purified PDC were determined using a Neubauer chamber.
Monocytes
Monocytes were isolated from PBMC via MACS using CD14 MicroBeads (Miltenyi Biotec).
PBMC were centrifuged at 440x g for 5min, supernatant was discarded and cells were washed
by re-suspension in MACS buffer and centrifugation at 440x g for 5min. Cells were then re-

Materials and Methods
26
suspended in MACS buffer and incubated with CD14 MicroBeads at 4 °C for 15min. Per one
million cells, 4µl MACS buffer and 1µl CD14 MicroBeads were used. Cells were then
washed, re-suspended in 1ml MACS buffer and applied to a LS MACS column that had been
placed in a MACS separator and equilibrated with 3ml MACS buffer. Magnetically labeled
monocytes were retained within the column, while unlabeled cells could flow through. After
three washing steps with 3ml MACS buffer, the column was removed from the separator and
monocytes were eluted using 10ml MACS buffer. Flow through was used as
monocyte-depleted PBMC for depletion experiments. Numbers of purified monocytes were
determined using a Neubauer chamber.
Natural killer cells
NK cells were isolated from PBMC via MACS using the NK Cell Isolation Kit (Miltenyi
Biotec). PBMC were centrifuged at 440x g for 5min, supernatant was discarded and cells
were washed by re-suspension in MACS buffer and centrifugation at 440x g for 5min. Cells
were then re-suspended in MACS buffer and incubated with a biotinylated antibody cocktail
against lineage markers of non-NK cells at 4°C for 10min. Per one million cells, 4µl MACS
buffer and 1µl antibody cocktail were used. Cells were then further incubated with
MicroBead-coupled secondary antibodies directed against biotin at 4°C for 15min, using 3µl
MACS buffer and 2µl secondary antibodies per one million cells. Cells were then washed,
re-suspended in 1ml MACS buffer and applied to a LS MACS column that had been placed in
a MACS separator and equilibrated with 3ml MACS buffer. Magnetically labeled non-NK
cells were retained within the column, while unlabeled NK cells could flow through.
Thereafter, three washing steps with 3ml MACS buffer were performed. Numbers of purified
NK cells were determined using a Neubauer chamber.
4.2.2 Determination of cell numbers
For determination of PBMC numbers, one volume of cell suspension was mixed with one
volume of Turks solution (3% C2H4O2) to lyse erythrocytes still present within PBMC, and
with two volumes of Trypan blue to exclude dead cells from the count. This resulted in a 1:4
dilution of the cells. A Neubauer chamber was filled with the suspension, and cells were
counted within four sixteen-square fields, of which one is equivalent to 0.1µl.
The number of cells per ml was calculated as depicted below.
cells / ml = cells counted x104
For counting of purified cells or cell lines, one volume of cell suspension was mixed with one
volume Trypan blue, leading to a 1:2 dilution of cells, and cells were counted within two
sixteen-square fields.

Materials and Methods
27
FIG. 5. Sixteen-square field of a Neubauer chamber. The cell number counted within this sixteen-square field
is equivalent to the cell number per 0.1µl and is multiplied with 104 to obtain the cell number per ml.
4.2.3 Herpes simplex virus type 1 stocks
Generation
For the generation of HSV-1 stocks, Vero cells were cultured in 650ml cell culture flasks. A
confluent monolayer was inoculated with HSV-1 stock in a volume of 20ml per cell culture
flask. After incubation at 37°C for 2h, cell cultures were washed with 50ml warm DMEM and
then given 50ml supplemented DMEM per tissue culture flask. After incubation at 37°C for
3d, infected cells were re-suspended. After two freeze-and-thaw cycles, lysates were
centrifuged at 440x g for 5min and supernatants were harvested. Cell-free supernatants were
either purified over a sucrose gradient or used directly. Directly used lysates were filtered
through a 0.22µm filter. Aliquots were stored at -80°C.
Purification
Cell-free supernatants were filled into 38.5ml polyallomer tubes and centrifuged in an
ultracentrifuge at 50,000x g for 90min at 4°C. Supernatants were discarded; virus pellets were
incubated in the residual liquid overnight at 4°C, re-suspended and pooled and dounced
twenty times. A continuous gradient from 30% to 15% sucrose was filled into a 38.5ml ultra
clear tube, re-suspended virus was layered upon the sucrose gradient and centrifuged at
50,000x g for 30min at 4°C. The virus ring that was visible within the sucrose gradient when
exposed to strong light of a microscope was collected, given into a 38.5ml polyallomer tube,
filled up to 38.5ml with virus standard buffer and centrifuged at 78,000x g for 90min at 4°C.
Supernatant was discarded; the virus pellet was incubated in the residual liquid for 1h at 4°C,
re-suspended in RPMI 1640 and filtered through a 0.22µm filter (HSVINF). Part of he purified
virus stock was inactivated by exposure to ultraviolet light (HSVUV). Aliquots were stored at
-80°C.
UV-inactivation
Virus stocks were inactivated using an UV crosslinker (Vilber Lourmat). They were irradiated
in an open cell culture plate five times to a final dose of 1J/cm2, with shaking of the plate
between irradiation rounds. Complete UV-inactivation was proven by inoculation of Vero
cells with undiluted virus stock resulting in lack of cytopathic effect and hence lack of viral
infection in the cell culture after 3d of incubation at 37°C.

Materials and Methods
28
Determination of the TCID50/ml
For the determination of the 50% tissue culture infective dose (TCID50)/ml of a virus stock,
Vero cells from one 50ml cell culture flask were re-suspended in 25ml supplemented DMEM
and seeded into three 96 well plates using 75µl per well. The virus stock was pre-diluted
1:100, 1:1,000 and 1:10,000 for the first, second and third plate, respectively, and a 1:4
dilution series was achieved for each pre-dilution by pipetting 25µl pre-diluted virus stock
into the eight wells of the first row, mixing, pipetting 25µl from the first row into the second
row and so on, until the last row of each plate was filled. Cell cultures were incubated at 37°C
for 3d and then screened for cytopathic effect indicating infection. The TCID50/ml was
calculated according to the method of estimating fifty percent endpoints published by Reed
and Muench (L.J.REED and H.MUENCH, 1938). For this purpose, the threshold between the
last row with 50% or more infected wells and the first row with less than 50% infected wells
was determined for each 96 well plate. The TCID50/ml for each plate was calculated as
depicted below and a mean TCID50/ml was determined.
e = ( a / ( a + b ) ) x100
f = ( c / ( c + d ) ) x100
P = ( e - 50% ) / ( e - f )
TCID50 / ml = D R + P
x C x ( 1 / V)
a: (total of infected wells below the threshold and in the last row above the threshold) x2
b: (total of non-infected wells above the threshold) x2
c: (total of infected wells below the threshold) x2
d: (total of non-infected wells above the threshold and in the first row below the threshold) x2
e: percentage of infected wells above the threshold
f: percentage of infected wells below the threshold
D: applied dilution series of virus stock
R: last row above the threshold
P: proportional distance
V: applied volume of virus stock (ml)
C: applied pre-dilution of virus stock
4.2.4 PDC supernatants
Generation
PDC supernatants (PDC-SN) were generated by stimulation of PDC with HSVINF. A total of
5x105 PDC were cultured in 500µl supplemented RPMI 1640 containing 20ng/ml rhIL-3 in
24 well plates and inoculated with 1x106 TCID50/ml HSV-1. After incubation at 37°C for 3h
PDC were harvested and centrifuged at 590x g for 10min. PDC were washed with DPBS, re-

Materials and Methods
29
suspended in 100µl trypsin EDTA, and after incubation at 37°C for 15min, PDC were washed
again and cultured in 500µl supplemented RPMI 1640 containing 20ng/ml rhIL-3 at 37°C for
18h. PDC-SN were then harvested and stored at -20°C. IFN-2a/2b concentrations were
determined using the IFN- Matched Antibody Pairs (Affymetrix eBioscience).
Determination of inhibitory potential on HSV-1 replication
In order to determine the potential of PDC-SN to inhibit HSV-1 replication in target cells,
human foreskin fibroblasts (HFF) were cultured in 24 well plates, using 1x105 cells in 500µl
supplemented DMEM per culture, and inoculated with a green fluorescing HSV-1 (HSVGFP)
at an MOI of 0.01 and 0.001. After incubation at 37°C for 2h, virus-containing media of cell
cultures were exchanged with fresh media containing either no PDC-SN or PDC-SN at
concentrations of different IFN-2a/2b concentrations. After incubation at 37°C for 24h and
48h, cells were harvested for FACS analysis. Cells were analyzed for infection and viability.
Stimulation of NK cells
A total of 2.5x105 NK cells were cultured in 24 well plates in 500µl supplemented RPMI
1640, inoculated with PDC-SN or recombinant human IFN-2b (rhIFN-) (Invitrogen), using
comparable IFN-2a/2b concentrations, incubated at 37°C for 3 to 18h and harvested for
FACS analysis. For neutralization of type I IFN activity, 15µg/ml anti ()IFN-/ receptor
(IFN-R) antibody was added to the cell culture. Activation of cells was determined by
surface expression of CD69.
4.2.5 Stimulation and infection of cells with HSV-1
Stimulation of PBMC
A total of 1x106 PBMC or PBMC depleted of monocytes or PBMC depleted of PDC were
cultured in 24 well plates in 500µl supplemented RPMI 1640 and inoculated with 1x106
TCID50/ml HSVUV and HSVINF, 0.75µM CpG-A, and 100U/ml rhIL-2. Mock served as
control. For neutralization experiments, IL-1, TNF-, IFN-R, and their respective
isotype controls were added to cell cultures at a concentration of 15µg/ml before stimulation.
PBMC were incubated for 12 to 18h at 37°C and then harvested for FACS analysis. Activated
cells were determined by surface expression of CD69, by degranulation and by secretion of
IFN- and TNF-.
Stimulation of NK cells in the presence of PDC and HFF
A total of 2x105 NK cells were cultured in 24 well plates in 500µl supplemented RPMI 1640
without any further cell population, with PDC in the donor-specific ratio to NK cells, with
1x105 HFF or with both PDC and HFF. Cell cultures were inoculated with 1x10
6 TCID50/ml

Materials and Methods
30
HSVUV and HSVINF, mock served as control. After incubation at 37°C for 24h, cells were
harvested for FACS analysis. NK cells were analyzed for surface expression of CD69 and
CD56.
Infection of monocytes
A total of 5x105 monocytes were cultured in 24 well plates in 500µl supplemented RPMI
1640, inoculated with HSVGFP, HSVUV and HSVINF at a MOI of 1, incubated at 37°C for 24
and 48h and harvested for FACS analysis. For neutralization experiments, IFN-R and an
isotype control were added to cell cultures at a concentration of 15µg/ml before inoculation.
Cells were analyzed for infection, viability and expression of lineage markers and monocyte
markers, as well as MHC class I (MHC-I) molecules and stress-induced molecules.
Infection of cells for quantitative polymerase chain reaction (PCR)
A total of 1x105 monocytes and HFF were cultured in 24 well plates in 500µl supplemented
RPMI 1640 and DMEM, respectively, and inoculated with HSVGFP or an infectious, but non-
replicative HSV-1 variant (HSVd106S) at a MOI of 1. After incubation at 37°C for 2h, cells
were washed once with DPBS, incubated with 100µl trypsin EDTA at 37°C for 10min and
then re-suspended to separate cells from each other and from the well surface. After addition
of 100µl supplemented cell culture medium cells were centrifuged at 590x g for 10min, re-
suspended in 500µl supplemented cell culture medium and cultured in fresh 24 well plates.
After incubation at 37°C for 24, 48, 72 and 120h, supernatants were harvested and stored at
-20°C.
4.2.6 FACS analysis of cells
Degranulation assay
For the determination of degranulation, 5µl fluorescing CD107a antibody per cell culture
was added 1.5h before harvesting the cells. In case of degranulation, the antibody could bind
to CD107a, which is a protein lining the membranes of endosomal and secretory vesicles and
being temporarily exposed on the surface of a degranulating cell.
Harvesting of cells
Primary cells were put on ice for 10min and then re-suspended thoroughly to remove all cells
from the well surface. HFF were washed once with DPBS, incubated with 100µl trypsin
EDTA at 37°C for 10min and then re-suspended to separate cells from each other and from
the well surface.

Materials and Methods
31
Cytokine secretion assays
Secretion of IFN- and TNF- by PBMC was determined using the IFN- Secretion Assay
Detection Kit and the TNF- Secretion Assay Detection Kit (Miltenyi Biotec). Harvested
cells were centrifuged at 590x g for 10min, washed once with 900µl cold cytokine buffer, re-
suspended in 90µl cold supplemented RPMI 1640 and after addition of 10µl cytokine catch
reagent antibody incubated on ice for 5min. After addition of 900µl warm supplemented
RPMI 1640, cells were incubated at 37°C for 45min in a micro tube shaker. Thereafter, cells
were put on ice for a few seconds, 1ml of cold cytokine buffer was added, and cells were
centrifuged and labeled for FACS analysis, starting with the blocking step.
Labeling of cells for FACS analysis
Harvested cells were centrifuged at 590x g for 10min, washed once with 900µl FACS buffer,
then re-suspended in 100µl FACS buffer and incubated at 4°C for 10min in the presence of
3µl FcR blocking reagent. Blocked cells were incubated with antibodies against specific cell
surface and activation markers at 4°C for 20min, washed with 3ml FACS buffer and re-
suspended in 100 - 180µl 4% PFA for fixation.
Live-dead staining of cells
Viability of cells was determined using a Fixable Violet Dead Cell Stain Kit (Invitrogen).
Cells were washed once with FACS buffer, resuspended in 100µl FACS buffer and incubated
with 0.5µg dye at 4°C for 20min, washed with 3ml FACS buffer and re-suspended in 100 -
180µl 4% PFA for fixation.
FACS analysis
Cells were analyzed in an LSRII (BD Biosciences), equipped with FACSDiva Software (BD
Biosciences) for automatic compensation and measurement of probes. Results of the
measurements were evaluated using the FCS Express 3 Software (De Novo Software).
4.2.7 Determination of secreted cytokines within supernatants
IFN-2a/2b
IFN-2a/2b (IFN-) concentrations were determined using the Human IFN- Matched
Antibody Pairs (Affymetrix eBioscience). Microwell plates were coated with 100µl coating
solution per well, sealed with an adhesive cover and incubated at 4°C overnight. After
washing once with 300µl washing buffer per well, the plate was blocked with 200µl assay
buffer per well, sealed with an adhesive cover and incubated either at room temperature for 2h
or at 4°C overnight. Standard was prepared by dilution of concentrated standard protein with
assay buffer to reach 1ng/ml standard protein. HRP-conjugate was prepared by dilution of

Materials and Methods
32
5.5µl concentrated HRP-conjugate with assay buffer to a final volume of 5.5ml. The
microwell plate was washed twice with 300µl washing buffer and filled with assay buffer:
Wells of rows 1 and 2 were filled with 100µl assay buffer for standard dilution and all other
wells were filled with 50µl assay buffer and 50µl probe. Samples were appropriately diluted
to measure within the linear range of the assay. A 1:2 dilution series of standard protein in
rows 1 and 2 was achieved by pipetting 100µl of 1ng/ml standard into the first two wells,
mixing the first dilution and pipetting 100µl of it into the next two wells. This procedure was
repeated until the penultimate wells were reached, 100µl of the last dilution were discarded,
and the last two wells filled with assay buffer served as blank controls. After addition of 50µl
HRP-conjugate per well, the plate was sealed with an adhesive cover and incubated at room
temperature for 2h on a microplate shaker. Substrate solution was prepared 30min before
continuation of the protocol by mixing equal volumes of H2O2 and tetramethylbenzidine.
After washing the plate three times, 100µl substrate solution per well was added and the plate
was incubated at room temperature for about 10min, avoiding direct exposure to light and
monitoring the color development of the standard. When the standard with the highest
concentration had developed a dark blue color, 100µl stop solution per well was added and
the plate was measured at 450nm with reference at 650nm.
sCD40L
sCD40L concentrations were determined using the Human sCD40L Matched Antibody Pairs
(Affymetrix eBioscience). Microwell plates serving as sample plates were coated with 100µl
coating solution per well, covered with an adhesive film and incubated at 4°C overnight. After
washing once with 300µl washing buffer per well, the sample plate was blocked with 200µl
assay buffer per well, sealed with an adhesive cover and incubated either at room temperature
for 2h or at 4°C overnight. Standard was prepared by dilution of concentrated standard protein
with assay buffer to reach 20ng/ml standard protein. HRP-conjugate was prepared by dilution
of 11µl concentrated HRP-conjugate with assay buffer to a final volume of 11ml. Wells of
rows 1 and 2 of a dilution plate were filled with 100µl sample diluent for further standard
dilution and all other wells were filled with 80µl sample diluent and 20µl of 1:5-diluted
plasma probes. A 1:2 dilution series of standard protein was achieved by pipetting 100µl of
20ng/ml standard into the first two wells, mixing the first dilution and pipetting 100µl of it
into the next two wells. This procedure was repeated until the penultimate wells were reached,
100µl of the last dilution were discarded, and the last two wells filled with sample diluent
served as blank controls. Last, 100µl HRP-conjugate were added per well. The sample plate
was washed twice with 300µl washing buffer. After transfer of 150µl from the wells of the
dilution plate into the sample plate, the sample plate was sealed with an adhesive cover and
incubated at room temperature for 2h on a microplate shaker. Substrate solution was prepared
30min before continuation of the protocol by mixing equal volumes of H2O2 and
tetramethylbenzidine. After washing the plate three times, 100µl substrate solution per well

Materials and Methods
33
was added and the plate was incubated at room temperature for about 10min, avoiding direct
exposure to light and monitoring the color development of the standard. When the highest
standard had developed a dark blue color, 100µl stop solution per well was added and plate
was measured at 450nm with reference at 650nm.
Th1/Th2 cytokines
Cytokines secreted into supernatants were analyzed using the Th1/Th2 11plex RTU
FlowCytomix Multiplex kit (Affymetrix eBioscience). For 96 samples, assay buffer was
prepared by dilution of 50ml 10x assay buffer with deionized H2O to a final volume of 500ml.
Bead mix was prepared by dilution of 1.5ml 2x bead mix with reagent dilution buffer to a
final volume of 3ml. Bead mix was centrifuged at 3,000x g for 5min, supernatant was
discarded, beads were re-suspended in 3ml reagent dilution buffer and mixed well. Biotin
conjugate mix was prepared by dilution of 3.5ml 2x biotin conjugate mix with reagent
dilution buffer to a final volume of 7ml. Standard protein was reconstituted in 200µl assay
buffer, mixed well and completely resolved within 10 - 30min. A 1:3 standard dilution series
was achieved by mixing 50µl standard with 100µl assay buffer, repeating this procedure with
the resulting dilution five times. Assay buffer served as negative control. FACS tubes were
filled with 25µl of the samples, standard dilution series and negative control. Standard
dilution series and negative control were used in duplicate, the standard with the highest
concentration was used in triplicate for instrument setup. After addition of 25µl bead mix and
50µl biotin conjugate mix per tube, probes were incubated at room temperature in the dark for
2h. Streptavidin-PE solution was prepared by dilution of 200µl concentrated streptavidin-PE
with 6,050µl assay buffer. After two washing steps of the probes (addition of 1ml assay buffer
per tube, centrifugation at 355x g for 5min and discarding of the supernatant), 50µl
streptavidin-PE solution per tube was added and probes were incubated at room temperature
in the dark for 1h. After two more washing steps, 500µl assay buffer was added per tube and
probes were ready for FACS analysis. Instrument was setup using the setup beads and the
standard with the highest concentration to adjust FSC / SSC parameters, to create regions for
the different bead populations, that were defined by different size and APC fluorescence
intensity, and to adjust voltage of PE emission, so that the bead population of the negative
control was visible far left in the plot, while the bead population of the highest standard was
visible far right in the plot. After analysis of the standard, probes were measured. Results of
the measurement were evaluated using the FlowCytomixPro software (Affymetrix
eBioscience).

