strukturen organischer moleküle (rademacher:strukturen o-bk) || methoden der strukturanalyse
TRANSCRIPT

l Methoden der Struktur-analyse
In diesem Kapitel werden die vier wichtigsten Methoden zur Bestimmung dergeometrischen Struktur von Molekülen - Mikrowellenspektroskopie, Elektronen-beugung, Röntgenstrukturanalyse und Neutronenbeugung - behandelt. Anderephysikalische Methoden wie NMR-, IR-, Raman-, UV- und Massenspektroskopiesind als Hilfsmittel bei der Aufklärung der Konstitution einer Verbindung und beimStudium seiner Konformationseigenschaften unerläßlich, sie gestatten aber nur inAusnahmefällen die Ermittlung exakter Strukturparameter. Tabelle 1-1 gibt eineknappe Übersicht über die wichtigsten experimentellen Methoden der Struktur-analyse. Auf einige wird in Kap. 3 und 5 näher eingegangen.
Zunächst sind jedoch einige Begriffserläuterungen und allgemeine Angaben zumbesseren Verständnis der nachfolgenden Abschnitte angebracht.
1.1 Die Gestalt eines Moleküls und ihreBeschreibung
1.1.1 Einleitung
In der Chemie gibt es kaum einen wichtigeren Begriff als den der Molekülstruktur,denn die Eigenschaften einer Verbindung sind eng mit der Art und Weise ihrerAtomverknüpfung korreliert. Die Vorstellung von der Gestalt organischer Molekülewurden im 19. Jahrhundert entwickelt. Allerdings war auch die Meinung, daß einMolekül keine physikalische Realität besäße, sondern lediglich ein nützlicher Begriffsei, noch lange verbreitet. Auch wenn in jüngster Zeit die Diskussion über die Frage,ob einem Molekül eine reale Gestalt zukommt, wieder auflebte (s. z. B. Weininger,1984), läßt sich doch ohne Einschränkung feststellen, daß die Fortschritte im Ver-ständnis chemischer Vorgänge untrennbar mit der Kenntnis der Molekülstrukturverbunden sind. Das Strukturdenken in der organischen Chemie (Weißbach, 1971)manifestiert sich in ihrer derzeit wohl wichtigsten Disziplin, der Stereochemie, alsder dritten Dimension in der organischen Chemie (Bassindale, 1984).
Der Begriff chemische Struktur wurde 1861 von dem russischen Chemiker A. M.Butlerow eingeführt; die Stereochemie stammt aus dem Jahre 1890 von V. Meyer.
Strukturen organischer Moleküle. Paul RademacherCopyright © 1987 VCH Verlagsgesellschaft mbH, WeinheimISBN: 3-527-26545-7

2 l Methoden der Strukturanalyse
Schon A. Kekule hatte erkannt, daß Kohlenstoff vierwertig ist, und zwischen 1858und 1865 seine Strukturtheorie entwickelt, mit der er die Konstitution und vieleEigenschaften organischer Verbindungen sehr gut erklären konnte. Diese Theoriegestattete es, Strukturformeln zu schreiben, und erlaubte damit die Unterscheidungverschiedener Verbindungsklassen nach den vorhandenen funktionellen Gruppen.Entscheidend erweitert wurden diese Vorstellungen durch J.H. van't Hoffund J. A.LeBel, die 1874 unabhängig voneinander die tetraedrische Anordnung der Valenzendes vierbindigen Kohlenstoffs beschrieben (Ramsay, 1981; Weyer, 1974).
Damit war das Konzept der dreidimensionalen Natur von Molekülen entwickeltund auch die Unterscheidung von Stereoisomeren möglich geworden. Daß Molekülekeine starren Gebilde sind, sondern auch innere Bewegungsmöglichkeiten besitzen,zeigten insbesondere die Untersuchungen an Cyclohexan und anderen Alicyclen vonH. Sachse (1890), E. Mohr (1918), W. Hückel (1925) und O. Hassel (1943), sowieD. Bartons Arbeiten an Steroiden (1950). Die moderne Strukturtheorie wurde jedocherst nach der Aufklärung des Atombaus und der Deutung der chemischen Bindungdurch die Quantentheorie möglich.