Materials and Methods
34
4.2.8 Quantification of HSV-1 DNA
Isolation of viral DNA from cell culture supernatants
HSV-1 DNA was extracted from cell culture supernatants using the EZ1 Virus Mini Kit v2.0
together with the EZ1 Advanced XL robotic workstation (both Qiagen, Hilden, DE) according
to the manufacturer’s recommendations. A total of 200µl of supernatant was used for
extraction and DNA was eluted into 120µl of volume. Isolation of HSV-1 DNA was
performed by the diagnostic services of the Institute of Microbiology and Hygiene,
Regensburg.
Quantitative PCR
Absolute quantification of HSV-1 DNA was performed by realtime amplification of a
sequence within the HSV-1 glycoprotein G. HSV-1 DNA concentration within each sample
was determined with reference to standard controls containing defined copies of HSV-1 DNA.
The mastermix contained forward and reverse primers and VIC-/FAM-TAMRA-labeled
Taqman probes for HSV-1 and HSV-2 (Metabion, Martinsried, DE). 5µl of each sample was
added to 25µl mastermix and amplified in duplicates. Samples were analyzed using the
StepOnePlus Real-Time PCR System (Applied Biosystems, Darmstadt, DE). Initial
denaturation at 95°C for 10min was followed by 45 cycles of annealing and extension at 60°C
for 1min and denaturation at 95°C for 15sec. Quantification of HSV-1 DNA was performed
by the diagnostic services of the Institute of Microbiology and Hygiene, Regensburg.
4.2.9 Virological analysis of hyperproliferative lesions
A swab from the hyperproliferative lesions was analyzed using the RealArt HSV-1/2 PCR kit
according to the manufacturer’s recommendations (Qiagen). For the analysis of
papillomavirus DNA, E1 consensus primers were used for amplification, sequencing, and
GenBank alignement (Iftner et al., 2003). All analyses were performed by the diagnostic
services of the Institute of Clinical and Molecular Virology, Erlangen, Germany. These
studies together with the immunological analyses of the patient’s PBMC were approved by
the Ethical Committee of the Medical Faculty, Friedrich-Alexander-Universität Erlangen-
Nürnberg (Ref. no. 3375).
4.2.10 Statistical analysis
Statistical analysis was carried out using the online tool VassarStats Statistical Computation
(http://www.vassarstats.net/). For comparison of two samples the Student’s t-test was applied,
for comparison of three or more samples the Tukey HSD test was applied to account for
multiple comparisons.

Results
35
5 Results
5.1 Stimulation of PBMC with HSV-1 leads to NK cell activation
In order to analyze the potential of HSV-1 to induce NK cell activation and effector functions
within the PBMC context, we stimulated PBMC with infectious (HSVINF) and UV-inactivated
(HSVUV) HSV-1, CpG-A-ODN (CpG-A), a toll-like receptor 9 (TLR-9) agonist, and IL-2
(FIG. 6A). CpG-A served as representative of PDC-dependent NK cell activation (Hemmi et
al., 2000) and IL-2 was used for direct NK cell activation (Trinchieri et al., 1984). All stimuli
significantly up-regulated the activation marker CD69 on NK cells compared to the mock
control (p<0.01) (FIG. 6B, C). Comparison of two different time points displayed diverse
kinetics of NK cell activation by the different stimuli. At 12h post stimulation (p.s.) HSVINF
induced significantly stronger NK cell CD69 up-regulation than all other stimuli, while at 18h
p.s. the significant difference to HSVINF was lost for CpG-A and HSVUV, and reduced for
IL-2 (p<0.05). Altogether, the data indicate faster NK cell activation by HSVINF than by the
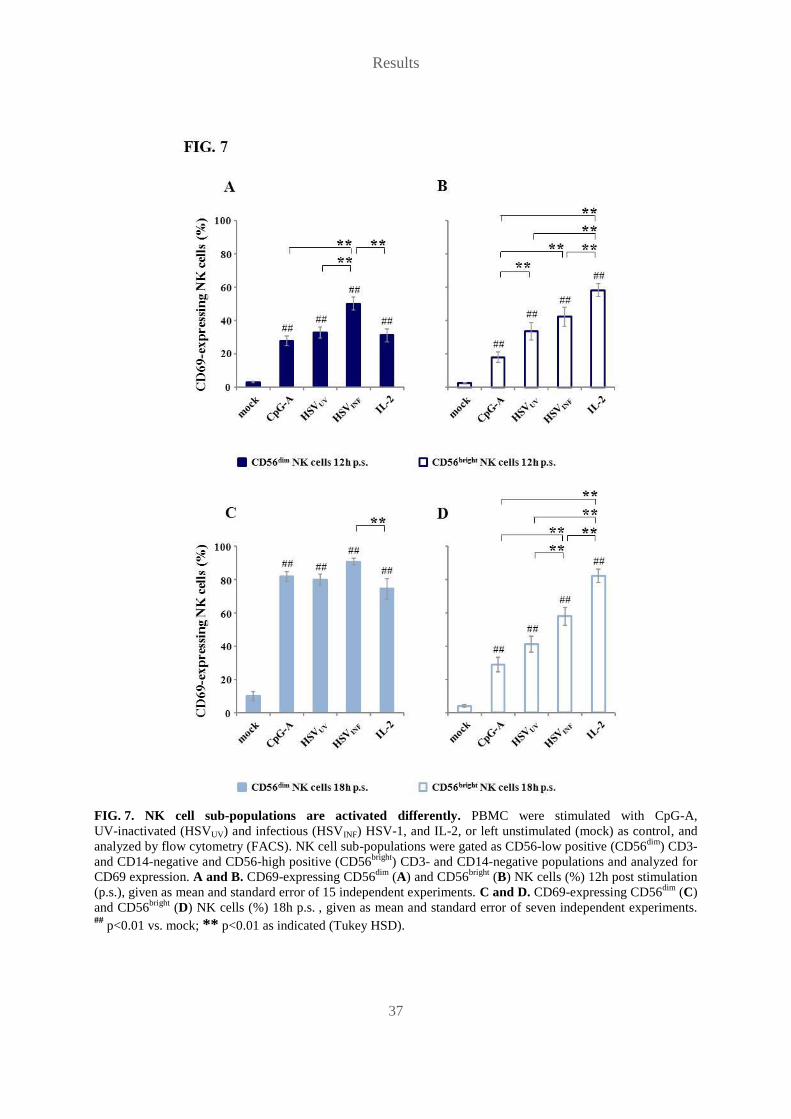
other stimuli.
All stimuli activated both CD56dim
and CD56bright
NK cells significantly compared to the
mock control at 12h and 18h p.s. (p<0.01), but activation of the CD56bright
population varied
between stimuli. CD56dim
cells were activated faster by HSVINF than by any other stimulus
(FIG. 7A), but significant differences between HSVINF and CpG-A as well as HSVUV did not
persist (FIG. 7C). IL-2 proved to be the strongest stimulus for CD69 up-regulation on
CD56bright
NK cells with significant differences to all other stimuli at both time points
(p<0.01) (FIG. 7B, D). Both HSVINF and HSVUV activated CD56bright
NK cells to a greater
extent than CpG-A, but the difference to CpG-A persisted only for HSVINF at both time points
(p<0.01), while it was lost for HSVUV from 12h (p<0.01) to 18h p.s. Furthermore, at 18h p.s.
HSVINF-induced activation of CD56bright
NK cells was significantly stronger than HSVUV-
induced activation of CD56bright
NK cells (p<0.05). The discrepancy in kinetics and activation
of CD56bright
NK cells between HSVINF and HSVUV indicates that viral infectivity might be
important for HSV-1-induced NK cell activation.

Results
36
FIG. 6. HSV-1 induces CD69 up-regulation on NK cells. PBMC were stimulated with CpG-A, UV-inactivated
(HSVUV) and infectious (HSVINF) HSV-1, and IL-2, or left unstimulated (mock) as control, and analyzed by flow
cytometry (FACS). NK cells were gated as CD56-positive CD3- and CD14-negative population and analyzed for
CD69 expression. A. Representative FACS plot of NK cells 12h and 18h post stimulation (p.s.) B and C. CD69-
expressing NK cells (%) 12h (B) and 18h (C) p.s. , given as mean and standard error of 15 (B) and seven (C)
independent experiments. ##
p<0.01 vs. mock; * p<0.05, ** p<0.01 as indicated (Tukey HSD).
Results
37
FIG. 7. NK cell sub-populations are activated differently. PBMC were stimulated with CpG-A,
UV-inactivated (HSVUV) and infectious (HSVINF) HSV-1, and IL-2, or left unstimulated (mock) as control, and
analyzed by flow cytometry (FACS). NK cell sub-populations were gated as CD56-low positive (CD56dim
) CD3-
and CD14-negative and CD56-high positive (CD56bright
) CD3- and CD14-negative populations and analyzed for
CD69 expression. A and B. CD69-expressing CD56dim
(A) and CD56bright
(B) NK cells (%) 12h post stimulation
(p.s.), given as mean and standard error of 15 independent experiments. C and D. CD69-expressing CD56dim
(C)
and CD56bright
(D) NK cells (%) 18h p.s. , given as mean and standard error of seven independent experiments. ##
p<0.01 vs. mock; ** p<0.01 as indicated (Tukey HSD).

Results
38
5.2 Only infectious HSV-1 induces NK cell effector functions
We next wanted to know, if CD69 up-regulation reflected induction of NK cell effector
functions, so we investigated NK cell IFN- secretion, using a Cytokine Secretion Assay
Detection Kit (Miltenyi Biotec), and NK cell degranulation, detecting CD107a surface
expression. CD107a, also called lysosomal-associated membrane protein-1 (LAMP-1), lines
the membranes of endosomal and secretory vesicles, like cytolytic granules, and is normally
not expressed on the outer cellular membrane. Upon release of cytolytic granules, CD107a is
temporarily present on the cell surface, therefore serving as an indicator of degranulation. The
correlation of CD107a surface expression with cytokine secretion and in particular
cytotoxicity has been demonstrated for NK cells (Alter et al., 2004). NK cell effector
functions were measured at 12h p.s. (FIG. 8A). CpG-A and HSVUV failed to induce NK cell
IFN- secretion as well as degranulation, whereas IL-2 stimulated IFN- secretion (p<0.05)
(FIG. 8B), but no degranulation (FIG. 8C). HSVINF induced significant IFN- secretion by NK
cells compared to the mock control (p<0.01), and also compared to CpG-A (p<0.01), HSVUV
(p<0.05) and IL-2 (p<0.05) (FIG. 8B). HSVINF was also the only stimulus leading to
significant degranulation compared to the mock control and all other stimuli (p<0.01)
(FIG. 8C). The fact that only HSVINF, not HSVUV, was able to induce NK cell effector
functions further supports the relevance of HSV-1 infectivity for full activation of NK cells
and their effector functions. Interestingly, HSVINF-induced effector functions were not
restricted to one of the NK cell subpopulations, as reported in the literature, namely IFN-
secretion mainly by CD56bright
cells and degranulation, indicating cytotoxicity, mostly by
CD56dim
cells (Cooper et al., 2001a). NK cell effector functions rather seemed evenly
distributed between both subpopulations (FIG. 8A), without significant differences in IFN-
secretion and degranulation between CD56bright
cells and CD56dim
cells (data not shown).
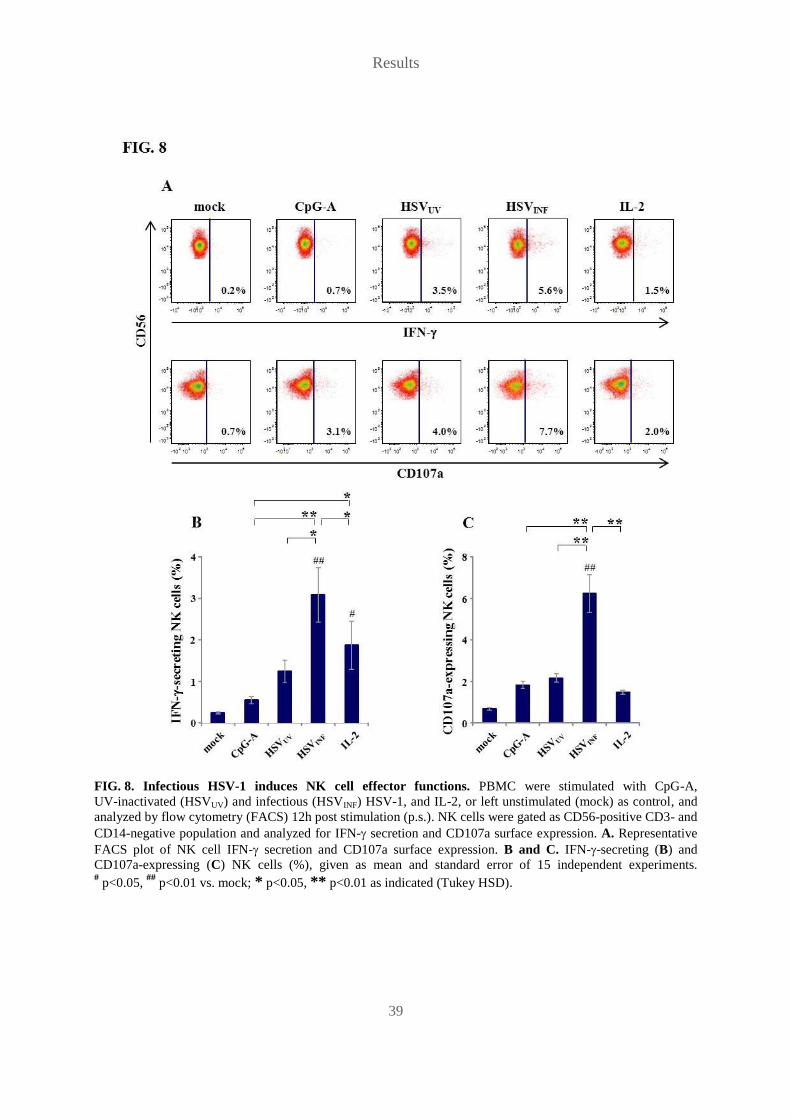
Results
39
FIG. 8. Infectious HSV-1 induces NK cell effector functions. PBMC were stimulated with CpG-A,
UV-inactivated (HSVUV) and infectious (HSVINF) HSV-1, and IL-2, or left unstimulated (mock) as control, and
analyzed by flow cytometry (FACS) 12h post stimulation (p.s.). NK cells were gated as CD56-positive CD3- and
CD14-negative population and analyzed for IFN- secretion and CD107a surface expression. A. Representative
FACS plot of NK cell IFN- secretion and CD107a surface expression. B and C. IFN--secreting (B) and
CD107a-expressing (C) NK cells (%), given as mean and standard error of 15 independent experiments.
# p<0.05,
## p<0.01 vs. mock; * p<0.05, ** p<0.01 as indicated (Tukey HSD).
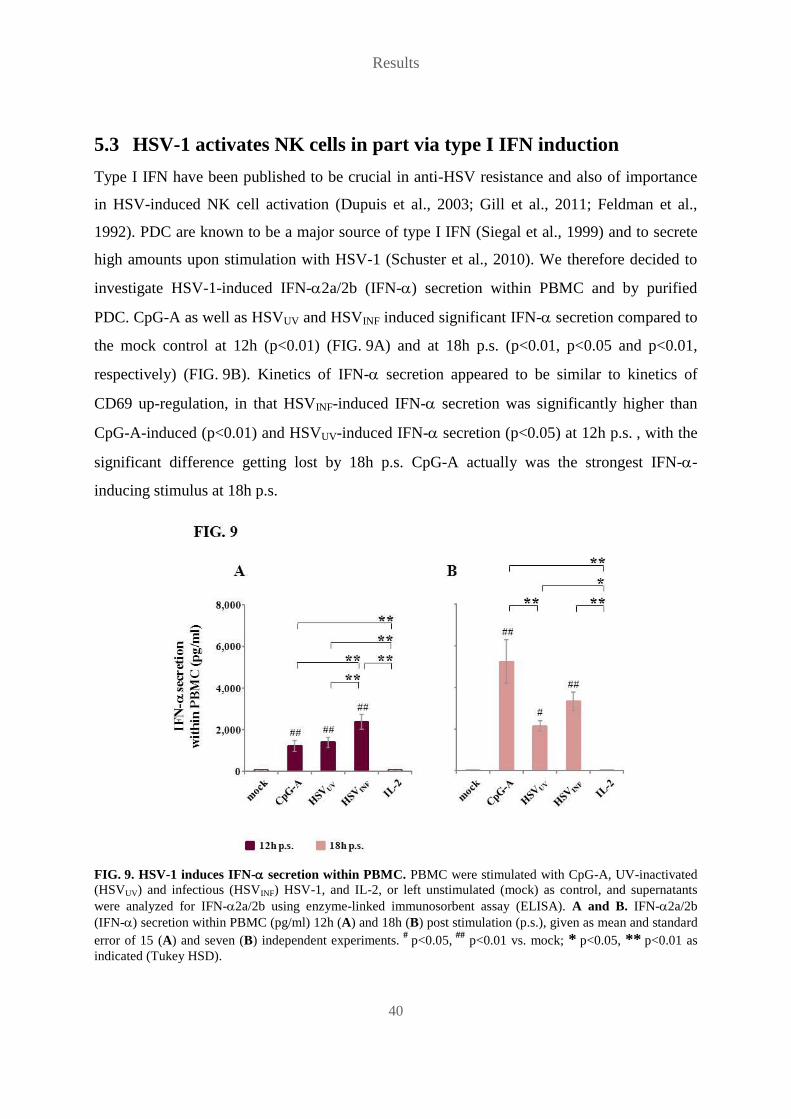
Results
40
5.3 HSV-1 activates NK cells in part via type I IFN induction
Type I IFN have been published to be crucial in anti-HSV resistance and also of importance
in HSV-induced NK cell activation (Dupuis et al., 2003; Gill et al., 2011; Feldman et al.,
1992). PDC are known to be a major source of type I IFN (Siegal et al., 1999) and to secrete
high amounts upon stimulation with HSV-1 (Schuster et al., 2010). We therefore decided to
investigate HSV-1-induced IFN-2a/2b (IFN-) secretion within PBMC and by purified
PDC. CpG-A as well as HSVUV and HSVINF induced significant IFN- secretion compared to
the mock control at 12h (p<0.01) (FIG. 9A) and at 18h p.s. (p<0.01, p<0.05 and p<0.01,
respectively) (FIG. 9B). Kinetics of IFN- secretion appeared to be similar to kinetics of
CD69 up-regulation, in that HSVINF-induced IFN- secretion was significantly higher than
CpG-A-induced (p<0.01) and HSVUV-induced IFN- secretion (p<0.05) at 12h p.s. , with the
significant difference getting lost by 18h p.s. CpG-A actually was the strongest IFN--
inducing stimulus at 18h p.s.
FIG. 9. HSV-1 induces IFN- secretion within PBMC. PBMC were stimulated with CpG-A, UV-inactivated
(HSVUV) and infectious (HSVINF) HSV-1, and IL-2, or left unstimulated (mock) as control, and supernatants
were analyzed for IFN-2a/2b using enzyme-linked immunosorbent assay (ELISA). A and B. IFN-2a/2b
(IFN-) secretion within PBMC (pg/ml) 12h (A) and 18h (B) post stimulation (p.s.), given as mean and standard
error of 15 (A) and seven (B) independent experiments. #
p<0.05, ##
p<0.01 vs. mock; * p<0.05, ** p<0.01 as
indicated (Tukey HSD).

Results
41
Stimulation of 5x105 purified PDC with HSVINF led to a mean IFN- secretion into
supernatants of about 10ng/ml (data not shown), and we decided to examine the potential of
PDC-derived supernatants (PDC-SN) to stimulate CD69 up-regulation on purified NK cells.
Kinetic studies revealed a rapid effect of PDC-SN on NK cells. Supernatants containing an
INF-2a/2b concentration of 20pg/ml lead to significant CD69 up-regulation compared to the
mock control within 12h (p<0.05) and supernatants of 40 or 80pg/ml INF-2a2b did this even
within 3h (p<0.05 and p<0.01, respectively) (FIG 10A). This demonstrates that PDC-SN of
low IFN- concentrations are sufficient to activate NK cells within few hours. Interestingly,
NK cell CD69 up-regulation mediated by PDC-SN seemed to occur in two phases, namely
between 3 and 6h and between 12 and 18h p.s. , with a plateau between 6 and 12h p.s.
In order to figure out if NK cell activation induced by PDC-SN was due to IFN- or other
type I IFN, we compared PDC-SN with recombinant human IFN-2b (rhIFN-) containing
equal IFN-2a/2b concentrations and further used an antibody against the IFN-/ receptor
(IFN-R) to block any impact of type I IFN. Stimulation of purified NK cells with serial
dilutions of PDC-SN and rhIFN- confirmed IFN- as potent stimulus for NK cell activation,
with saturation beginning at IFN-2a/2b concentrations between 32 and 64pg/ml (FIG. 10B).
The effect of PDC-SN on CD69 up-regulation was more potent than the effect of rhIFN-.
Neutralization of IFN-R diminished CD69 up-regulation caused by PDC-SN significantly
(p<0.05), proving type I IFN as major soluble factors in PDC-dependent NK cell activation
after stimulation with HSV-1. CD69 up-regulation by rhIFN- was reduced clearly, but not
significantly (FIG. 10C).
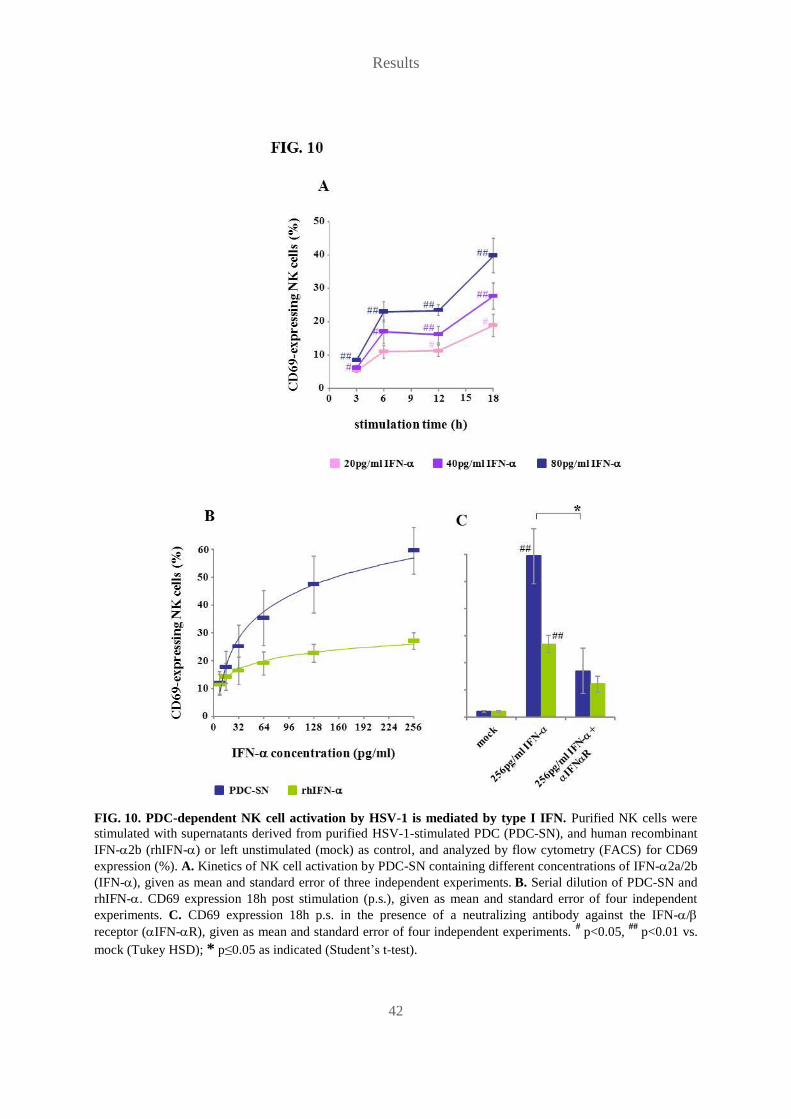
Results
42
FIG. 10. PDC-dependent NK cell activation by HSV-1 is mediated by type I IFN. Purified NK cells were
stimulated with supernatants derived from purified HSV-1-stimulated PDC (PDC-SN), and human recombinant
IFN-2b (rhIFN-) or left unstimulated (mock) as control, and analyzed by flow cytometry (FACS) for CD69
expression (%). A. Kinetics of NK cell activation by PDC-SN containing different concentrations of IFN-2a/2b
(IFN-), given as mean and standard error of three independent experiments. B. Serial dilution of PDC-SN and
rhIFN-. CD69 expression 18h post stimulation (p.s.), given as mean and standard error of four independent
experiments. C. CD69 expression 18h p.s. in the presence of a neutralizing antibody against the IFN-/
receptor (IFN-R), given as mean and standard error of four independent experiments. # p<0.05,
## p<0.01 vs.
mock (Tukey HSD); * p≤0.05 as indicated (Student’s t-test).

Results
43
5.4 TNF- plays a major role in HSV-1-induced NK cell activation
PDC-SN seemed to be more potent in activating NK cells than rhIFN-, and within PBMC
more cell populations are present besides PDC, so other cytokines besides type I IFN could be
involved in HSV-1-induced NK cell activation. We therefore decided to check supernatants of
CpG-A- and HSV-1-stimulated PBMC for further cytokines using a Human Th1/Th2 11plex
bead array (Affymetrix eBioscience). Neither stimulus caused significant levels of IL-12,
IFN-, IL-2, IL-10, IL-4, IL-5 or TNF- within supernatants. Significant IL-6 secretion
compared to the mock control was induced by CpG-A (p<0.01) (data not shown), while IL-8
was secreted in all samples including the mock control (FIG. 11). Two cytokines, namely
IL-1 and TNF-, were significantly increased in HSVINF-stimulated PBMC compared to the
mock control and to HSVUV-stimulated PBMC (p<0.05). These findings suggest that IL-1
and TNF- might be involved in the stimulation of NK cell activation and in particular NK
cell effector functions by HSVINF.
FIG. 11. Infectious HSV-1 induces secretion of IL-1 and TNF-. PBMC were stimulated for 18h with
CpG-A, UV-inactivated (HSVUV) and infectious (HSVINF) HSV-1, or left unstimulated (mock) as control, and
supernatants were analyzed for cytokines using an 11plex bead array (Affymetrix eBioscience). Secretion of
IL-8, IL-1, and TNF- within PBMC (pg/ml), given as mean and standard error of seven independent
experiments. #
p<0.05 vs. mock; * p<0.05 as indicated (Tukey HSD).

Results
44
In order to investigate the effect of PDC-derived type I IFN, and also of IL-1 and TNF-,
more closely, neutralization experiments were conducted. PBMC were stimulated with
CpG-A, HSVUV and HSVINF in the presence of antibodies against IFN-R, IL-1 and TNF-
or the respective isotype controls. At 12h p.s. NK cell CD69 up-regulation, IFN- secretion
and degranulation as well as IFN- secretion within PBMC were determined. Neutralization
of TNF- significantly decreased CD69 up-regulation induced by CpG-A, HSVUV and
HSVINF (p<0.01) (FIG. 12A), and it also significantly reduced HSVINF-induced IFN-
secretion (p<0.05) (FIG. 12B), while it did not affect NK cell degranulation (FIG. 12C).
Blocking of IFN-R significantly diminished CpG-A- and HSVUV-induced CD69 up-
regulation (p=0.05 and p<0.01, respectively), but had only a minimal effect on HSVINF-
induced CD69 up-regulation (FIG. 12A) and no inhibitory effect on NK cell effector
functions (FIG. 12B, C). In fact, neutralization of the IFN-R even increased IFN- secretion,
although not significantly (FIG. 12B). In contrast, neutralization of IL-1 did neither
influence NK cell activation nor NK cell effector functions. IFN- secretion was reduced by
neutralization of IFN-R, in consistence with the known autokrine loop (Marie et al., 1998),
and interestingly, also by neutralization of TNF- as well as IL-1 (FIG. 12D). Reduction of
IFN- levels was distinct, although only significant for CpG-A (p<0.05 for TNF- and
IFN-R). Simultaneous neutralization of TNF- and IFN-R did not result in increased
effects on HSVINF-induced CD69 up-regulation (FIG. 13A), degranulation (FIG. 13C) or
IFN- secretion (FIG. 13D), while the increase of IFN- secretion observed after IFN-R
neutralization was abolished by combination of both antibodies (FIG. 13B). These findings
indicate a crucial role for TNF- in HSVINF-induced NK cell activation and IFN- secretion,
whereas it is negligible in HSVINF-induced NK cell degranulation. Besides, all three cytokines
seem to be required for the secretion of large amounts of IFN- upon stimulation with either
CpG-A or HSV-1. Furthermore, combined neutralization of TNF- and IFN-R suggests
opposed functions of TNF- and type I IFN in IFN- induction by HSVINF.
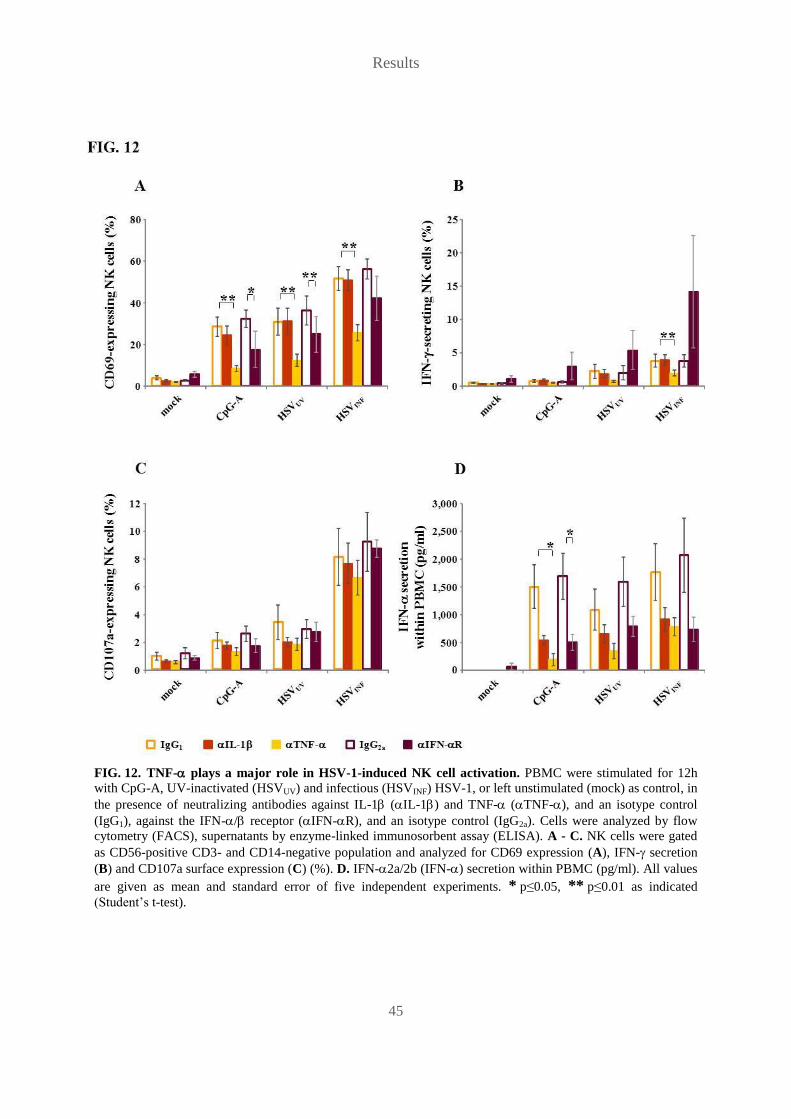
Results
45
FIG. 12. TNF- plays a major role in HSV-1-induced NK cell activation. PBMC were stimulated for 12h
with CpG-A, UV-inactivated (HSVUV) and infectious (HSVINF) HSV-1, or left unstimulated (mock) as control, in
the presence of neutralizing antibodies against IL-1 (IL-1) and TNF- (TNF-), and an isotype control
(IgG1), against the IFN-/ receptor (IFN-R), and an isotype control (IgG2a). Cells were analyzed by flow
cytometry (FACS), supernatants by enzyme-linked immunosorbent assay (ELISA). A - C. NK cells were gated
as CD56-positive CD3- and CD14-negative population and analyzed for CD69 expression (A), IFN- secretion
(B) and CD107a surface expression (C) (%). D. IFN-2a/2b (IFN-) secretion within PBMC (pg/ml). All values
are given as mean and standard error of five independent experiments. * p≤0.05, ** p≤0.01 as indicated
(Student’s t-test).
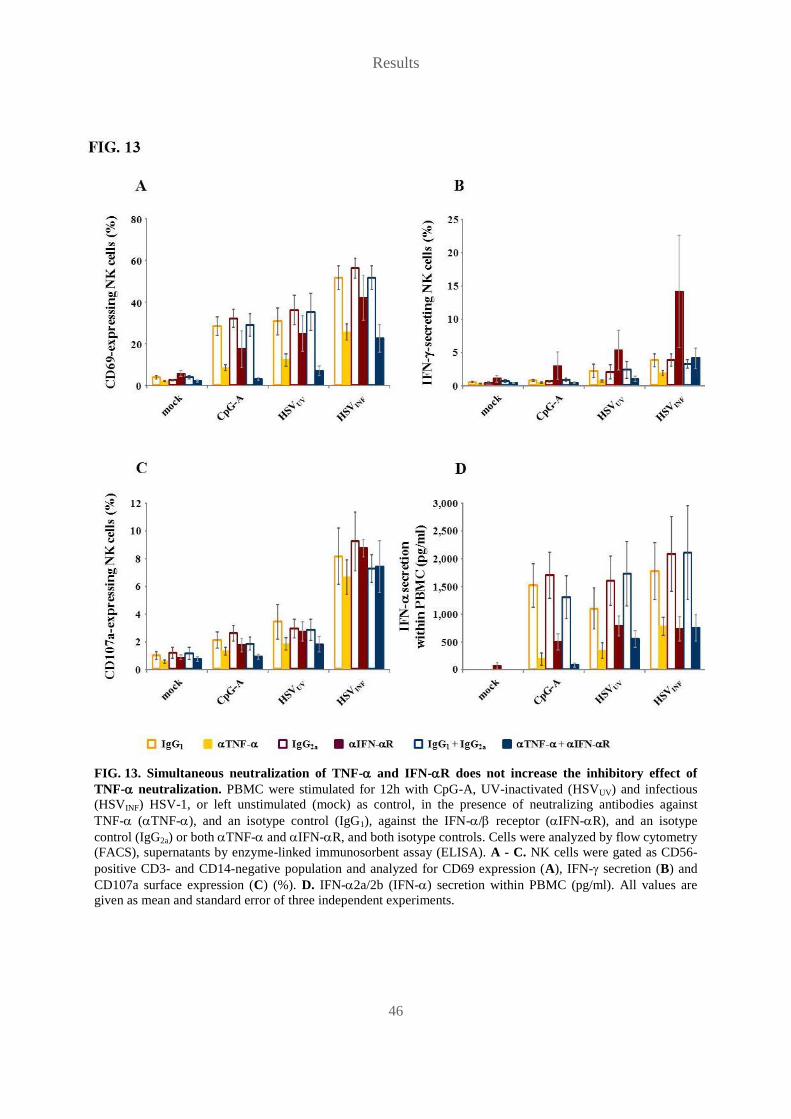
Results
46
FIG. 13. Simultaneous neutralization of TNF- and IFN-R does not increase the inhibitory effect of
TNF- neutralization. PBMC were stimulated for 12h with CpG-A, UV-inactivated (HSVUV) and infectious
(HSVINF) HSV-1, or left unstimulated (mock) as control, in the presence of neutralizing antibodies against
TNF- (TNF-), and an isotype control (IgG1), against the IFN-/ receptor (IFN-R), and an isotype
control (IgG2a) or both TNF- and IFN-R, and both isotype controls. Cells were analyzed by flow cytometry
(FACS), supernatants by enzyme-linked immunosorbent assay (ELISA). A - C. NK cells were gated as CD56-
positive CD3- and CD14-negative population and analyzed for CD69 expression (A), IFN- secretion (B) and
CD107a surface expression (C) (%). D. IFN-2a/2b (IFN-) secretion within PBMC (pg/ml). All values are
given as mean and standard error of three independent experiments.

Results
47
5.5 Monocytes contribute to HSV-1-induced TNF- production
Since TNF- seemed to be the key cytokine in HSVINF-induced NK cell activation within
PBMC, we were interested in which cell populations might be responsible for TNF-
production and performed a Cytokine Secretion Assay (Miltenyi Biotec) to detect
TNF--secreting cells within PBMC. We analyzed TNF- secretion within seven different
cell populations, namely PDC, monocytes, B cells, NK cells, T cells, CD4+ T cells and
CD8+ T cells, upon stimulation with CpG-A, HSVUV and HSVINF. We first looked at TNF-
secretion within the individual cell populations and could identify PDC and monocytes as
major TNF- sources with significant secretion upon stimulation with CpG-A (p<0.01),
HSVUV (p<0.01 for PDC and p<0.05 for monocytes) and HSVINF compared to mock (p<0.01)
(FIG. 14A). In addition, CpG-A stimulated TNF- secretion within the B cell population
(p<0.01), while HSVINF induced TNF- secretion within B cells (p<0.01), NK cells (p<0.01),
T cells (p<0.05), CD4+ T cells (p<0.01) and CD8
+ T cells (p<0.05). A significant
difference in TNF- secretion between HSVUV and HSVINF stimulation was observed within
monocytes (p<0.01). We next decided to identify total TNF- secretion within PBMC. For
this purpose we multiplied TNF- secretion within each cell population (FIG. 14A) with the
frequency of the respective cell population within PBMC (FIG. 14B), resulting in each cell
population’s TNF- secretion within PBMC, and combined TNF- secretion of all cell
populations, to get total TNF- secretion within PBMC (FIG. 14C). Interestingly, only
CpG-A and HSVINF induced significant overall TNF- secretion within PBMC compared to
the mock control (p<0.01), and HSVINF-induced TNF- secretion also differed significantly
from HSVUV-induced TNF- secretion (p<0.05), confirming the results of the bead array
(FIG. 11). Notably, monocytes appeared to be key producers of TNF- in this analysis
(FIG. 14C).
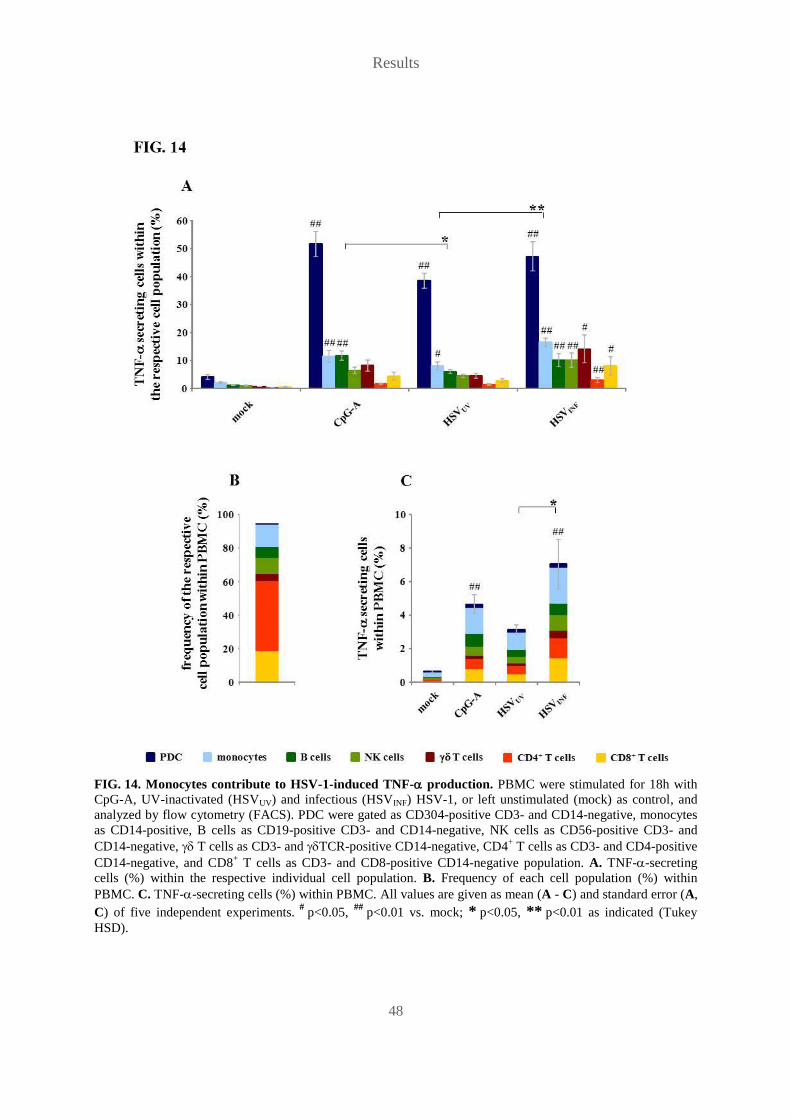
Results
48
FIG. 14. Monocytes contribute to HSV-1-induced TNF- production. PBMC were stimulated for 18h with
CpG-A, UV-inactivated (HSVUV) and infectious (HSVINF) HSV-1, or left unstimulated (mock) as control, and
analyzed by flow cytometry (FACS). PDC were gated as CD304-positive CD3- and CD14-negative, monocytes
as CD14-positive, B cells as CD19-positive CD3- and CD14-negative, NK cells as CD56-positive CD3- and
CD14-negative, T cells as CD3- and TCR-positive CD14-negative, CD4+ T cells as CD3- and CD4-positive
CD14-negative, and CD8+ T cells as CD3- and CD8-positive CD14-negative population. A. TNF--secreting
cells (%) within the respective individual cell population. B. Frequency of each cell population (%) within
PBMC. C. TNF--secreting cells (%) within PBMC. All values are given as mean (A - C) and standard error (A,
C) of five independent experiments. #
p<0.05, ##
p<0.01 vs. mock; * p<0.05, ** p<0.01 as indicated (Tukey
HSD).