Heute stehen uns mehrere experimentelle Methoden zur Strukturanalyse zur Ver-fügung (Tab. 1-1). Genaue Strukturparameter - Bindungslängen, Bindungswinkel,Torsionswinkel - werden vor allem mit Hilfe der Mikrowellenspektroskopie sowie
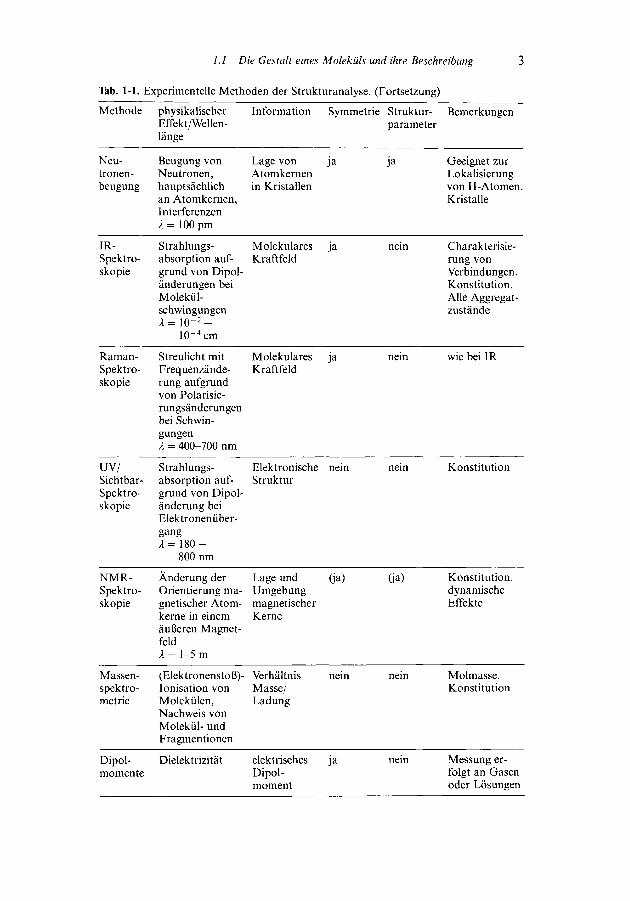
Tab. 1-1. Experimentelle Methoden der Strukturanalyse.
Methode
Mikro-wellen-spektro-skopie
Elek-tronen-beugung
Röntgen-beugung
physikalischerEffekt/Wellen-länge
Rotations-spektroskopie:Strahlungs-absorption auf-grund von Dipol-änderungenbei der Rotation1 = 0.1 — 30 cm
Beugung vonElektronen anAtomkernenund Elektronen,InterferenzenA = 10- 100 pm
Beugung vonRöntgenstrahlen,hauptsächlichdurch die Elek-tronen, Inter-ferenzenA = 10-
lOOOpm
Information
Mittelwertvon r~2
Intra-molekulareAtom-abstände
Elektronen-dichte inKristallen
Symmetrie Struktur- BemerkungenParameter
ja ja Moleküle müs-sen permanen-tes Dipolmo-ment besitzen.Analyse schwie-rig bei großenMolekülen mitniedrigerSymmetrie.Gase
ja ja schwierig beigroßen Mole-külen mit nied-riger Symmetrie.Gase
ja ja Lokalisierungvon H-Atomenschwierig.Kristalle
1.1 Die Gestalt eines Moleküls und ihre Beschreibung
Tab. 1-1. Experimentelle Methoden der Strukturanalyse. (Fortsetzung)
Methode
Neu-tronen-beugung
IR-Spektro-skopie
Raman-Spektro-skopie
UV/Sichtbar-Spektro-skopie
NMR-Spektro-skopie
Massen-spektro-metrie
Dipol-momente
physikalischerEffekt/Wellen-länge
Beugung vonNeutronen,hauptsächlichan Atomkernen,InterferenzenA = 100 pm
Strahlungs-absorption auf-grund von Dipol-änderungen beiMolekül-schwingungenA = io-2-
10-4cm
Streulicht mitFrequenzände-rung aufgrundvon Polarisie-rungsänderungenbei Schwin-gungenX = 400-700 nm
Strahlungs-absorption auf-grund von Dipol-änderung beiElektronenüber-gangA =180-
800 nm
Änderung derOrientierung ma-gnetischer Atom-kerne in einemäußeren Magnet-feldA = 1-5 m
(Elektronenstoß)-lonisation vonMolekülen,Nachweis vonMolekül- undFragmentionen
Dielektrizität
Information Symmetrie Struktur-parameter
Lage von ja jaAtomkernenin Kristallen
Molekulares ja neinKraftfeld
Molekulares ja neinKraftfeld
Elektronische nein neinStruktur
Lage und (ja) (ja)UmgebungmagnetischerKerne
Verhältnis nein neinMasse/Ladung
elektrisches ja neinDipol-moment
Bemerkungen
Geeignet zurLokalisierungvon H- Atomen.Kristalle
Charakterisie-rung vonVerbindungen.Konstitution.Alle Aggregat-zustände
wie bei IR
Konstitution
Konstitution,dynamischeEffekte
Molmasse.Konstitution
Messung er-folgt an Gasenoder Lösungen

4 l Methoden der Strukturanalyse
der Elektronen-, Röntgen- und Neutronenbeugung ermittelt. Teilstrukturen odereinzelne Details der Konstitution, Konfiguration oder Konformation können aus denInfrarot-, Raman-, NMR- und Massenspektren, aber auch mit anderen physikali-schen Methoden gewonnen werden. Auch die Frage nach der Symmetrie einesMoleküls läßt sich mit spektroskopischen Methoden, ohne Bestimmung der exaktenStrukturparameter, beantworten.
1.1.2 Beschreibung der Topologie eines Moleküls
Zur Beschreibung der räumlichen Struktur eines Moleküls kann man z. B. diekartesischen Koordinaten aller Atome, alle intramolekularen Atomabstände oder dieValenzkoordinaten verwenden. Letztere stellen sämtliche Bindungslängen und -win-kel sowie die Diederwinkel (Torsionswinkel) dar. In jedem Fall werden für ein n-atomiges Molekül 3n — 6 Strukturparameter benötigt, es sei denn das Molekül besitztSymmetrie, wodurch sich die Anzahl der Parameter verringert.
Beispiel 1-1:
Die Struktur eines vieratomigen, nicht ebenen Moleküls
kann man auf verschiedene Weisen beschreiben. Wählt man dazu kartesische Koordinaten, so wirdzweckmäßigerweise das Atom A in den Ursprung, Atom B auf die x- Achse und Atom C in die x-y-Ebene gelegt. Man benötigt dann sechs von Null verschiedene Koordinaten.
kartesische Koordinaten intramolekulare Atomabständex y z
ABCD
0 0*B 0xc yc
•^D yr>
000ZD
A—BA-C B—CA-D B-D C—D
yD ZDValenzkoordinatenBindungslängen: A—B, B—C, C—DBindungswinkel: A—B—C, B—C—DTorsionswinkel: A—B—C—D
Auch durch die sechs intramolekularen Atomabstände ist die Struktur eindeutig definiert, undandere Daten wie Bindungs- und Torsionswinkel können berechnet werden. Letztere bilden mit dendrei Bindungslängen die Valenzkoordinaten, die sich als Strukturparameter am besten z.B. zumVergleich mit anderen Molekülen eignen.
Zur Beschreibung der geometrischen Struktur des Methans, CH4, genügt wegender Tetraedersymmetrie (rd) ein Parameter, nämlich die C—H-Bindungslänge. DieSymmetrieeigenschaften eines Moleküls können zwar die Gleichheit einzelner Bin-dungslängen, nie aber deren tatsächlichen Wert angeben. Bindungswinkel sind je-doch oft durch die Symmetrie auch in ihrem Wert festgelegt. So sind mit der

L l Die Gestalt eines Moleküls und ihre Beschreibung 5
Tetraedersymmetrie des Methans beliebige Werte der C—H-Bindungslänge verein-bar; als H—C—H-Winkel kommt jedoch nur der Tetraederwinkel (109.5°) in Frage.Analog bedingt die D6h-Symmetrie des Benzols einen C—C—C-Bindungswinkelvon 120°.