Results
49
5.6 Monocytes can be infected by HSV-1
Next, we decided to engage in HSV-1 infection experiments, because NK cells are known to
recognize infected cells as target cells (Vivier, 2006). Monocyte activation seemed to be
particularly influenced by HSV-1 infectivity, and several working groups could already
demonstrate infection of mononuclear phagocytes (Daniels et al., 1978; Albers et al., 1989).
Since preliminary experiments of PBMC infected with a virus isolate (HSVGFP) expressing a
GFP-VP22 fusion protein also hinted at monocytes as HSV-1 target cells within PBMC (data
not shown), we conducted infection experiments with isolated monocytes. We noticed that
monocytes, which had been purified using magnetic beads specific for CD14, were only in
part positive for CD14 after being in cell culture for 24h or 48h (FIG. 15). Labeling of freshly
isolated monocytes evidenced a purity of about 95%, and staining of monocytes for lineage
markers as well as specific phagocyte markers demonstrated a contamination by other cells of
less than 5% after cultivation (data not shown). Thus, monocytes appear to down-regulate
CD14 when being cultured.
FIG. 15. Monocytes down-regulate CD14 upon cultivation. Monocytes were purified by magnetic-activated
cell sorting (MACS) using CD14-coupled beads and analyzed by flow cytometry (FACS) for CD14 expression.
Representative FACS plot of monocytes immediately post purification (p.p.) and 24h and 48h p.p.
Monocytes were infected with HSVGFP and analyzed for green fluorescence. HSVINF was used
as infectious non-fluorescent and HSVUV as non-infectious non-fluorescent control virus. In
fact, monocytes infected with HSVGFP exhibited significant green fluorescence compared to
the mock control and the two non-fluorescent viruses HSVUV and HSVINF at 24h (p<0.01) and
48h post infection (p.i.) (p<0.01 for mock and HSVUV, n.s. for HSVINF), demonstrating
infection of monocytes (FIG. 16A). The percentage of HSVGFP-infected monocytes declined

Results
50
from 24h to 48h p.i. , indicating rather abortive than productive HSV-1 infection, in
concordance with observations of several working groups (Daniels et al., 1978; Albers et al.,
1989; Bruun et al., 1998). In order to determine productivity of monocyte infection we
analyzed supernatants of HSV-1-infected monocytes and HSV-1-infected human foreskin
fibroblasts (HFF) as control cells for HSV-1 DNA, using quantitative PCR. Cells were
infected with HSVGFP and with another HSV-1 variant (HSVd106S), which is infectious, but
non-replicative (Liu et al., 2009). Infected cells were cultured for up to 5 days (FIG. 16B).
HSV-1 DNA increased over time in supernatants of HSVGFP-infected HFF, whereas it
declined in supernatants of HSVd106S-infected HFF, corresponding to the replication capacities
of HSVGFP and HSVd106S. In contrast, HSV-1 DNA dropped in supernatants of HSVd106S- as
well as HSVGFP-infected monocytes. These results confirm non-productive infection of
monocytes by HSV-1.
FIG. 16. Monocytes are non-productively infected by HSV-1. A. Purified monocytes were infected with
UV-inactivated (HSVUV), infectious (HSVINF), and infectious GFP-expressing (HSVGFP) HSV-1, or left
uninfected (mock) as control, and analyzed by flow cytometry (FACS) for green fluorescence (%) 24h and 48h
post infection (p.i.). Values are given as mean and standard error of eleven independent experiments. ##
p<0.01
vs. mock; ** p<0.01 as indicated (Tukey HSD). B. Purified monocytes of three different donors and human
foreskin fibroblasts (HFF) were infected with infectious GFP-expressing (HSVGFP) and an infectious but
replication-deficient GFP-expressing (HSVd106S) HSV-1 and cultivated for different time periods. Supernatants
of the indicated time points were analyzed by quantitative PCR for viral load (copies/ml). Values of monocytes
are given as mean of three different donors.

Results
51
5.7 Monocytes up-regulate MHC-I upon exposure to infectious HSV-1
Since HSV-1 has been published to down-regulate MHC-I molecules via ICP47 (Hill et al.,
1995; Früh et al., 1995), which might be responsible for HSVINF-induced NK cell activation
(Huard and Früh, 2000), we checked for expression of classical HLA-ABC and non-classical
HLA-E. Monocytes inoculated with HSVINF and HSVGFP exhibited significant HLA-ABC up-
regulation compared to the mock control at 24h and 48h p.i. (p<0.01), with rising kinetics
from 24h to 48h p.i. (p<0.01) (FIG. 17A). In contrast, HSVUV did not induce HLA-ABC up-
regulation, but behaved like the mock control with significant differences to both HSVINF and
HSVGFP at 24h and 48h p.i. (p<0.01). HLA-E was regulated in a similar manner to
HLA-ABC, with overall up-regulation being induced by HSVINF and HSVGFP (FIG. 17B).
Although plotting green fluorescence against HLA-ABC and HLA-E expression indicated
MHC-I down-regulation in few infected monocytes, overall up-regulation of MHC-I in
monocyte cultures was much more distinct (FIG. 18).
FIG. 17. Monocytes up-regulate MHC-I upon exposure to infectious HSV-1. Purified monocytes were
infected with UV-inactivated (HSVUV), infectious (HSVINF), and infectious GFP-expressing (HSVGFP) HSV-1, or
left uninfected (mock) as control, and analyzed by flow cytometry (FACS) for MHC-I expression (MFI) 24h and
48h post infection (p.i.). A. Fold change of HLA-ABC expression, given as mean and standard error of eleven
independent experiments. ##
p<0.01 vs. mock; ** p<0.01 as indicated (Tukey HSD). B. Fold change of HLA-E
expression, given as mean of two (HSVUV, HSVINF) and three (HSVGFP) independent experiments.
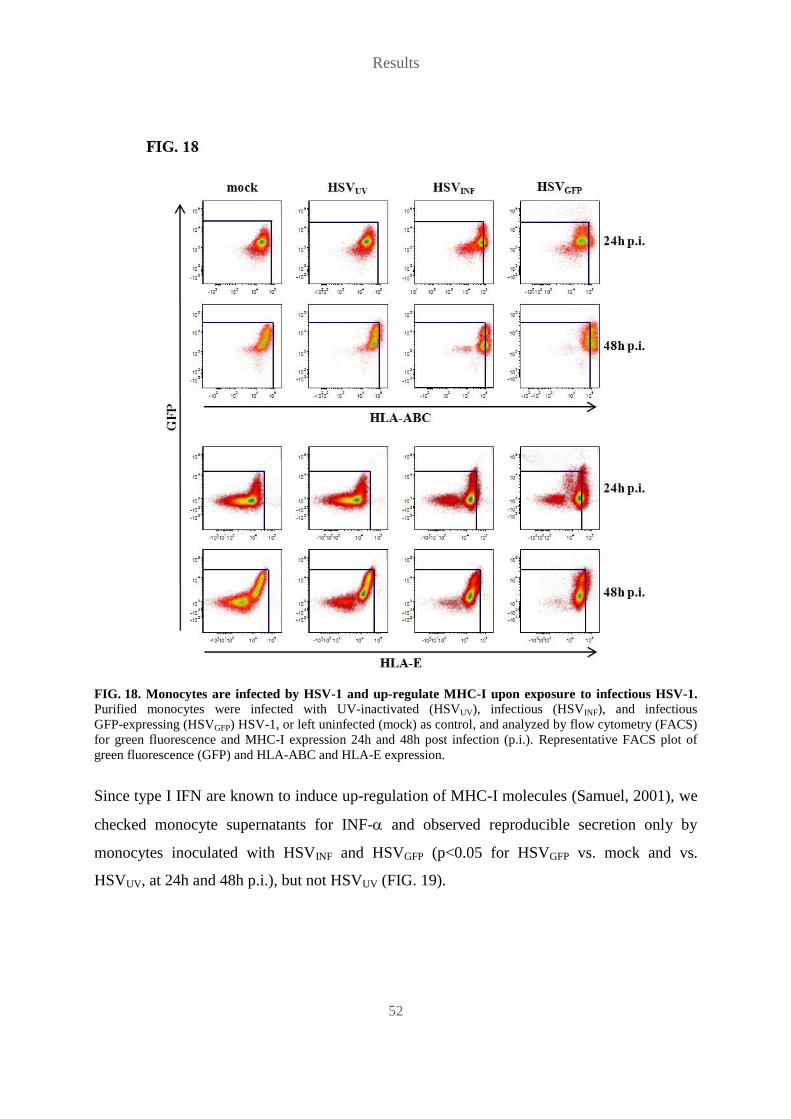
Results
52
FIG. 18. Monocytes are infected by HSV-1 and up-regulate MHC-I upon exposure to infectious HSV-1. Purified monocytes were infected with UV-inactivated (HSVUV), infectious (HSVINF), and infectious
GFP-expressing (HSVGFP) HSV-1, or left uninfected (mock) as control, and analyzed by flow cytometry (FACS)
for green fluorescence and MHC-I expression 24h and 48h post infection (p.i.). Representative FACS plot of
green fluorescence (GFP) and HLA-ABC and HLA-E expression.
Since type I IFN are known to induce up-regulation of MHC-I molecules (Samuel, 2001), we
checked monocyte supernatants for INF- and observed reproducible secretion only by
monocytes inoculated with HSVINF and HSVGFP (p<0.05 for HSVGFP vs. mock and vs.
HSVUV, at 24h and 48h p.i.), but not HSVUV (FIG. 19).

Results
53
FIG. 19. Infectious HSV-1 induces IFN- secretion by monocytes. Purified monocytes were infected with
UV-inactivated (HSVUV), infectious (HSVINF), and infectious GFP-expressing (HSVGFP) HSV-1, or left
uninfected (mock) as control, and supernatants were analyzed for IFN-2a/2b using enzyme-linked
immunosorbent assay (ELISA). IFN-2a/2b (IFN-) secretion at 24h and 48h post infection (p.i.), given as
mean and standard error of ten independent experiments. #
p<0.05 vs. mock; * p<0.05 as indicated (Tukey
HSD).
In order to test the hypothesis, that type I IFN were responsible for HLA-ABCE up-regulation
after HSV-1 infection, we performed neutralization experiments, where we infected
monocytes with HSVGFP in the presence of IFN-R and the isotype control (IgG2a).
Comparing IFN-R with IgG2a revealed distinct effects of type I IFN on monocyte infection
as well as HLA-ABCE regulation (FIG. 20).

Results
54
FIG. 20. Type I IFN suppress HSV-1 infection of monocytes and induce up-regulation of MHC-I. Purified
monocytes were infected with infectious GFP-expressing HSV-1 (HSVGFP) in the presence of a neutralizing
antibody against the IFN-/ receptor (IFN-R) and an isotype control (IgG2a) and analyzed by flow
cytometry (FACS) for green fluorescence and MHC-I expression 24h and 48h post infection (p.i.).
Representative FACS plot of green fluorescence (GFP) and HLA-ABC and HLA-E expression.
Neutralization of IFN-R increased monocyte infection at 24h (p=0.05) and 48h (p<0.05) p.i.
(FIG. 21A) and prevented up-regulation of HLA-ABC at 24h (n.s.) and 48h (p<0.05) p.i.
(FIG. 21B) as well as HLA-E at 24h (p<0.05) and 48h (p<0.01) p.i. (FIG. 21C). Furthermore,
IFN-R neutralization significantly diminished IFN- secretion at 24h and 48h p.i. (p<0.05)
(FIG. 21D), once again confirming the positive feedback loop for IFN- production (Marie et
al., 1998) (FIG. 12D). These results prove type I IFN as cause of HSV-1-induced HLA-ABCE
up-regulation by monocytes and further propose type I IFN as potential restriction factors for
productive HSV-1 infection and replication in monocytes.

Results
55
FIG. 21. Type I IFN suppress HSV-1 infection of monocytes, induce up-regulation of MHC-I and trigger
IFN- secretion. Purified monocytes were infected with infectious GFP-expressing HSV-1 (HSVGFP) in the
presence of a neutralizing antibody against the IFN-/ receptor (IFN-R) and an isotype control (IgG2a), or
left uninfected (mock) as control, and analyzed by flow cytometry (FACS) 24h and 48h post infection (p.i.),
supernatants were analyzed using enzyme-linked immunosorbent assay (ELISA). A. Green fluorescent
monocytes (%). B. Fold change of HLA-ABC expression (MFI). C. Fold change of HLA-E expression (MFI). D.
IFN-2a/2b (IFN-) secretion. All values are given as mean and standard error of three independent
experiments. * p≤0.05, ** p≤0.01 as indicated (Student’s t-test).

Results
56
5.8 HSVd106S affects monocytes similar to HSVGFP
GFP is coupled to the tegument protein VP22 in HSVGFP and is thus present within viral
particles, so HSVGFP particles themselves fluoresce. Consequently, green fluorescing
monocytes may not be infected monocytes expressing GFP, but monocytes with fluorescing
viral particles sticking to them. We therefore repeated our infection experiments with
HSVd106S, which carries the GFP gene under the control of a human cytomegalovirus
(HCMV) promoter. HSVd106S particles do not fluoresce, so monocytes can only fluoresce
when they have been infected and express GFP. Infection of monocytes with HSVd106S had
effects similar to infection with HSVGFP. HSVd106S induced significant fluorescence at 24h p.i.
(p<0.05) (FIG. 22A), that was lost at 48h p.i. HSVd106S infection induced up-regulation of
HLA-ABC at 24h and 48h p.i. (p<0.01) (FIG. 22B) and of HLA-E at 24h (p<0.05) and 48h
p.i. (FIG. 22C). HSVd106S-infected monocytes secreted even higher amounts of IFN- than
HSVGFP-infected monocytes (FIG. 22D). Similarities in fluorescence as well as IFN-
induction and HLA-ABCE up-regulation induced by both viruses argue for an actual infection
of monocytes by HSV-1.

Results
57
FIG. 22. HSVd106S affects monocytes similar to HSVGFP. Purified monocytes were infected with infectious
GFP-expressing (HSVGFP) and an infectious but replication-deficient GFP-expressing (HSVd106S) HSV-1, or left
uninfected (mock) as control, and analyzed by flow cytometry (FACS) 24h and 48h post infection (p.i.),
supernatants were analyzed using enzyme-linked immunosorbent assay (ELISA). A. Green fluorescent
monocytes (%). B. Fold change of HLA-ABC expression (MFI). C. Fold change of HLA-E expression (MFI). D.
IFN-2a/2b (IFN-) secretion. All values are given as mean and standard error of six (A, D), five (B), and three
(C) independent experiments. # p≤0.05,
## p≤0.01 HSVd106S vs. mock (Student’s t-test).

Results
58
Altogether, infection experiments demonstrated monocytes as target cells for HSVINF,
suggesting them as crucial cell population in HSVINF-induced NK cell activation not only via
TNF- secretion, but via recognition of infected monocytes by NK cells. MHC-I down-
regulation by infected monocytes is a possible mechanism, yet according to our studies
unlikely. Furthermore, up-regulation of MHC class I polypeptide-related sequence (MIC) A
or B, which would be recognized by activating NK cell receptors (Vivier, 2006), could be
excluded in preliminary experiments (FIG. 23).
FIG. 23. HSV-1 does not induce up-regulation of MHC class I polypeptide-related sequence (MIC)A or B.
Purified monocytes were infected with infectious GFP-expressing HSV-1 (HSVGFP), or left uninfected (mock) as
control, and analyzed by flow cytometry (FACS) for MICA/B expression 24h post infection (p.i.).
Representative FACS plot of MICA/B expression.

Results
59
5.9 Monocytes mediate NK cell effector functions upon HSV-1
infection within the PBMC context
In order to investigate the actual contribution of monocytes and also of PDC to
HSVINF-induced NK cell activation within the PBMC context, we conducted cell depletion
experiments, comparing non-depleted PBMC with monocyte- or PDC-depleted PBMC.
Depletion of monocytes as well as PDC decreased CD69 up-regulation, although to a variable
extent (FIG. 24A): CpG-A-induced CD69 up-regulation was significantly reduced only by
monocyte depletion (p<0.05), while it was diminished by both monocyte and PDC depletion
in the case of HSVUV (p<0.01) and HSVINF (p<0.01 and p<0.05, respectively). For HSVINF
stimulation the inhibitory effect of monocyte depletion was significantly stronger than the
effect of PDC depletion (p<0.01), which argues for monocytes to be more important in NK
cell activation than PDC when HSV-1 is infectious. HSVINF-induced NK cell effector
functions were both affected by cell depletion in the same manner. While depletion of PDC
had no effect on either effector function, depletion of monocytes prevented both IFN-
secretion (FIG. 24B) and degranulation (FIG. 24C). These results confirm PDC as important
cell population in NK cell activation by HSV-1, and they furthermore reveal monocytes as
key accessory cells in HSVINF-caused NK cell activation, and as indispensable cell population
for the induction of NK cell effector functions within the PBMC context. Interestingly, both
cell populations seem to be crucial for CpG-A- as well as HSV-1-stimulated IFN-
production (FIG. 24D). Depletion of monocytes as well as PDC reduced secretion of IFN-
induced by CpG-A, HSVUV (p<0.01 for monocyte depletion and p<0.05 for PDC depletion)
and HSVINF (p<0.01).

Results
60
FIG. 24. Monocytes mediate NK cell effector functions upon HSV-1 infection within the PBMC context.
PBMC were left non-depleted (PBMC) or were depleted of monocytes (PBMC monocytes) or of PDC
(PBMC PDC) and stimulated for 12h with CpG-A, UV-inactivated (HSVUV) and infectious (HSVINF) HSV-1,
or left unstimulated (mock) as control. Cells were analyzed by flow cytometry (FACS), supernatants by enzyme-
linked immunosorbent assay (ELISA). A - C. NK cells were gated as CD56-positive CD3- and CD14-negative
population and analyzed for CD69 expression (A), IFN- secretion (B) and CD107a surface expression (C) (%).
D. IFN-2a/2b (IFN-) secretion within PBMC (pg/ml). All values are given as mean and standard error of eight
independent experiments. * p<0.05, ** p<0.01 as indicated (Tukey HSD).

Results
61
5.10 PDC serve as crucial accessory cell population in NK cell
activation by HSV-1-infected HFF
Donaghy et al. demonstrated the presence of PDC within recurrent genital herpes lesions and
their co-localization with NK cells (Donaghy et al., 2009), so we wanted to investigate the
role of PDC as accessory cell population in NK cell activation within infected tissue. In order
to simulate the situation of NK cell activation within tissue, we conducted experiments, in
which we inoculated NK cells with HSVUV and HSVINF and co-cultivated them with human
foreskin fibroblasts (HFF) in the absence and in the presence of PDC. NK cell-PDC ratios in
assays were adjusted to their physiological ratio within PBMC of the respective donors. At
24h p.s. , CD69 up-regulation on NK cells was measured. Clearly, HSV-1 does not activate
NK cells in a direct manner, since NK cells did not up-regulate CD69 in response to either
HSVUV or HSVINF, when cultured alone (FIG. 25A). Stimulation of NK cells with HSVUV and
in particular HSVINF in the presence of PDC led to moderate but not significant CD69
up-regulation. NK cells stimulated with HSV-infected HFF alone also slightly up-regulated
CD69, however, CD69 up-regulation was not significant. In contrast, when co-cultivated with
both HFF and PDC, NK cells stimulated with HSVUV as well as HSVINF significantly up-
regulated CD69 compared to the mock control (p<0.01). Checking IFN- levels, we observed
that PDC secreted considerably more IFN- in the presence of HFF (FIG. 25B). These results
indicate PDC as important accessory cells for NK cell activation within HSV-1-infected
tissue, possibly via secretion of type I IFN, and further suggest a dependence of PDC on a
sufficient cell density, and hence possible interactions with other cells, within the cell culture
to secrete high amounts of IFN- in response to HSV-1 stimulation, as observed by
Rönnblom et al. (Rönnblom et al., 1988).