1.1.3 Molekülschwingungen und Strukturparameter
Da die Atome innerhalb des Moleküls stets Schwingungen um ihre Gleich-gewichtspositionen ausführen, muß z.B. bei der präzisen Angabe einer Bindungs-länge berücksichtigt werden, daß der Gleichgewichtsabstand re zweier Atome einenetwas kleineren Wert hat als der über die Schwingungen gemittelte mittlere AbstandV
Im weiteren Sinne gehören zur Struktur eines Moleküls auch die Elektronenvertei-lung und das Kraftfeld, in dem sich die Atome bewegen. Durch eine Normalkoordi-naten-Analyse unter Einbeziehung der Schwingungsspektren isotopomerer Mole-küle kann man für die einzelnen Bindungen und Winkel Kraftkonstanten ermitteln.Die Elektronenverteilung läßt sich experimentell bestimmen (s. Abschn. 1.4.7 und1.5.3). Über die Stabilität und Reaktivität eines Moleküls geben die Bildungswärme,die Bindungsdissoziationsenergien sowie die Polarität einzelner Bindungen Auf-schluß. Die elektronische Struktur des Moleküls wird im Rahmen der MO-Theoriemit Hilfe von Molekülorbitalen beschrieben, deren Gestalt und Energie eng mit derReaktivität verknüpft sind.
Die physikalischen Methoden der Strukturanalyse beruhen auf einer Wechselwir-kung zwischen Materie und elektromagnetischer Strahlung. Bei den spektroskopi-schen Methoden ändert sich durch diese Wechselwirkung die Energie der Strahlung,während bei den Beugungsmethoden eine Lichtstreuung weitgehend ohne Energie-übertragung erfolgt, die sogar bei der ungeordneten Verteilung der Moleküle in derGasphase zu charakteristischen Interferenzerscheinungen führt.
Bei experimentellen Strukturdaten gibt man die Standardabweichung als ein Maßfür die Genauigkeit an. Diese läßt sich folgendermaßen erklären: Wird ein Para-meter r wiederholt gemessen, so schwanken die einzelnen Meßwerte um einenMittelwert f. Die Wahrscheinlichkeit p{ für das Auftreten eines bestimmten Meß-wertes r i läßt sich mit einer Gaußschen Glockenkurve beschreiben, deren Integralden Wert l besitzt. Ein Maß für die Streuung der Meßwerte um r ist die Varianz a2
(Gl. 1-1), deren Wurzel o Standardabweichung genannt wird. Die mit ± angegebe-nen Fehlergrenzen sind in der Regel ein Mehrfaches von er.
^lAta-r-)2 (1-1)i
Bindungslängen werden üblicherweise auf einige Zehntel pm (oder 10~3 Ä) ange-geben. Die Ergebnisse verschiedener Methoden lassen sich jedoch nicht unmittelbarmiteinander vergleichen. Hierfür sind hauptsächlich zwei Ursachen zu nennen:Einerseits werden mit den verschiedenen Methoden unterschiedliche physikalischeEigenschaften gemessen, und andererseits gibt es verschiedene Definitionen derBindungslänge.

6 l Methoden der Strukturanalyse
Die experimentellen Unterschiede sollen hier für die Elektronen- und Röntgenbeu-gung verdeutlicht werden. Während Röntgenstrahlen hauptsächlich an den Elektro-nen gestreut werden, erfolgt die Streuung des Elektronenstrahls an den Atomkernenund den Rumpfelektronen. Da die Elektronen nicht genau sphärisch um die Kerneverteilt sind, müssen unterschiedliche Atomabstände resultieren. Dies macht sichinsbesondere bei Wasserstoff-Atomen bemerkbar: für X—H-Bindungen werden beider Röntgenanalyse wesentlich kleinere Längen gefunden als bei den anderen Me-thoden (Näheres s. Abschn. 1.4 und 1.5).
Weiter ist zu berücksichtigen, daß bei der Röntgenkristallographie der Abstandzwischen den mittleren Atomlagen (hier gleichgesetzt mit den Maxima der Elektro-nendichte), bei der Elektronenbeugung aber der mittlere intramolekulare Atomab-stand bestimmt wird. Daß diese Größen nicht genau übereinstimmen, zeigt diefolgende vereinfachte Betrachtung:
In einem linearen Molekül A—B—C werden die Atome A und C als unbeweglichangesehen, während B senkrecht zur Molekülachse schwingen soll. Die mittlerePosition von B liegt wie gezeichnet auf der Achse, und A—B ist der Abstandzwischen den mittleren Atomlagen von A und B, wie er sich bei der Röntgenstruktur-analyse ergibt. A — B ist aber nicht der mittlere Abstand zwischen A und B, den manbei der Elektronenbeugung mißt, sondern tatsächlich der Minimalwert. Deshalbwird bei dieser Methode eine größere Länge für die A—B-Bindung gefunden.
Die Auswirkung der Schwingungen zeigt sich bei der Elektronenbeugung weiter-hin im sog. Schrumpfeffekt (Bastiansen-Morino-Effekt), der bewirkt, daß für nichtge-bundene Abstände kleinere Werte gefunden werden, als für eine starre Anordnungzu erwarten wäre. Dies läßt sich wiederum am Beispiel des linearen MolekülsA—B—C verdeutlichen. Durch die Knickschwingung wird der mittlere Abstandzwischen den Atomen A und C kleiner als die Summe der Bindungsabstände:A - - - C < A—B + B—C. Für den Abstand zwischen den beiden äußersten Kohlen-stoff-Atomen des Butatriens z.B. beträgt dieser Schrumpfparameter 1.3 pm.
In Tab. 1-2 sind die bei Strukturanalysen verwendeten intramolekularen Ab-standsparameter aufgeführt. Bei der Elektronenbeugung werden Atomabstände alsra> r
g? ra und ra angegeben; die Mikrowellenspektroskopiker verwenden Bezeichnun-
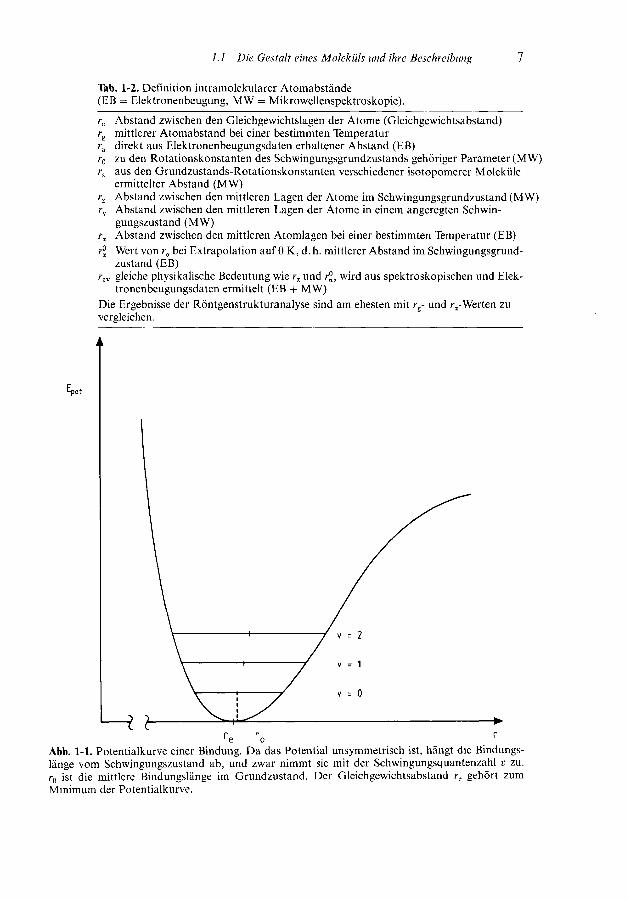
gen wie r0, rs und rz. Beide Methoden führen zu den Größen rav und re. Davon kannman letztere (re) am leichtesten erklären, obwohl ihre experimentelle Bestimmungschwierig ist. Der Gleichgewichtsabstand re entspricht dem Kernabstand in einemstarren Modell, also dem Minimum der Potentialkurve (Abb. 1-1). Dieser Wertergibt sich auch bei quantentheoretischen Berechnungen.