Results
62
FIG. 25. PDC serve as crucial accessory cell population in NK cell activation by HSV-1-infected HFF.
Purified NK cells were cultivated alone or together with purified PDC, with HFF, or with HFF and PDC, and
stimulated for 24h with UV-inactivated (HSVUV) and infectious (HSVINF) HSV-1, or left unstimulated (mock) as
control. Cells were analyzed by flow cytometry (FACS), supernatants by enzyme-linked immunosorbent assay
(ELISA). A. NK cells were gated as CD56-positive CD3- and CD14-negative population and analyzed for CD69
expression (%). B. IFN-2a/2b (IFN-) secretion within cell culture (pg/ml). All values are given as mean and
standard error of three independent experiments. ##
p<0.01 vs. mock (Tukey HSD).
Interestingly, NK cells co-cultivated with HSV-infected HFF expressed less CD56 than
HSV-stimulated NK cells cultured without HFF. The decrease in CD56 expression was even
more obvious, when PDC were present (FIG. 26A). This effect was caused only by HSVINF,
not by HSVUV (FIG. 26B). CD56 expression on NK cells stimulated with HSVINF was
significantly lower compared to mock (p<0.05) and HSVUV (p<0.05). Apparently, HSV
infection of and / or replication within HFF induces NK cells to down-regulate CD56, the
effect being boosted by PDC.

Results
63
FIG. 26. NK cells co-cultivated with HSV-1-infected HFF down-regulate CD56. Purified NK cells were
cultivated alone or together with purified PDC, with HFF, or with HFF and PDC, and stimulated for 24h with
UV-inactivated (HSVUV) and infectious (HSVINF) HSV-1, or left unstimulated (mock) as control. NK cells were
gated as CD56-positive CD3- and CD14-negative population and analyzed by flow cytometry (FACS) for CD56
expression (MFI). A. Representative FACS plot of CD56 expression on NK cells after stimulation with HSVINF.
B. Fold change of CD56 expression, given as mean and standard error of three independent experiments.
# p<0.05 vs. mock; * p<0.05 as indicated (Tukey HSD).

Results
64
5.11 PDC supernatants inhibit HSV-1 replication in HFF
Type I IFN are known to lead to an antiviral state of virus-infected and -susceptible cells
(ISAACS and LINDENMANN, 1957). Since PDC are key producers of type I IFN (Siegal et
al., 1999; Cella et al., 1999) and furthermore have been shown to suppress HSV-2 replication
upon vaginal infection (Lund et al., 2006), we decided to examine the potential of PDC-SN to
inhibit HSV-1 replication in HFF. For this purpose, HFF were infected with HSVGFP at an
MOI of 0.001 and 0.01 and cultivated for 24h and 48h in the absence and presence of
PDC-SN corresponding to IFN-2a/2b levels of 20pg/ml and 200pg/ml. Infection rates were
determined via GFP expression in HFF. The first observation we made was the wide range of
infection rates in HFF cultured without PDC-SN (FIG. 28A), varying for MOI 0.001 between
0.1% and 2.6% at 24h and between 0.5% and 82.0% at 48h, for MOI 0.01 between 0.1% and
19.5% at 24h and between 19.3% and 99.4% at 48h p.i. Obviously, productivity of HSV-1
replication depends on the current state and condition of the infected cell, which seems to be
variable for HFF. However, when infected HFF were cultured in the presence of PDC-SN,
infection rates were reduced compared to infection rates in the absence of PDC-SN (FIG. 27).
Relative reduction of infection rates was significant at 24h p.i. for MOI 0.01 at 20pg/ml and
200pg/ml (p<0.05) (FIG. 28B), at 48h p.i. for MOI 0.001 at 200pg/ml (p<0.05) and for MOI
0.01 at 20pg/ml and 200pg/ml (p<0.01) (FIG. 28C). These results evidence the potential of
PDC to directly inhibit HSV-1 replication in target cells via secretion of antiviral cytokines,
most likely type I IFN.

Results
65
FIG. 27. HSV-1 replication in HFF decreases in the presence of PDC supernatants. HFF were infected with
infectious GFP-expressing HSV-1 (HSVGFP) at a MOI of 0.001 and a MOI of 0.01, in the absence of PDC
supernatants (w/o PDC-SN) and in the presence of PDC supernatants containing 20pg/ml and 200pg/ml
IFN-2a/2b (IFN-), or left uninfected (mock) as control, and analyzed by flow cytometry (FACS) for green
fluorescence 24h and 48h post infection (p.i.). Representative FACS plot of green fluorescence (GFP).

Results
66
FIG. 28. PDC supernatants inhibit HSV-1 replication in HFF. HFF were infected with infectious
GFP-expressing HSV-1 (HSVGFP) at a MOI of 0.001 and a MOI of 0.01, in the absence of PDC supernatants
(w/o PDC-SN) and in the presence of PDC supernatants containing 20pg/ml and 200pg/ml IFN-2a/2b (IFN-),
or left uninfected (mock) as control, and analyzed by flow cytometry (FACS) for green fluorescence 24h and
48h post infection (p.i.). A. HSV-1 replication in HFF w/o PDC-SN, shown as green fluorescent HFF (%) 24h
and 48h p.i. B and C. HSV-1 replication in the presence of PDC-SN, shown as fold change of green fluorescent
HFF (%) compared to infection w/o PDC-SN 24h (B) and 48h (C) p.i. All values are given as mean and standard
error of three independent experiments. #
p<0.05, ##
p<0.01 vs. w/o PDC-SN (Tukey HSD).

Results
67
5.12 PDC-NK cell interactions are hampered in an HIV-1-infected
woman suffering from persisting genital ulcers
Human immunodeficiency virus type 1 (HIV-1) infection leads to a decrease in numbers as
well as function of PDC, leading to reduced IFN- secretion (Feldman et al., 2001; Schmidt
et al., 2005; Schmidt et al., 2006). It furthermore causes a defective crosstalk between PDC
and NK cells via functional defects of PDC and also NK cells (Reitano et al., 2009), on which
antiretroviral therapy has only minimal effects (Benlahrech et al., 2011). We therefore
analyzed PDC and NK cell activation in PBMC of an African woman infected with HIV-1
and suffering from immune reconstitution inflammatory syndrome (IRIS). Three months after
viral load decline and CD4+ T cell increase due to successful antiretroviral treatment she
developed painful genital ulcers due to HSV-2, which were only temporarily resolved by
several courses of aciclovir, topical application of imiquimod and a radical bilateral
vulvectomy (Strehl et al., 2012). Repeated virological analysis of the hyperproliferative
lesions revealed human papilloma virus type 54 (HPV-54) infection in addition to HSV-2
infection.
PBMC of this patient were stimulated with CpG-A, a TLR-7 agonist (S-27609), HSVUV and
HSVINF and analyzed at 18h p.s. for IFN- secretion, expression of markers for PDC
migration (CCR7), activation (CD80) and maturation (CD83) as well as NK cell activation
(CD69). Up-regulation of CCR7, CD80 and CD83 on stimulated but also on mock-cultivated
PDC suggested pre-stimulation of PDC in vivo (FIG. 29A). Activation of the patient’s NK
cells upon stimulation was severely impaired (FIG. 29B), as well as IFN- secretion within
PBMC (FIG. 29C), compared to a healthy control donor. These results suggest that impaired
IFN- production by PDC and subsequently reduced activation of NK cells contributed to the
patient’s disease.
Silencing of peripheral IFN- responses in HIV-1 infection has been associated with
enhanced interaction of CD40 on PDC with CD40 ligand (CD40L), a co-stimulatory
molecule, which is up-regulated upon immune activation (Donhauser et al., 2012). CD40L
levels transiently increase with the CD4+ T cell recovery upon antiretroviral therapy, which
might boost IFN-susceptible opportunistic infections in IRIS. Therefore, we retrospectively
analyzed levels of soluble (s)CD40L in the plasma of our patient (P1) and four other patients

Results
68
suffering from opportunistic infections (P2 - P5). Indeed, sCD40L levels in eight consecutive
plasma samples of P1 after initiation of antiretroviral treatment were significantly higher than
in cross-sectional samples of 52 untreated HIV-1-infected patients (p<0.001) (Donhauser et
al., 2012), and they were also higher than in the samples of P2 - P5 (P<0.01) (FIG. 29D).
Thus, suppression of TLR-7- and TLR-9-induced IFN- production by elevated sCD40L
levels may have contributed to the unusual and treatment-refractory genital ulcers in P1.
Alternatively, enhanced sCD40L levels may reflect prior in vivo stimulation caused by HSV-2
and HPV-54 infections. Altogether, these data indicate important interactions of PDC and NK
cells, which are hampered in immunosuppressed individuals and thus may lead to inefficient
control of persistent viral infections, such as human papilloma virus and herpes simplex virus
infections.
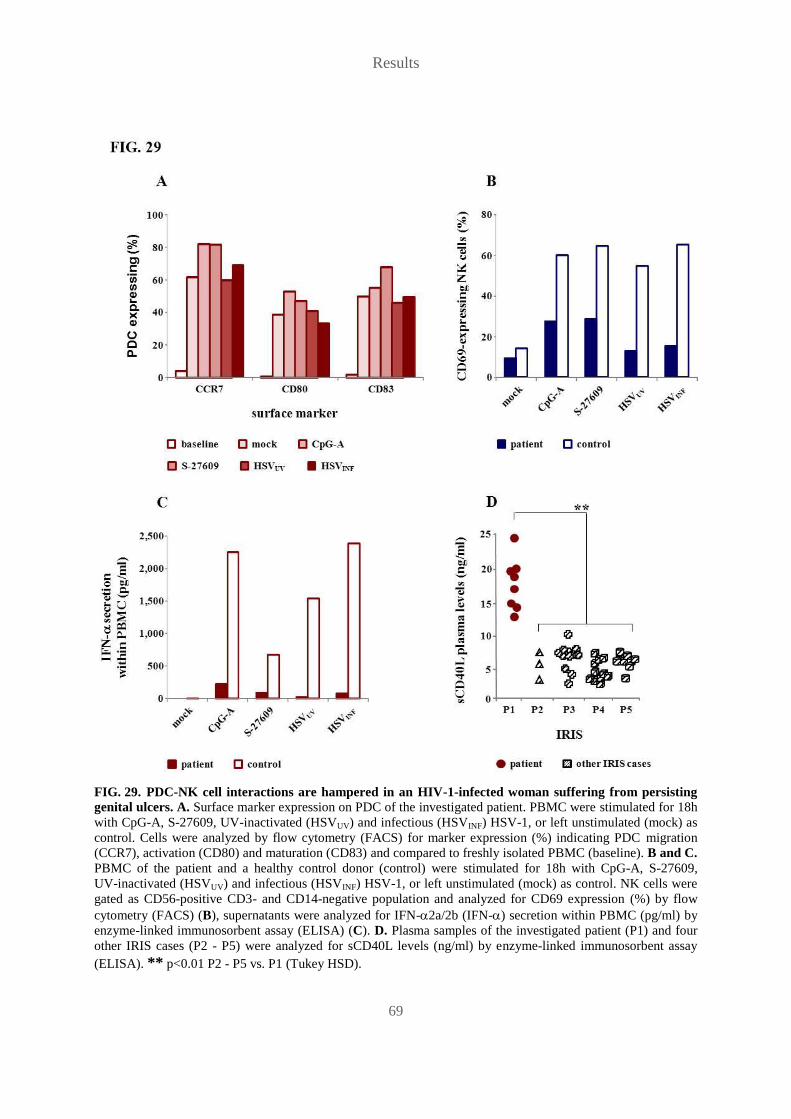
Results
69
FIG. 29. PDC-NK cell interactions are hampered in an HIV-1-infected woman suffering from persisting
genital ulcers. A. Surface marker expression on PDC of the investigated patient. PBMC were stimulated for 18h
with CpG-A, S-27609, UV-inactivated (HSVUV) and infectious (HSVINF) HSV-1, or left unstimulated (mock) as
control. Cells were analyzed by flow cytometry (FACS) for marker expression (%) indicating PDC migration
(CCR7), activation (CD80) and maturation (CD83) and compared to freshly isolated PBMC (baseline). B and C.
PBMC of the patient and a healthy control donor (control) were stimulated for 18h with CpG-A, S-27609,
UV-inactivated (HSVUV) and infectious (HSVINF) HSV-1, or left unstimulated (mock) as control. NK cells were
gated as CD56-positive CD3- and CD14-negative population and analyzed for CD69 expression (%) by flow
cytometry (FACS) (B), supernatants were analyzed for IFN-2a/2b (IFN-) secretion within PBMC (pg/ml) by
enzyme-linked immunosorbent assay (ELISA) (C). D. Plasma samples of the investigated patient (P1) and four
other IRIS cases (P2 - P5) were analyzed for sCD40L levels (ng/ml) by enzyme-linked immunosorbent assay
(ELISA). ** p<0.01 P2 - P5 vs. P1 (Tukey HSD).

Discussion
70
6 Discussion
Our analysis of human NK cell activation and induction of effector functions by different
stimuli revealed HSV-1 as potent and fast inducer of NK cell activation within the PBMC
context. Interestingly, we observed several differences concerning the stimulating potential
between HSVINF and HSVUV, first, on the level of NK cell activation and second, on the level
of cytokine secretion within PBMC (FIG. 30).
HSVINF induced overall NK cell activation significantly faster than HSVUV (FIG. 6B) and also
caused significantly stronger activation within the CD56bright
NK cell subset (FIG. 7D). In
addition, only HSVINF, and not HSVUV, induced significant NK cell IFN- secretion (FIG. 8B)
and degranulation (FIG. 8C). These results support the finding of Fitzgerald-Bocarsly and
colleagues that HSV-1-inoculated HFF were only lysed by human PBMC when the virus used
was infectious (Fitzgerald-Bocarsly et al., 1991), but stand in contrast to another study by
Ahmad et al., in which NK activity of HSV-1-stimulated human PBMC was similar for
infectious and UV-inactivated virus (Ahmad et al., 2000). This discrepancy could be caused
by the use of different methods. Ahmad et al. investigated the increase of basic lytic activity
against the NK cell target K562, whereas Fitzgerald-Bocarsly et al. analyzed lysis of HFF,
and we detected induction of degranulation in the absence of cytotoxicity-inducing target
cells.
Although both HSVINF and HSVUV caused secretion of exceedingly high amounts of IFN-
within PBMC, HSVINF-induced IFN- secretion was significantly faster and slightly stronger
than HSVUV-induced IFN- secretion (FIG. 9). Furthermore, in our study HSVINF stimulated
significant secretion of IL-1 and TNF-, while HSVUV failed to induce these two cytokines
within PBMC (FIG. 11), contradicting an early study published by Gosselin et al., who saw
similar TNF- induction in human PBMC by infectious and UV-irradiated HSV-1 (Gosselin
et al., 1992). Our observations indicate an effect of viral infectivity on the induction of pro-
inflammatory cytokines as well as on NK cell activation within the PBMC context and
suggest the necessity for viral infectivity in the induction of NK cell effector functions
(FIG. 30), either via induction of different cytokines, or via direct recognition of virus-
infected cells by NK cells as targets, or both.

Discussion
71
FIG. 30. Infectious and UV-inactivated HSV-1 exhibit different stimulation potentials within the PBMC
context. Both infectious and UV-inactivated HSV-1 are able to induce IFN-2a/2b (IFN-) secretion within
PBMC and CD69 up-regulation on NK cells, whereas only infectious HSV-1 causes secretion of TNF- and
IL-1 within PBMC and NK cell effector functions IFN- secretion and degranulation indicated by CD107a
surface expression. These findings suggest the importance of viral infectivity in complete activation of effector
NK cells by HSV-1.
NK cell effector functions were evenly distributed between the two NK cell subsets
(FIG. 8A), contradicting a previous concept of strict classification of CD56dim
and CD56bright
NK cells into a mainly cytotoxic and a major cytokine secreting subset, respectively (Cooper
et al., 2001a). However, Vivier proposed to rather define the CD56dim
and CD56bright
NK cell
subsets as “target cell responsive” and “cytokine responsive”, respectively, both possessing
the ability for cytotoxicity as well as cytokine secretion, depending on the stimulus (Vivier,
2006). Recently, De Maria et al. described CD56dim
NK cells as rapid producers of IFN-
upon antibody-mediated stimulation of natural killer receptors (De et al., 2011). We report

Discussion
72
here for the first time that infectious HSV-1 is a potent stimulus for IFN- secretion by
CD56dim
NK cells within the PBMC context.
INF- has been published to be of importance in HSV-induced NK cell activation (Gill et al.,
2011; Feldman et al., 1992) and several groups identified it as main cytokine in the induction
of NK cell activation after stimulation of human PDC with influenza virus, CpG, and poly
(I:C) (Benlahrech et al., 2009; Gerosa et al., 2005; Marshall et al., 2006; Romagnani et al.,
2005). Stimulation of purified NK cells with supernatants of HSVINF-stimulated PDC
(PDC-SN) demonstrated time- and dose-dependent induction of CD69 on NK cells by
PDC-SN (FIG. 10A). Interestingly, NK cell activation occurred in two phases, suggesting that
the initial and subsequent CD69 up-regulation were induced by two different mechanisms.
Further studies are required to identify the underlying mechanisms. The fact that
neutralization of the IFN-/ receptor significantly decreased PDC-SN-induced NK cell
activation (FIG. 10C) proves type I IFN as key cytokines in PDC-induced NK cell activation
after stimulation with HSV-1 (FIG. 31), consistent with PDC-induced NK cell activation after
stimulation with CpG-A (Benlahrech et al., 2009; Gerosa et al., 2005; Marshall et al., 2006;
Romagnani et al., 2005). However, NK cell activation by PDC-SN was slightly stronger than
NK cell activation by rhIFN- (FIG. 10B), and other groups described the involvement of
further cytokines, in particular TNF-, which collaborated with IFN- in PDC-induced NK
cell activation (Gerosa et al., 2005; Marshall et al., 2006; Romagnani et al., 2005). Actually,
we detected TNF- not only in PDC supernatants (data not shown), but also in PBMC
supernatants (FIG. 11).

Discussion
73
FIG. 31. PDC-dependent NK cell activation by HSV-1 is mediated by type I IFN. HSV-1-stimulated PDC
secrete high amounts of IFN-2a/2b (IFN-) and other type I IFN. Stimulation of purified NK cells with PDC
supernatants leads to CD69 up-regulation which is inhibited by a neutralizing antibody against the IFN-/
receptor. This identifies type I IFN as key cytokines in PDC-dependent NK cell activation by HSV-1.
TNF- has been shown to play an essential role in HSV infection in vivo. TNF- knockout
mice exhibited decreased survival rates in acute corneal HSV-1 infections and increased
reactivation rates after UV light stimulation (Minami et al., 2002), and lethal encephalitis after
intranasal HSV-1 infection (Sergerie et al., 2007). In our studies we demonstrated TNF- as
critical cytokine for CpG-A- and HSV-1-induced human NK cell activation (FIG. 12A) and
also for HSVINF-caused IFN- secretion (FIG. 12B) within the PBMC context. Cooper et al.
described the ability of IL-1 to co-stimulate IFN- production of CD56bright
NK cells together
with IL-12 or, in particular, IL-15 (Cooper et al., 2001b), however, we did not detect any
direct influence of IL-1 on NK cell activation or induction of effector functions
(FIG. 12A, B, C).
Type I IFN, which played a major role in PDC-mediated NK cell activation (FIG. 10),
appeared to be less important within the PBMC context. IFN-R neutralization only
significantly inhibited NK cell activation induced by CpG-A and HSVUV, but not by HSVINF
(FIG. 12A), and had no influence on degranulation (FIG. 12C). On the contrary, HSVINF-
induced IFN- secretion was increased by IFN-R neutralization (FIG. 12B), suggesting a
rather inhibitory influence of high amounts of type I IFN on NK cell IFN- secretion.
Similarly, Cousens et al. observed inhibition of murine IL-12 and subsequently IFN-

Discussion
74
secretion by type I IFN upon viral infection and bacterial stimulation (Cousens et al., 1997).
IFN- secretion after simultaneous neutralization of TNF- and IFN-R (FIG. 13B) points to
opposed functions of TNF- and type I IFN in IFN- induction by HSVINF, namely that
TNF- induces, and type I IFN rather inhibit HSV-1-induced IFN- production by NK cells.
We could observe a strict dependence of IFN- levels on type I IFN upon stimulation of
PBMC with GpG-A and HSV-1 (FIG. 12D) and also upon infection of monocytes with
HSV-1 (FIG. 21D), which is in concordance with the already published autocrine loop (Marie
et al., 1998). Interestingly, TNF- and IL-1 also strongly influenced IFN- secretion
(FIG. 12D). This suggests that secretion of high amounts of IFN- demands a positive
feedback loop consisting of a tight crosstalk of IFN--producing cells with each other and / or
other immune cells via production of type I IFN, TNF-, and IL-1 (FIG 32). Actually,
TNF- has been shown to induce secretion of low amounts of type I IFN, particularly IFN-,
in both human and mouse macrophages (Yarilina et al., 2008). Jimbo et al. demonstrated
IL-1 to be involved in a positive feedback loop increasing its own secretion by intervertebral
disc cells and also secretion of other inflammatory mediators like IL-6 and cyclooxygenase
(COX)-2 (Jimbo et al., 2005).

Discussion
75
FIG. 32. Secretion of high amounts of IFN-2a/2b (IFN-) within PBMC demands a positive feedback
loop involving type I IFN, TNF- and IL-1. HSV-1-induced secretion of IFN- is greatly diminished by
neutralization of the IFN-/ receptor as well as TNF- and IL-1, suggesting that not only type I IFN, but also
TNF- and IL-1 are involved in a positive feedback loop leading to secretion of high IFN-2a/2b (IFN-)
amounts after HSV-1 simulation of PBMC.
TNF- secretion assays revealed PDC and monocytes as potent TNF- sources upon
stimulation with HSVINF (FIG. 14A). Considering the much higher frequency of monocytes
within PBMC (FIG. 14B), monocytes have to be regarded as the most numerous TNF-
producers in the blood. Obviously, TNF- secretion by PDC did not depend on viral
infectivity, in contrast to monocytic TNF- secretion, which was significantly increased by
viral infectivity (FIG. 14A). Interestingly, with 16% the percentage of TNF--secreting
monocytes (FIG. 14A) was clearly higher than the percentage of infected monocytes of 2%
(FIG. 16A), which argues against TNF- secretion mainly by infected monocytes. It rather
suggests that infection of few monocytes stimulates a number of uninfected bystander
monocytes to secrete TNF-. The difference in the induction of TNF- secretion that we
observed between HSVINF and HSVUV on the PBMC level (FIG. 14C) could explain why
HSVUV was unable to mediate significant IFN- secretion by NK cells; neutralization of
TNF- significantly diminished NK cell IFN- secretion induced by HSVINF (FIG. 12B).

Discussion
76
These observations indicate that induction of certain NK cell effector functions, like IFN-
secretion, requires the secretion of pro-inflammatory cytokines within PBMC, which in turn
depends on viral infectivity. Induction of other NK cell effector functions, like cytotoxicity
indicated by degranulation, is independent at least from the cytokines investigated in our
study, but also depends on viral infectivity.
Ahmad et al. reported IL-15 as crucial cytokine in HSV-1-induced NK activity of human
PBMC, but in their study, infectivity of HSV-1 was not required for the induction of NK
activity (Ahmad et al., 2000), and in a later study Ahmad et al. showed that HSV-1-induced
up-regulation of IL-15 gene expression in monocytic cells was independent of viral infectivity
(Ahmad et al., 2007). We tested PBMC supernatants for secreted IL-15, but did not detect any
after HSV-1 stimulation (data not shown). Furthermore, the observations in the two other
studies would preclude IL-15 as critical factor for NK cell effector functions, since in our
studies only HSVINF induced NK cell effector functions, whereas HSVUV failed to induce
them (FIG 8). IL-18, which we did not investigate, might play a role in HSVINF-induced NK
cell activation and effector functions, since it has already been demonstrated to contribute to
NK cell activation in HSV-1-infected mice (Barr et al., 2007; Reading et al., 2007).
Infection experiments evidenced monocytes as target cells for HSV-1 which are infected, yet
to a very small percentage and without allowing productive viral replication (FIG. 16)
(FIG. 33), in concordance to prior studies of monocyte infection by HSV-1 (Bruun et al.,
1998; Daniels et al., 1978). Ineffective infection of and replication in monocytes was in part
due to monocytic type I IFN secretion, since IFN-R blocking significantly increased
infection rates in purified cells, in particular 48h p.i. (FIG. 21A).
Similarly, type I IFN were published to suppress HSV-1 replication in vitro in Vero cells,
HEp-2 cells, and fibroblasts (Härle et al., 2001; Noisakran et al., 2000) and to play a crucial
role in resistance to HSV infections in vivo (Dupuis et al., 2003; Casrouge et al., 2006).
Another restriction factor for HSV-1 replication in monocytes might be SAM domain and HD
domain-containing protein 1 (SAMHD1), which was recently shown to inhibit HSV-1
replication in differentiated macrophage cell lines (Kim et al., 2013).
Interestingly, IFN- secretion by monocytes depended on HSV-1 infectivity (FIG. 19), in
concordance to the observation of Melchjorsen et al., that cytokine induction by HSV in

Discussion
77
human monocyte-derived cells is dependent on virus replication (Melchjorsen et al., 2006).
This stands in contrast to IFN- secretion by PDC, which is induced by HSVINF as well as
HSVUV (Schuster et al., 2010) (FIG. 9). This difference is probably due to the fact that HSV-1
is able to infect monocytes (FIG. 16A) (Bruun et al., 1998; Daniels et al., 1978), but not PDC
(Schuster et al., 2010), and due to different recognition molecules involved; TLR-9 is
responsible for HSV-1 recognition in PDC (Krug et al., 2004), but not in monocytes, where
melanoma differentiation-associated protein 5 (MDA5) was shown to be the primary mediator
of HSV-1 recognition in macrophages (Melchjorsen et al., 2010). MDA5, a retinoic acid-
inducible gene (RIG)-I-related protein, senses viral RNA with a helicase domain and mediates
the induction of an antiviral response within the infected cell (Yoneyama et al., 2005). Since
the presence of herpesviral RNA requires the initiation of viral replication and therefore viral
infectivity, UV-inactivated HSV-1 should not be recognized by MDA5, explaining the lack of
IFN- induction in monocytes by HSVUV (FIG. 19). Interestingly, HSVd106S induced stronger
IFN- secretion than both infectious wildtype isolates (FIG. 22D). This might be due to a
deletion within the ICP27 gene of HSVd106S (Liu et al., 2009). Melchjorsen et al. determined
ICP27 as a factor counteracting cytokine induction in monocyte-derived cells by HSV
(Melchjorsen et al., 2006).

Discussion
78
FIG. 33. Monocytes are non-productively infected by HSV-1 and up-regulate MHC-I in a type I IFN-
dependent manner. HSV-1 is able to infect monocytes, but without production of new viral particles. HSV-1-
infected monocytes secrete low amounts of IFN-2a/2b (IFN-). Neutralization of the IFN-/ receptor
prevents MHC-I up-regulation induced by infectious HSV-1 (HSVINF) and increases infection rates in
monocytes. These data demonstrate non-productive infection of monocytes by HSV-1 leading to type I IFN-
dependent up-regulation of MHC-I. Furthermore, type I IFN prove to be involved in preventing productive
HSV-1 infection of and replication in monocytes.
Depletion experiments confirmed PDC as crucial IFN- source (FIG. 24D), and furthermore
as potent mediators of CpG-A- and HSV-induced NK cell activation (FIG. 24A) within the
PBMC context. PDC-induced NK cell activation was at least in part due to IFN- production
(FIG. 10, FIG. 12A, FIG. 24D). However, NK cell effector functions did not depend on PDC
(FIG. 24B, C), which stands in contrast to an early study, that indicated a supporting role for
the so called “IFN-producing cells (IPC)” in NK cell-mediated lysis of HSV-1-infected
fibroblasts (Feldman et al., 1992). In addition, our studies showed the importance of
monocytes in NK cell activation (FIG. 24A) and also in IFN- secretion (FIG. 24D).
Monocytes may account for high IFN- levels in different ways. They could contribute
directly by secretion of IFN- itself, as observed for infected monocytes by us (FIG. 19) and
also by others (Linnavuori and Hovi, 1983). However, isolated monocytes only reacted with
IFN- production to infectious, not to UV-inactivated HSV-1, while depletion of monocytes
from PBMC diminished IFN- levels upon stimulation with both HSVINF and HSVUV.
Another possibility would be an indirect contribution of monocytes via secretion of IL-1 and
TNF-, thereby further stimulating CpG-A- and HSV-1-induced IFN- secretion by PDC.
Cytokine neutralization experiments (FIG. 12D) suggest this way of monocyte involvement in

Discussion
79
IFN- production. Furthermore, early studies suggested the dependence of an IFN- response
to HSV-1 on close contact and interactions of IFN-producing cells with other cells within the
cell culture (Rönnblom et al., 1988) and the potential of PBMC-derived cytokines to enhance
HSV-1-induced IFN- secretion by IFN-producing cells (Cederblad and Alm, 1990). Also,
Megjugorac et al. investigated interactions between PDC and HSV-infected monocyte-
derived (mo) DC and could demonstrate induction of IFN- secretion from PDC by HSV-
infected moDC (Megjugorac et al., 2007). Most importantly, we identified monocytes as
indispensable cell population in the induction of NK cell effector functions by HSVINF within
the PBMC context (FIG. 24B, C). Our findings within PBMC appear similar to a study of
PDC-induced NK cell activation, in which NK cell CD69 up-regulation and IFN- production
were induced by soluble factors, whereas degranulation and cytotoxicity were only observed
after direct contact with CpG-stimulated PDC (Benlahrech et al., 2009). In contrast to
Benlahrech et al. we did not find IFN- as major soluble factor for NK cell IFN- production,
but could determine TNF- secretion as important mechanism in the induction of IFN-
(FIG. 12B), which is probably due to the fact that our analyses were conducted with whole
PBMC, not purified PDC and NK cells. The exact process, in which degranulation was
induced, remained elusive.
It is very possible that infected monocytes are directly recognized by NK cells as target cells.
NK cell activation and induction of effector functions could be mediated through various
possible mechanisms. Induction of NK cell cytotoxicity via down-regulation of HLA-C
molecules on productively infected cells was demonstrated for both HSV-1 and HSV-2
(Elboim et al., 2013; Huard and Früh, 2000). Yet, in our studies we observed an overall up-
regulation of HLA-ABC and HLA-E on monocytes inoculated with HSV-1 (FIG. 17,
FIG. 18). MHC-I up-regulation was due to monocytic IFN- secretion (FIG. 19), as
demonstrated by neutralization experiments (FIG. 20, FIG. 21B, C). Unfortunately, we were
not able to investigate HLA-A, HLA-B and HLA-C separately, because no specific antibodies
are available. But the fact that only a minority of HSVGFP-infected monocytes showed
decreased MHC-I expression (FIG. 18) makes recognition of infected monocytes via MHC-I
down-regulation very unlikely. MICA has been shown to be up-regulated on TLR-stimulated
monocytes (Kloss et al., 2008), but MICA/MICB was not induced by HSV-1 in our

Discussion
80
experiments (FIG. 23). Thus, we could exclude direct NK cell activation by infected
monocytes via expression of these stress molecules.
Fitzgerald-Bocarsly et al. demonstrated the expression of immediate early genes as sufficient
to induce NK cell-mediated lysis of HSV-1-infected fibroblasts (Fitzgerald-Bocarsly et al.,
1991), and Chisholm et al. identified ICP0 as effective to trigger lysis of HSV-1-infected cells
by NK cells via the natural cytotoxicity receptors (NCR) NKp30, NKp44, and NKp46
(Chisholm et al., 2007). However, the molecules induced by ICP0 and serving as ligands to
the NCR were not identified in this study. A possible candidate might be B7H6, a molecule
expressed on tumor cells that triggers NK cell cytotoxicity and cytokine secretion via
interaction with NKp30 (Brandt et al., 2009), which was shown to be induced on human
monocytes upon stimulation with TLR ligands and pro-inflammatory cytokines such as IL-1
and TNF- (Matta et al., 2013). Other molecules involved in NK cell-monocyte/macrophage
interaction could be macrophage-expressed CD48 and NK cell-expressed 2B4, which are
involved in NK cell activation after LPS stimulation (Nedvetzki et al., 2007), or macrophage-
expressed activation-induced C-type lectin (AICL) and NK cell-expressed NKp80, which are
involved in NK cell activation after TLR ligand stimulation (Welte et al., 2006), or the
glucocorticoid-induced tumor necrosis factor receptor-ligand (GITRL), which was described
to be involved in the induction of NK cell cytotoxicity by CpG-stimulated PDC (Hanabuchi et
al., 2006) and was shown to be induced on monocytes by staphylococcal enterotoxin B
(Cardona et al., 2006).
While PDC did not mediate NK cell effector functions within the PBMC context (FIG. 24B,
C), they influenced NK cell activation (FIG. 24A). Outside the PBMC context they were
indispensable for NK cell activation, since HSV-1 did not activate purified NK cells directly
(FIG. 25A) (FIG. 34). This observation stands in contrast to a study by Kim et al., where HSV
glycoprotein (g)D peptides directly activated NK cells (Kim et al., 2012). In our co-culture
experiments with HSV-exposed HFF and PDC, neither cell type induced significant CD69 up-
regulation on NK cells by its own. Only in combination, HSV-exposed HFF and PDC
succeeded to strongly activate NK cells (FIG. 25A), which was either caused by synergistic
effects of both cell types on NK cells or mediated by IFN-, which was produced by PDC in
high amounts only in the presence of HFF (FIG. 25B). Dependence of high IFN- production

Discussion
81
by PDC on co-culture with HFF might, similar to IFN- production within PBMC
(FIG. 24D), be due to the need of PDC for close contact to and possible interactions with
other cell populations (Rönnblom et al., 1988). Interestingly, HSV-1-infected HFF somehow
caused down-regulation of CD56 on NK cells (FIG 34), with the effect being enhanced by
PDC (FIG. 26). The significance of this finding remains unclear.
FIG. 34. NK cell activation by HSV-1-infected HFF depends on PDC. HSV-1 does not activate NK cells in a
direct manner, but demands the presence of cells that are stimulated (PDC) or infected (HFF) by HSV-1. HSV-1-
infected HFF induce significant CD69 up-regulation on NK cells only in the presence of PDC, possibly in a type
I IFN-dependent manner, suggesting PDC as important accessory cells for NK cell activation within HSV-1-
infected tissue. Interestingly, NK cells stimulated with HSV-1-infected HFF down-regulate CD56, particularly in
the presence of PDC.
Besides influencing immune responses to HSV-1, PDC might also play a crucial role in
HSV-1 infection by inhibiting viral replication in HSV-1-susceptible cells via secretion of
antiviral cytokines, thereby limiting spread of virus progeny and protecting tissue from
immense damage. We observed an inhibitory effect of PDC-SN in infection experiments with
HFF (FIG. 35). Addition of PDC-SN to HSVGFP-infected HFF clearly decreased HSV-1
replication, evident from reduced green fluorescence of HFF (FIG. 27, FIG. 28B, C).

Discussion
82
Inhibition of HSV-1 replication was most likely mediated by type I IFN, as observed in
monocytes (FIG. 21A), and also demonstrated by others (Härle et al., 2001; Noisakran et al.,
2000).
FIG. 35. PDC supernatants inhibit HSV-1 replication in HFF. Viral infection of and replication in HFF are
diminished in the presence of supernatants derived from HSV-1-stimulated PDC. The inhibitory effect of PDC
supernatants is possibly mediated by type I IFN. These findings evidence the role of PDC as suppressors of
spread of HSV-1 infection within tissue by suppressing viral replication in HSV-1-susceptible cells via secretion
of antiviral cytokines.
Studies of stimulation of PDC and NK cells from HIV-1-infected individuals propose a role
for defective PDC-NK cell interactions in HIV-1-induced immune suppression (Conry et al.,
2009; Reitano et al., 2009), allowing opportunistic or IRIS-related infections. In our case
study, an HIV-1-infected patient suffering from vaginal hyperproliferative lesions due to
HSV-2 and HPV-54 infections exhibited severe functional deficits of PDC as well as NK cells
within the PBMC context. NK cells were only minimally activated by HSV-1 (FIG. 29B)
which appeared to be due to impaired IFN- secretion by PDC (FIG. 29C). Impaired IFN-
secretion upon stimulation with HSV-1 and TLR-7 and TLR-9 agonists might have been
caused by increased CD40-CD40L interactions, as demonstrated by Donhauser et al.
(Donhauser et al., 2012), since sCD40L levels were elevated in the patient (FIG. 29D). Our
findings suggest a role for impaired PDC-NK cell interactions in the severe and treatment-
refractory course of disease in the patient, emphasizing the importance of PDC-NK cell
crosstalk for efficient control of herpesviral infections.

Discussion
83
Altogether, our data propose a model in which the induction of high IFN- levels by HSV-1
within PBMC demands a tight crosstalk between PDC and monocytes involving a positive
feedback loop influenced by type I IFN, TNF- and IL-1 (FIG. 32). Secretion of IL-1 does
not directly influence NK cells, whereas type I IFN and TNF- secreted by both PDC and
monocytes mediate NK cell activation (FIG. 36). IFN- secretion and NK cell activation
alone do not depend on HSV-1 infectivity, whereas only HSVINF, not HSVUV, further induces
NK cell INF- secretion as well as degranulation (FIG. 30). Monocytes, in contrast to PDC,
play a key role in the induction of both NK cell effector functions. HSV-1-induced IFN-
secretion by NK cells is independent of type I IFN, but involves TNF- (FIG. 36), which is
only produced in sufficient amounts in response to HSVINF, not HSVUV. In contrast, NK cell
degranulation is independent of all three cytokines tested and either involves other cytokines
produced by monocytes or is mediated by direct cell:cell interactions between NK cells and
monocytes (FIG. 36). Presumably, NK cells recognize HSV-1-infected monocytes as target
cells via mechanisms other than monocytic MHC-I down-regulation or MICA/MICB
expression. While monocytes are particularly important in the activation of effector NK cells,
PDC appear to contribute to immune control early during infection by protecting HSV-1-
susceptible tissue as they suppress viral replication via secreted antiviral cytokines, probably
type I IFN, and therefore limit viral spread (FIG. 36).
Our data may stimulate further studies investigating cell surface molecules as well as
cytokines involved in the crosstalk between PDC, monocytes and NK cells. Deciphering the
mechanisms that induce functional effector NK cells is important as all three cell types are
among the first cells to infiltrate herpetic lesions and thereby may contribute to the efficient
control of primary and recurrent herpes simplex virus infections.

Discussion
84
FIG. 36. Monocytes mediate HSV-1-induced activation of effector NK cells, while PDC limit HSV-1
replication within infected tissue. While depletion of PDC as well as monocytes greatly diminishes secretion of
IFN-2a/2b (IFN-) and CD69 up-regulation on NK cells, only depletion of monocytes prevents NK cell
effector functions IFN- secretion and degranulation, identifying monocytes as crucial accessory cell population
for HSV-1-induced activation of effector NK cells within PBMC. Both type I IFN and TNF- are involved in
CD69 up-regulation, whereas only TNF- impacts IFN- secretion. Degranulation is independent of type I IFN,
TNF- and IL-1 and is possibly mediated by infected monocytes via direct cell contact. The ability of PDC
supernatants to inhibit HSV-1 replication in HFF proves PDC as important cell population in the limitation of
spread of infection within tissue via secretion of antiviral cytokines.

Abbreviations
85
7 Abbreviations
Abbreviation Full length spelling
AC accessory cell(s)
ADCC antibody-dependent cellular cytotoxicity
AICL activation-induced C-type lectin
APC allophycocyanin
ARN acute retinal necrosis
B bone marrow-derived
BDCA blood dendritic cell antigen
BSA bovine serum albumin
C Celsius
CCL chemokine (C-C motif) ligand
CD cluster of differentiation
cm centimeter
CXCL chemokine (C-X-C motif) ligand
Cy cyanine
D day(s)
DMEM Dulbecco`s Modified Eagle Medium
DNA deoxyribonucleic acid
DNAM DNAX accessory molecule
DPBS phosphate buffered saline without calcium or magnesium
ELISA enzyme-linked immunosorbent assay
EDTA ethylenediaminetetraacetic acid
FACS fluorescence-activated cell sorting
FasL Fas ligand
Fc fragment, cristallizable
FcRIIIA low affinity Fc receptor IIIA
FCS fetal calf serum
FITC fluorescein isothiocyanate
FSC forward scatter
g glycoprotein
GFP green fluorescent protein
GITRL glucocorticoid-induced tumor necrosis factor receptor-ligand
h hour(s)
HIV human immunodeficiency virus
HFF human foreskin fibroblast(s)
HLA human leukocyte antigen

Abbreviations
86
Abbreviation Full length spelling
HPV human papilloma virus
HRP horseradish peroxidase
HSV herpes simplex virus
ICP infected cell polypeptide
IFN interferon(s)
Ig immunoglobulin
IL interleukin
IPC interferon producing cell(s)
IRIS immune reconstitution inflammatory syndrome
J Joule
KIR killer cell immunoglobulin-like receptor
L ligand
l liter
MACS magnetic-activated cell sorting
MDA5 melanoma differentiation-associated protein 5
MFI median fluorescence intensity
MHC major histocompatibility complex
MIC MHC class I polypeptide-related sequence
min minute(s)
µl microliter
ml milliliter
µm micrometer
mo monocyte-derived
MOI multiplicity of infection
n nano
NCAM neural cell adhesion molecule
NCR natural cytotoxicity receptor(s)
NKG2D natural killer group 2, member D
NO nitric oxide
NK cell natural killer cell
OD optical density
ODN oligodeoxynucleotides
PBMC peripheral blood mononuclear cell(s)
PBS phosphate buffered saline
PDC plasmacytoid dendritic cell(s)
PE phycoerythrin
PFA paraformaldehyde

Abbreviations
87
Abbreviation Full length spelling
p.i. post infection
p.p. post purification
p.s. post stimulation
R receptor
rh recombinant human
RIG-I retinoic acid-inducible gene I
ROS reactive oxygen species
RPMI 1640 Roswell Park Memorial Institute 1640 Medium
s soluble
SSC sideward scatter
T thymus-derived
TCID tissue culture infective dose
Th T helper
TLR toll-like receptor
TNF tumor necrosis factor
TRAIL TNF-related apoptosis-inducing ligand
Tris tris(hydroxymethyl)aminomethane
ULBP UL-16-binding protein
UV ultraviolet light
VSB virus standard buffer
VZV varicella zoster virus
W Watt

References
88
8 References
Abdul-Careem,M.F., Lee,A.J., Pek,E.A., Gill,N., Gillgrass,A.E., Chew,M.V., Reid,S., and
Ashkar,A.A. (2012). Genital HSV-2 infection induces short-term NK cell memory. PLoS. One. 7,
e32821.
Ahmad,A., Sharif-Askari,E., Fawaz,L., and Menezes,J. (2000). Innate immune response of the human
host to exposure with herpes simplex virus type 1: in vitro control of the virus infection by enhanced
natural killer activity via interleukin-15 induction. J. Virol. 74, 7196-7203.
Ahmad,R., Ennaciri,J., Cordeiro,P., El,B.S., and Menezes,J. (2007). Herpes simplex virus-1 up-
regulates IL-15 gene expression in monocytic cells through the activation of protein tyrosine kinase
and PKC zeta/lambda signaling pathways. J. Mol. Biol. 367, 25-35.
Albers,I., Kirchner,H., and Domke-Opitz,I. (1989). Resistance of human blood monocytes to infection
with herpes simplex virus. Virology 169, 466-469.
Alter,G., Malenfant,J.M., and Altfeld,M. (2004). CD107a as a functional marker for the identification
of natural killer cell activity. J. Immunol. Methods 294, 15-22.
Arase,H., Arase,N., and Saito,T. (1995). Fas-mediated cytotoxicity by freshly isolated natural killer
cells. J. Exp. Med. 181, 1235-1238.
Ashkar,A.A. and Rosenthal,K.L. (2003). Interleukin-15 and natural killer and NKT cells play a critical
role in innate protection against genital herpes simplex virus type 2 infection. J. Virol. 77, 10168-
10171.
Barr,D.P., Belz,G.T., Reading,P.C., Wojtasiak,M., Whitney,P.G., Heath,W.R., Carbone,F.R., and
Brooks,A.G. (2007). A role for plasmacytoid dendritic cells in the rapid IL-18-dependent activation of
NK cells following HSV-1 infection. Eur. J. Immunol. 37, 1334-1342.
Bauer,S., Groh,V., Wu,J., Steinle,A., Phillips,J.H., Lanier,L.L., and Spies,T. (1999). Activation of NK
cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 285, 727-729.
Benlahrech,A., Donaghy,H., Rozis,G., Goodier,M., Klavinskis,L., Gotch,F., and Patterson,S. (2009).
Human NK Cell Up-regulation of CD69, HLA-DR, Interferon gamma Secretion and Cytotoxic
Activity by Plasmacytoid Dendritic Cells is Regulated through Overlapping but Different Pathways.
Sensors. (Basel) 9, 386-403.
Benlahrech,A., Gotch,F., Kelleher,P., and Patterson,S. (2011). Loss of NK stimulatory capacity by
plasmacytoid and monocyte-derived DC but not myeloid DC in HIV-1 infected patients. PLoS. One.
6, e17525.
Bernard Roizman, David M.Knipe, and Richard J.Whitley (2007a). Herpes Simplex Viruses: Clinical
Manifestations of Disease. In Fields Virology, David M.Knipe and Peter M.Howley, eds.
LippincottWilliams &Wilkins), pp. 1878-1885.
Bernard Roizman, David M.Knipe, and Richard J.Whitley (2007b). Herpes Simplex Viruses:
Epidemiology. In Fields Virology, David M.Knipe and Peter M.Howley, eds. LippincottWilliams
&Wilkins), pp. 1871-1876.

References
89
Bernard Roizman, David M.Knipe, and Richard J.Whitley (2007c). Herpes Simplex Viruses: Latent
Infection. In Fields Virology, David M.Knipe and Peter M.Howley, eds. LippincottWilliams
&Wilkins), pp. 1862-1869.
Bernard Roizman, David M.Knipe, and Richard J.Whitley (2007d). Herpes Simplex Viruses: Structure
and Sequence Arrangement of Viral DNA. In Fields Virology, David M.Knipe and Peter M.Howley,
eds. LippincottWilliams &Wilkins), pp. 1826-1830.
Bernard Roizman, David M.Knipe, and Richard J.Whitley (2007e). Herpes Simplex Viruses: Virion
Structure. In Fields Virology, David M.Knipe and Peter M.Howley, eds. LippincottWilliams
&Wilkins), pp. 1825-1826.
Brandt,C.S., Baratin,M., Yi,E.C., Kennedy,J., Gao,Z., Fox,B., Haldeman,B., Ostrander,C.D., Kaifu,T.,
Chabannon,C., Moretta,A., West,R., Xu,W., Vivier,E., and Levin,S.D. (2009). The B7 family member
B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J. Exp.
Med. 206, 1495-1503.
Bruun,T., Kristoffersen,A.K., Rollag,H., and Degre,M. (1998). Interaction of herpes simplex virus
with mononuclear phagocytes is dependent on the differentiation stage of the cells. APMIS 106, 305-
314.
Cardona,I.D., Goleva,E., Ou,L.S., and Leung,D.Y. (2006). Staphylococcal enterotoxin B inhibits
regulatory T cells by inducing glucocorticoid-induced TNF receptor-related protein ligand on
monocytes. J. Allergy Clin. Immunol. 117, 688-695.
Casrouge,A., Zhang,S.Y., Eidenschenk,C., Jouanguy,E., Puel,A., Yang,K., Alcais,A., Picard,C.,
Mahfoufi,N., Nicolas,N., Lorenzo,L., Plancoulaine,S., Senechal,B., Geissmann,F., Tabeta,K.,
Hoebe,K., Du,X., Miller,R.L., Heron,B., Mignot,C., de Villemeur,T.B., Lebon,P., Dulac,O.,
Rozenberg,F., Beutler,B., Tardieu,M., Abel,L., and Casanova,J.L. (2006). Herpes simplex virus
encephalitis in human UNC-93B deficiency. Science 314, 308-312.
Cederblad,B. and Alm,G.V. (1990). Infrequent but efficient interferon-alpha-producing human
mononuclear leukocytes induced by herpes simplex virus in vitro studied by immuno-plaque and
limiting dilution assays. J. Interferon Res. 10, 65-73.
Cella,M., Jarrossay,D., Facchetti,F., Alebardi,O., Nakajima,H., Lanzavecchia,A., and Colonna,M.
(1999). Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type
I interferon. Nat. Med. 5, 919-923.
Cheng,H., Tumpey,T.M., Staats,H.F., van,R.N., Oakes,J.E., and Lausch,R.N. (2000). Role of
macrophages in restricting herpes simplex virus type 1 growth after ocular infection. Invest
Ophthalmol. Vis. Sci. 41, 1402-1409.
Chisholm,S.E., Howard,K., Gomez,M.V., and Reyburn,H.T. (2007). Expression of ICP0 is sufficient
to trigger natural killer cell recognition of herpes simplex virus-infected cells by natural cytotoxicity
receptors. J. Infect. Dis. 195, 1160-1168.
Colonna,M., Trinchieri,G., and Liu,Y.J. (2004). Plasmacytoid dendritic cells in immunity. Nat.
Immunol. 5, 1219-1226.

References
90
Conry,S.J., Milkovich,K.A., Yonkers,N.L., Rodriguez,B., Bernstein,H.B., Asaad,R., Heinzel,F.P.,
Tary-Lehmann,M., Lederman,M.M., and Anthony,D.D. (2009). Impaired plasmacytoid dendritic cell
(PDC)-NK cell activity in viremic human immunodeficiency virus infection attributable to
impairments in both PDC and NK cell function. J. Virol. 83, 11175-11187.
Cooper,M.A., Fehniger,T.A., and Caligiuri,M.A. (2001a). The biology of human natural killer-cell
subsets. Trends Immunol. 22, 633-640.
Cooper,M.A., Fehniger,T.A., Ponnappan,A., Mehta,V., Wewers,M.D., and Caligiuri,M.A. (2001b).
Interleukin-1beta costimulates interferon-gamma production by human natural killer cells. Eur. J.
Immunol. 31, 792-801.
Cousens,L.P., Orange,J.S., Su,H.C., and Biron,C.A. (1997). Interferon-alpha/beta inhibition of
interleukin 12 and interferon-gamma production in vitro and endogenously during viral infection.
Proc. Natl. Acad. Sci. U. S. A 94, 634-639.
Cunningham,A.L., Diefenbach,R.J., Miranda-Saksena,M., Bosnjak,L., Kim,M., Jones,C., and
Douglas,M.W. (2006). The cycle of human herpes simplex virus infection: virus transport and immune
control. J. Infect. Dis. 194 Suppl 1, S11-S18.
Dale,D.C., Boxer,L., and Liles,W.C. (2008). The phagocytes: neutrophils and monocytes. Blood 112,
935-945.
Dalloul,A., Oksenhendler,E., Chosidow,O., Ribaud,P., Carcelain,G., Louvet,S., Massip,P., Lebon,P.,
and Autran,B. (2004). Severe herpes virus (HSV-2) infection in two patients with myelodysplasia and
undetectable NK cells and plasmacytoid dendritic cells in the blood. J. Clin. Virol. 30, 329-336.
Daniels,C.A., Kleinerman,E.S., and Snyderman,R. (1978). Abortive and productive infections of
human mononuclear phagocytes by type I herpes simplex virus. Am. J. Pathol. 91, 119-136.
De,M.A., Bozzano,F., Cantoni,C., and Moretta,L. (2011). Revisiting human natural killer cell subset
function revealed cytolytic CD56(dim)CD16+ NK cells as rapid producers of abundant IFN-gamma
on activation. Proc. Natl. Acad. Sci. U. S. A 108, 728-732.
Diaz,M.O., Ziemin,S., Le Beau,M.M., Pitha,P., Smith,S.D., Chilcote,R.R., and Rowley,J.D. (1988).
Homozygous deletion of the alpha- and beta 1-interferon genes in human leukemia and derived cell
lines. Proc. Natl. Acad. Sci. U. S. A 85, 5259-5263.
Donaghy,H., Bosnjak,L., Harman,A.N., Marsden,V., Tyring,S.K., Meng,T.C., and Cunningham,A.L.
(2009). Role for plasmacytoid dendritic cells in the immune control of recurrent human herpes
simplex virus infection. J. Virol. 83, 1952-1961.
Donhauser,N., Pritschet,K., Helm,M., Harrer,T., Schuster,P., Ries,M., Bischof,G., Vollmer,J.,
Smola,S., and Schmidt,B. (2012). Chronic immune activation in HIV-1 infection contributes to
reduced interferon alpha production via enhanced CD40:CD40 ligand interaction. PLoS. One. 7,
e33925.
Dupuis,S., Jouanguy,E., Al-Hajjar,S., Fieschi,C., Al-Mohsen,I.Z., Al-Jumaah,S., Yang,K.,
Chapgier,A., Eidenschenk,C., Eid,P., Al,G.A., Tufenkeji,H., Frayha,H., Al-Gazlan,S., Al-Rayes,H.,
Schreiber,R.D., Gresser,I., and Casanova,J.L. (2003). Impaired response to interferon-alpha/beta and
lethal viral disease in human STAT1 deficiency. Nat. Genet. 33, 388-391.

References
91
Dzionek,A., Inagaki,Y., Okawa,K., Nagafune,J., Rock,J., Sohma,Y., Winkels,G., Zysk,M.,
Yamaguchi,Y., and Schmitz,J. (2002). Plasmacytoid dendritic cells: from specific surface markers to
specific cellular functions. Hum. Immunol. 63, 1133-1148.
Dzionek,A., Sohma,Y., Nagafune,J., Cella,M., Colonna,M., Facchetti,F., Gunther,G., Johnston,I.,
Lanzavecchia,A., Nagasaka,T., Okada,T., Vermi,W., Winkels,G., Yamamoto,T., Zysk,M.,
Yamaguchi,Y., and Schmitz,J. (2001). BDCA-2, a novel plasmacytoid dendritic cell-specific type II
C-type lectin, mediates antigen capture and is a potent inhibitor of interferon alpha/beta induction. J.
Exp. Med. 194, 1823-1834.
Elboim,M., Grodzovski,I., Djian,E., Wolf,D.G., and Mandelboim,O. (2013). HSV-2 specifically down
regulates HLA-C expression to render HSV-2-infected DCs susceptible to NK cell killing. PLoS.
Pathog. 9, e1003226.
Ellermann-Eriksen,S. (2005). Macrophages and cytokines in the early defence against herpes simplex
virus. Virol. J. 2, 59.
Elliott,G. and O'Hare,P. (1999). Live-cell analysis of a green fluorescent protein-tagged herpes
simplex virus infection. J. Virol. 73, 4110-4119.
Feldman,M., Howell,D., and Fitzgerald-Bocarsly,P. (1992). Interferon-alpha-dependent and -
independent participation of accessory cells in natural killer cell-mediated lysis of HSV-1-infected
fibroblasts. J. Leukoc. Biol. 52, 473-482.
Feldman,S., Stein,D., Amrute,S., Denny,T., Garcia,Z., Kloser,P., Sun,Y., Megjugorac,N., and
Fitzgerald-Bocarsly,P. (2001). Decreased interferon-alpha production in HIV-infected patients
correlates with numerical and functional deficiencies in circulating type 2 dendritic cell precursors.
Clin. Immunol. 101, 201-210.
Fields,M., Zheng,M., Zhang,M., and Atherton,S.S. (2006). Tumor necrosis factor alpha and
macrophages in the brain of herpes simplex virus type 1-infected BALB/c mice. J. Neurovirol. 12,
443-455.
Fitzgerald,P.A., Mendelsohn,M., and Lopez,C. (1985). Human natural killer cells limit replication of
herpes simplex virus type 1 in vitro. J. Immunol. 134, 2666-2672.
Fitzgerald-Bocarsly,P., Howell,D.M., Pettera,L., Tehrani,S., and Lopez,C. (1991). Immediate-early
gene expression is sufficient for induction of natural killer cell-mediated lysis of herpes simplex virus
type 1-infected fibroblasts. J. Virol. 65, 3151-3160.
Frank,G.M., Buela,K.A., Maker,D.M., Harvey,S.A., and Hendricks,R.L. (2012). Early responding
dendritic cells direct the local NK response to control herpes simplex virus 1 infection within the
cornea. J. Immunol. 188, 1350-1359.
Früh,K., Ahn,K., Djaballah,H., Sempe,P., van Endert,P.M., Tampe,R., Peterson,P.A., and Yang,Y.
(1995). A viral inhibitor of peptide transporters for antigen presentation. Nature 375, 415-418.
Gerosa,F., Gobbi,A., Zorzi,P., Burg,S., Briere,F., Carra,G., and Trinchieri,G. (2005). The reciprocal
interaction of NK cells with plasmacytoid or myeloid dendritic cells profoundly affects innate
resistance functions. J. Immunol. 174, 727-734.

References
92
Ghiasi,H., Perng,G., Nesburn,A.B., and Wechsler,S.L. (1999). Either a CD4(+)or CD8(+)T cell
function is sufficient for clearance of infectious virus from trigeminal ganglia and establishment of
herpes simplex virus type 1 latency in mice. Microb. Pathog. 27, 387-394.
Gill,N., Chenoweth,M.J., Verdu,E.F., and Ashkar,A.A. (2011). NK cells require type I IFN receptor
for antiviral responses during genital HSV-2 infection. Cell Immunol. 269, 29-37.
Gosselin,J., Flamand,L., D'Addario,M., Hiscott,J., and Menezes,J. (1992). Infection of peripheral
blood mononuclear cells by herpes simplex and Epstein-Barr viruses. Differential induction of
interleukin 6 and tumor necrosis factor-alpha. J. Clin. Invest 89, 1849-1856.
Habu,S., Akamatsu,K., Tamaoki,N., and Okumura,K. (1984). In vivo significance of NK cell on
resistance against virus (HSV-1) infections in mice. J. Immunol. 133, 2743-2747.
Hanabuchi,S., Watanabe,N., Wang,Y.H., Wang,Y.H., Ito,T., Shaw,J., Cao,W., Qin,F.X., and Liu,Y.J.
(2006). Human plasmacytoid predendritic cells activate NK cells through glucocorticoid-induced
tumor necrosis factor receptor-ligand (GITRL). Blood 107, 3617-3623.
Härle,P., Lauret,E., Pitha,P.M., De,M.E., and Carr,D.J. (2001). Expression of human and macaque
type I IFN transgenes interferes with HSV-1 replication at the transcriptional and translational levels:
IFN-beta is more potent than IFN-alpha 2. Virology 290, 237-248.
Hemmi,H., Takeuchi,O., Kawai,T., Kaisho,T., Sato,S., Sanjo,H., Matsumoto,M., Hoshino,K.,
Wagner,H., Takeda,K., and Akira,S. (2000). A Toll-like receptor recognizes bacterial DNA. Nature
408, 740-745.
Hill,A., Jugovic,P., York,I., Russ,G., Bennink,J., Yewdell,J., Ploegh,H., and Johnson,D. (1995).
Herpes simplex virus turns off the TAP to evade host immunity. Nature 375, 411-415.
Huard,B. and Früh,K. (2000). A role for MHC class I down-regulation in NK cell lysis of herpes
virus-infected cells. Eur. J. Immunol. 30, 509-515.
Iftner,A., Klug,S.J., Garbe,C., Blum,A., Stancu,A., Wilczynski,S.P., and Iftner,T. (2003). The
prevalence of human papillomavirus genotypes in nonmelanoma skin cancers of
nonimmunosuppressed individuals identifies high-risk genital types as possible risk factors. Cancer
Res. 63, 7515-7519.
ISAACS,A. and LINDENMANN,J. (1957). Virus interference. I. The interferon. Proc. R. Soc. Lond B
Biol. Sci. 147, 258-267.
Jawahar,S., Moody,C., Chan,M., Finberg,R., Geha,R., and Chatila,T. (1996). Natural Killer (NK) cell
deficiency associated with an epitope-deficient Fc receptor type IIIA (CD16-II). Clin. Exp. Immunol.
103, 408-413.
Jimbo,K., Park,J.S., Yokosuka,K., Sato,K., and Nagata,K. (2005). Positive feedback loop of
interleukin-1beta upregulating production of inflammatory mediators in human intervertebral disc
cells in vitro. J. Neurosurg. Spine 2, 589-595.
Johnson,A.J., Chu,C.F., and Milligan,G.N. (2008). Effector CD4+ T-cell involvement in clearance of
infectious herpes simplex virus type 1 from sensory ganglia and spinal cords. J. Virol. 82, 9678-9688.

References
93
Kadowaki,N., Antonenko,S., Lau,J.Y., and Liu,Y.J. (2000). Natural interferon alpha/beta-producing
cells link innate and adaptive immunity. J. Exp. Med. 192, 219-226.
Kägi,D., Ledermann,B., Burki,K., Seiler,P., Odermatt,B., Olsen,K.J., Podack,E.R., Zinkernagel,R.M.,
and Hengartner,H. (1994). Cytotoxicity mediated by T cells and natural killer cells is greatly impaired
in perforin-deficient mice. Nature 369, 31-37.
Kärre,K., Ljunggren,H.G., Piontek,G., and Kiessling,R. (1986). Selective rejection of H-2-deficient
lymphoma variants suggests alternative immune defence strategy. Nature 319, 675-678.
Kassim,S.H., Rajasagi,N.K., Ritz,B.W., Pruett,S.B., Gardner,E.M., Chervenak,R., and Jennings,S.R.
(2009). Dendritic cells are required for optimal activation of natural killer functions following primary
infection with herpes simplex virus type 1. J. Virol. 83, 3175-3186.
Kim,E.T., White,T.E., Brandariz-Nunez,A., az-Griffero,F., and Weitzman,M.D. (2013). SAMHD1
restricts herpes simplex virus 1 in macrophages by limiting DNA replication. J. Virol. 87, 12949-
12956.
Kim,M., Osborne,N.R., Zeng,W., Donaghy,H., McKinnon,K., Jackson,D.C., and Cunningham,A.L.
(2012). Herpes simplex virus antigens directly activate NK cells via TLR2, thus facilitating their
presentation to CD4 T lymphocytes. J. Immunol. 188, 4158-4170.
Kittan,N.A., Bergua,A., Haupt,S., Donhauser,N., Schuster,P., Korn,K., Harrer,T., and Schmidt,B.
(2007). Impaired plasmacytoid dendritic cell innate immune responses in patients with herpes virus-
associated acute retinal necrosis. J. Immunol. 179, 4219-4230.
Kloss,M., Decker,P., Baltz,K.M., Baessler,T., Jung,G., Rammensee,H.G., Steinle,A., Krusch,M., and
Salih,H.R. (2008). Interaction of monocytes with NK cells upon Toll-like receptor-induced expression
of the NKG2D ligand MICA. J. Immunol. 181, 6711-6719.
Kodukula,P., Liu,T., Rooijen,N.V., Jager,M.J., and Hendricks,R.L. (1999). Macrophage control of
herpes simplex virus type 1 replication in the peripheral nervous system. J. Immunol. 162, 2895-2905.
Koelle,D.M., Posavad,C.M., Barnum,G.R., Johnson,M.L., Frank,J.M., and Corey,L. (1998). Clearance
of HSV-2 from recurrent genital lesions correlates with infiltration of HSV-specific cytotoxic T
lymphocytes. J. Clin. Invest 101, 1500-1508.
Krug,A., Luker,G.D., Barchet,W., Leib,D.A., Akira,S., and Colonna,M. (2004). Herpes simplex virus
type 1 activates murine natural interferon-producing cells through toll-like receptor 9. Blood 103,
1433-1437.
L.J.REED and H.MUENCH (1938). A simple method of estimating fifty per cent
endpoints. American Journal of Hygiene 27, 493-497.
Lanier,L.L. (2005). NK cell recognition. Annu. Rev. Immunol. 23, 225-274.
Lanier,L.L., Testi,R., Bindl,J., and Phillips,J.H. (1989). Identity of Leu-19 (CD56) leukocyte
differentiation antigen and neural cell adhesion molecule. J. Exp. Med. 169, 2233-2238.
Leibson,P.J. (1997). Signal transduction during natural killer cell activation: inside the mind of a
killer. Immunity. 6, 655-661.

References
94
Leibson,P.J., Hunter-Laszlo,M., and Hayward,A.R. (1986). Inhibition of herpes simplex virus type 1
replication in fibroblast cultures by human blood mononuclear cells. J. Virol. 57, 976-982.
Leon,B., Lopez-Bravo,M., and Ardavin,C. (2007). Monocyte-derived dendritic cells formed at the
infection site control the induction of protective T helper 1 responses against Leishmania. Immunity.
26, 519-531.
Linnavuori,K. and Hovi,T. (1983). Restricted replication of herpes simplex virus in human monocyte
cultures: role of interferon. Virology 130, 1-9.
Liu,X., Broberg,E., Watanabe,D., Dudek,T., Deluca,N., and Knipe,D.M. (2009). Genetic engineering
of a modified herpes simplex virus 1 vaccine vector. Vaccine 27, 2760-2767.
Lodoen,M.B. and Lanier,L.L. (2006). Natural killer cells as an initial defense against pathogens. Curr.
Opin. Immunol. 18, 391-398.
Lund,J., Sato,A., Akira,S., Medzhitov,R., and Iwasaki,A. (2003). Toll-like receptor 9-mediated
recognition of Herpes simplex virus-2 by plasmacytoid dendritic cells. J. Exp. Med. 198, 513-520.
Lund,J.M., Linehan,M.M., Iijima,N., and Iwasaki,A. (2006). Cutting Edge: Plasmacytoid dendritic
cells provide innate immune protection against mucosal viral infection in situ. J. Immunol. 177, 7510-
7514.
Marie,I., Durbin,J.E., and Levy,D.E. (1998). Differential viral induction of distinct interferon-alpha
genes by positive feedback through interferon regulatory factor-7. EMBO J. 17, 6660-6669.
Marshall,J.D., Heeke,D.S., Abbate,C., Yee,P., and Van,N.G. (2006). Induction of interferon-gamma
from natural killer cells by immunostimulatory CpG DNA is mediated through plasmacytoid-
dendritic-cell-produced interferon-alpha and tumour necrosis factor-alpha. Immunology 117, 38-46.
Matta,J., Baratin,M., Chiche,L., Forel,J.M., Cognet,C., Thomas,G., Farnarier,C., Piperoglou,C.,
Papazian,L., Chaussabel,D., Ugolini,S., Vely,F., and Vivier,E. (2013). Induction of B7-H6, a ligand
for the natural killer cell-activating receptor NKp30, in inflammatory conditions. Blood 122, 394-404.
Megjugorac,N.J., Jacobs,E.S., Izaguirre,A.G., George,T.C., Gupta,G., and Fitzgerald-Bocarsly,P.
(2007). Image-based study of interferongenic interactions between plasmacytoid dendritic cells and
HSV-infected monocyte-derived dendritic cells. Immunol. Invest 36, 739-761.
Megjugorac,N.J., Young,H.A., Amrute,S.B., Olshalsky,S.L., and Fitzgerald-Bocarsly,P. (2004).
Virally stimulated plasmacytoid dendritic cells produce chemokines and induce migration of T and
NK cells. J. Leukoc. Biol. 75, 504-514.
Melchjorsen,J., Rintahaka,J., Soby,S., Horan,K.A., Poltajainen,A., Ostergaard,L., Paludan,S.R., and
Matikainen,S. (2010). Early innate recognition of herpes simplex virus in human primary
macrophages is mediated via the MDA5/MAVS-dependent and MDA5/MAVS/RNA polymerase III-
independent pathways. J. Virol. 84, 11350-11358.
Melchjorsen,J., Siren,J., Julkunen,I., Paludan,S.R., and Matikainen,S. (2006). Induction of cytokine
expression by herpes simplex virus in human monocyte-derived macrophages and dendritic cells is
dependent on virus replication and is counteracted by ICP27 targeting NF-kappaB and IRF-3. J. Gen.
Virol. 87, 1099-1108.

References
95
Metkar,S.S., Wang,B., guilar-Santelises,M., Raja,S.M., Uhlin-Hansen,L., Podack,E., Trapani,J.A., and
Froelich,C.J. (2002). Cytotoxic cell granule-mediated apoptosis: perforin delivers granzyme B-
serglycin complexes into target cells without plasma membrane pore formation. Immunity. 16, 417-
428.
Michael Ehrenstein, Claudia Mauri, Allan Mowat, and Andrey Shaw (2008a). Innate Immunity 2-23.
In Janeway's Immuno Biology, Kenneth Murphy, Paul Travers, and Mark Walport, eds. Garland
Science), pp. 83-86.
Michael Ehrenstein, Claudia Mauri, Allan Mowat, and Andrey Shaw (2008b). Innate Immunity 2-4. In
Janeway's Immuno Biology, Kenneth Murphy, Paul Travers, and Mark Walport, eds. Garland
Science), pp. 48-52.
Michael Ehrenstein, Claudia Mauri, Allan Mowat, and Andrey Shaw (2008c). Innate Immunity 2-6. In
Janeway's Immuno Biology, Kenneth Murphy, Paul Travers, and Mark Walport, eds. Garland
Science), pp. 54-59.
Michael Ehrenstein, Claudia Mauri, Allan Mowat, and Andrey Shaw (2008d). The Humoral Immune
Response 9-22. In Janeway's Immuno Biology, Kenneth Murphy, Paul Travers, and Mark Walport,
eds. Garland Science), pp. 411-412.
Michel,T., Hentges,F., and Zimmer,J. (2012). Consequences of the crosstalk between
monocytes/macrophages and natural killer cells. Front Immunol. 3, 403.
Minami,M., Kita,M., Yan,X.Q., Yamamoto,T., Iida,T., Sekikawa,K., Iwakura,Y., and Imanishi,J.
(2002). Role of IFN-gamma and tumor necrosis factor-alpha in herpes simplex virus type 1 infection.
J. Interferon Cytokine Res. 22, 671-676.
Mogensen,S.C. (1979). Role of macrophages in natural resistance to virus infections. Microbiol. Rev.
43, 1-26.
Morahan,P.S., Morse,S.S., and McGeorge,M.G. (1980). Macrophage extrinsic antiviral activity during
herpes simplex virus infection. J. Gen. Virol. 46, 291-300.
Nandakumar,S., Woolard,S.N., Yuan,D., Rouse,B.T., and Kumaraguru,U. (2008). Natural killer cells
as novel helpers in anti-herpes simplex virus immune response. J. Virol. 82, 10820-10831.
Nedvetzki,S., Sowinski,S., Eagle,R.A., Harris,J., Vely,F., Pende,D., Trowsdale,J., Vivier,E.,
Gordon,S., and Davis,D.M. (2007). Reciprocal regulation of human natural killer cells and
macrophages associated with distinct immune synapses. Blood 109, 3776-3785.
Nguyen,K.B., Salazar-Mather,T.P., Dalod,M.Y., Van Deusen,J.B., Wei,X.Q., Liew,F.Y.,
Caligiuri,M.A., Durbin,J.E., and Biron,C.A. (2002). Coordinated and distinct roles for IFN-alpha beta,
IL-12, and IL-15 regulation of NK cell responses to viral infection. J. Immunol. 169, 4279-4287.
Noisakran,S., Campbell,I.L., and Carr,D.J. (2000). IFN-alpha1 plasmid construct affords protection
against HSV-1 infection in transfected L929 fibroblasts. J. Interferon Cytokine Res. 20, 107-115.
Orange,J.S. (2002). Human natural killer cell deficiencies and susceptibility to infection. Microbes.
Infect. 4, 1545-1558.

References
96
Paya,C.V., Kenmotsu,N., Schoon,R.A., and Leibson,P.J. (1988). Tumor necrosis factor and
lymphotoxin secretion by human natural killer cells leads to antiviral cytotoxicity. J. Immunol. 141,
1989-1995.
Persson,C.M. and Chambers,B.J. (2010). Plasmacytoid dendritic cell-induced migration and activation
of NK cells in vivo. Eur. J. Immunol. 40, 2155-2164.
Philip E.Pellett and Bernard Roizman (2007). Herpesviridae: Nomenclature and Classification. In
Fields Virology, David M.Knipe and Peter M.Howley, eds. LippincottWilliams &Wilkins), pp. 1804-
1808.
Randolph,G.J., Inaba,K., Robbiani,D.F., Steinman,R.M., and Muller,W.A. (1999). Differentiation of
phagocytic monocytes into lymph node dendritic cells in vivo. Immunity. 11, 753-761.
Reading,P.C., Whitney,P.G., Barr,D.P., Wojtasiak,M., Mintern,J.D., Waithman,J., and Brooks,A.G.
(2007). IL-18, but not IL-12, regulates NK cell activity following intranasal herpes simplex virus type
1 infection. J. Immunol. 179, 3214-3221.
Reitano,K.N., Kottilil,S., Gille,C.M., Zhang,X., Yan,M., O'Shea,M.A., Roby,G., Hallahan,C.W.,
Yang,J., Lempicki,R.A., Arthos,J., and Fauci,A.S. (2009). Defective plasmacytoid dendritic cell-NK
cell cross-talk in HIV infection. AIDS Res. Hum. Retroviruses 25, 1029-1037.
Romagnani,C., Della,C.M., Kohler,S., Moewes,B., Radbruch,A., Moretta,L., Moretta,A., and Thiel,A.
(2005). Activation of human NK cells by plasmacytoid dendritic cells and its modulation by CD4+ T
helper cells and CD4+ CD25hi T regulatory cells. Eur. J. Immunol. 35, 2452-2458.
Rönnblom,L., Cederblad,B., Sandberg,K., and Alm,G.V. (1988). Determination of herpes simplex
virus-induced alpha interferon-secreting human blood leucocytes by a filter immuno-plaque assay.
Scand. J. Immunol. 27, 165-170.
Rutishauser,U. and Jessell,T.M. (1988). Cell adhesion molecules in vertebrate neural development.
Physiol Rev. 68, 819-857.
Samuel,C.E. (2001). Antiviral actions of interferons. Clin. Microbiol. Rev. 14, 778-809, table.
Schmidt,B., Ashlock,B.M., Foster,H., Fujimura,S.H., and Levy,J.A. (2005). HIV-infected cells are
major inducers of plasmacytoid dendritic cell interferon production, maturation, and migration.
Virology 343, 256-266.
Schmidt,B., Fujimura,S.H., Martin,J.N., and Levy,J.A. (2006). Variations in plasmacytoid dendritic
cell (PDC) and myeloid dendritic cell (MDC) levels in HIV-infected subjects on and off antiretroviral
therapy. J. Clin. Immunol. 26, 55-64.
Schuster,P., Boscheinen,J.B., Tennert,K., and Schmidt,B. (2011). The Role of Plasmacytoid Dendritic
Cells in Innate and Adaptive Immune Responses against Alpha Herpes Virus Infections. Adv. Virol.
2011, 679271.
Schuster,P., Donhauser,N., Pritschet,K., Ries,M., Haupt,S., Kittan,N.A., Korn,K., and Schmidt,B.
(2010). Co-ordinated regulation of plasmacytoid dendritic cell surface receptors upon stimulation with
herpes simplex virus type 1. Immunology 129, 234-247.

References
97
Sergerie,Y., Rivest,S., and Boivin,G. (2007). Tumor necrosis factor-alpha and interleukin-1 beta play
a critical role in the resistance against lethal herpes simplex virus encephalitis. J. Infect. Dis. 196, 853-
860.
Siegal,F.P., Kadowaki,N., Shodell,M., Fitzgerald-Bocarsly,P.A., Shah,K., Ho,S., Antonenko,S., and
Liu,Y.J. (1999). The nature of the principal type 1 interferon-producing cells in human blood. Science
284, 1835-1837.
Strehl,J.D., Mehlhorn,G., Koch,M.C., Harrer,E.G., Harrer,T., Beckmann,M.W., and Agaimy,A.
(2012). HIV-associated hypertrophic herpes simplex genitalis with concomitant early invasive
squamous cell carcinoma mimicking advanced genital cancer: case report and literature review. Int. J.
Gynecol. Pathol. 31, 286-293.
Swiecki,M., Wang,Y., Gilfillan,S., and Colonna,M. (2013). Plasmacytoid dendritic cells contribute to
systemic but not local antiviral responses to HSV infections. PLoS. Pathog. 9, e1003728.
Tanigawa,M., Bigger,J.E., Kanter,M.Y., and Atherton,S.S. (2000). Natural killer cells prevent direct
anterior-to-posterior spread of herpes simplex virus type 1 in the eye. Invest Ophthalmol. Vis. Sci. 41,
132-137.
Tough,D.F., Borrow,P., and Sprent,J. (1996). Induction of bystander T cell proliferation by viruses
and type I interferon in vivo. Science 272, 1947-1950.
Trinchieri,G. (1989). Biology of natural killer cells. Adv. Immunol. 47, 187-376.
Trinchieri,G., Matsumoto-Kobayashi,M., Clark,S.C., Seehra,J., London,L., and Perussia,B. (1984).
Response of resting human peripheral blood natural killer cells to interleukin 2. J. Exp. Med. 160,
1147-1169.
van,F.R. and Cohn,Z.A. (1968). The origin and kinetics of mononuclear phagocytes. J. Exp. Med. 128,
415-435.
Villadangos,J.A. and Young,L. (2008). Antigen-presentation properties of plasmacytoid dendritic
cells. Immunity. 29, 352-361.
Vivier,E. (2006). What is natural in natural killer cells? Immunol. Lett. 107, 1-7.
Vivier,E., Tomasello,E., Baratin,M., Walzer,T., and Ugolini,S. (2008). Functions of natural killer
cells. Nat. Immunol. 9, 503-510.
Voth,R., Rossol,S., Gallati,H., Pracht,I., Laubenstein,H.P., Hess,G., Muller,W.E., Schroder,H.C.,
Jochum,C., and Meyer zum Buschenfelde,K.H. (1988). In vivo and in vitro induction of natural killer
cells by cloned human tumor necrosis factor. Cancer Immunol. Immunother. 27, 128-132.
Welte,S., Kuttruff,S., Waldhauer,I., and Steinle,A. (2006). Mutual activation of natural killer cells and
monocytes mediated by NKp80-AICL interaction. Nat. Immunol. 7, 1334-1342.
Wolf,S.F., Temple,P.A., Kobayashi,M., Young,D., Dicig,M., Lowe,L., Dzialo,R., Fitz,L., Ferenz,C.,
Hewick,R.M., and . (1991). Cloning of cDNA for natural killer cell stimulatory factor, a heterodimeric
cytokine with multiple biologic effects on T and natural killer cells. J. Immunol. 146, 3074-3081.

References
98
Yarilina,A., Park-Min,K.H., Antoniv,T., Hu,X., and Ivashkiv,L.B. (2008). TNF activates an IRF1-
dependent autocrine loop leading to sustained expression of chemokines and STAT1-dependent type I
interferon-response genes. Nat. Immunol. 9, 378-387.
Yoneyama,M., Kikuchi,M., Matsumoto,K., Imaizumi,T., Miyagishi,M., Taira,K., Foy,E., Loo,Y.M.,
Gale,M., Jr., Akira,S., Yonehara,S., Kato,A., and Fujita,T. (2005). Shared and unique functions of the
DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J. Immunol. 175, 2851-
2858.
Zamai,L., Ahmad,M., Bennett,I.M., Azzoni,L., Alnemri,E.S., and Perussia,B. (1998). Natural killer
(NK) cell-mediated cytotoxicity: differential use of TRAIL and Fas ligand by immature and mature
primary human NK cells. J. Exp. Med. 188, 2375-2380.
Zhang,S.Y., Jouanguy,E., Sancho-Shimizu,V., von,B.H., Yang,K., Abel,L., Picard,C., Puel,A., and
Casanova,J.L. (2007). Human Toll-like receptor-dependent induction of interferons in protective
immunity to viruses. Immunol. Rev. 220, 225-236.

Publications
99
9 Publications
Vogel K, Thomann S, Vogel B, Schuster P, Schmidt B. Both plasmacytoid dendritic cells and
monocytes stimulate natural killer cells early during human HSV-1 infections. Immunology. 2014 Dec;
143(4):588-600.
Vogel B, Tennert K, Full F, Ensser A. Efficient generation of human natural killer cell lines by viral
transformation. Leukemia. 2014 Jan; 28(1):192-5.
Tennert K, Schneider L, Bischof G, Korn K, Harrer E, Harrer T, Schmidt B; German Competence
Network HIVAIDS. Elevated CD40 ligand silences α interferon production in an HIV-related immune
reconstitution inflammatory syndrome. AIDS. 2013 Jan 14; 27(2):297-9.
Schuster P, Boscheinen JB, Tennert K, Schmidt B. The Role of Plasmacytoid Dendritic Cells in
Innate and Adaptive Immune Responses against Alpha Herpes Virus Infections. Adv Virol. 2011;
2011:679271.