l .1 Die Gestalt eines Moleküls und ihre Beschreibung
Tab. 1-2. Definition intramolekularer Atomabstände(EB = Elektronenbeugung, MW = Mikrowellenspektroskopie).
re Abstand zwischen den Gleichgewichtslagen der Atome (Gleichgewichtsabstand)rg mittlerer Atomabstand bei einer bestimmten Temperaturra direkt aus Elektronenbeugungsdaten erhaltener Abstand (E B)r0 zu den Rotationskonstanten des Schwingungsgrundzustands gehöriger Parameter (MW)rs aus den Grundzustands-Rotationskonstanten verschiedener isotopomerer Moleküle
ermittelter Abstand (MW)rz Abstand zwischen den mittleren Lagen der Atome im Schwingungsgrundzustand (MW)rv Abstand zwischen den mittleren Lagen der Atome in einem angeregten Schwin-
gungszustand (MW)ra Abstand zwischen den mittleren Atomlagen bei einer bestimmten Temperatur (EB)rJJ Wert von ra bei Extrapolation auf 0 K, d. h. mittlerer Abstand im Schwingungsgrund-
zustand (EB)rav gleiche physikalische Bedeutung wie rz und rjj, wird aus spektroskopischen und Elek-
tronenbeugungsdaten ermittelt (EB + MW)Die Ergebnisse der Röntgenstrukturanalyse sind am ehesten mit rg- und ra-Werten zuvergleichen.
Abb. 1-1. Potentialkurve einer Bindung. Da das Potential unsymmetrisch ist, hängt die Bindungs-länge vom Schwingungszustand ab, und zwar nimmt sie mit der Schwingungsquantenzahl v zu.r0 ist die mittlere Bindungslänge im Grundzustand. Der Gleichgewichtsabstand re gehört zumMinimum der Potentialkurve.

8 l Methoden der Strukturanalyse
Die bei der Elektronenbeugung direkt erhaltenen r a-Werte werden durch Korrek-turen von einigen Zehntel pm in r° oder rg umgewandelt. Diese Werte vergleichtman üblicherweise mit den Bindungslängen der Röntgenbeugung. Für quantitativeZwecke sind jedoch auch bei den Röntgen-Ergebnissen Schwingungskorrekturenanzubringen. Diese müssen jedoch auch die Gitterschwingungen berücksichtigen.Bei einem solchen Vergleich darf man auch nicht außer acht lassen, daß in einemKristall die einzelnen Moleküle sich gegenseitig durch intermolekulare Wechselwir-kungen beeinflussen. Diese Packungseffekte sind nur schwer quantitativ im Hinblickauf die Molekülgeometrie zu erfassen. Signifikant dürften Unterschiede bei Bin-dungslängen, die im festen oder im gasförmigen Zustand gemessen wurden, nurdann sein, wenn sie größer als 0.5 pm sind.
Beispiel 1-2 1,4-Dicyanobenzol (Terephthalsäuredinitril):
Die Verbindung wurde in der Gasphase durch Elektronenbeugung und im festen Zustand röntgen-kristallographisch untersucht. Für die wichtigsten Bindungslängen wurden folgende Werte (in pm)gefunden (Colpapietro et al., 1984):
Gasa) Kristallb) Kristall, korrigiert^
c-cd)C-CNC=N
139.7 + 0.3145.4 + 0.5116.7 + 0.2
138.8(0.1)144.1 (0.1)114.5(0.2)
140.0 (0.2)144.5 (0.2)116.9(0.2)
a) rg-Werte mit Fehlergrenzenb),c) Ergebnisse ohne und mit Schwingungskorrektur, in Klammern Standardabwei-
chungend) Mittelwert im Ring
Unter Berücksichtigung der Fehlergrenzen (+) bzw. Standardabweichungen (in Klammern) sinddie in den ersten beiden Spalten aufgeführten Bindungsabstände signifikant verschieden. Nach derSchwingungskorrektur der Röntgen-Ergebnisse stimmen die Bindungslängen beider Methodenüberein.
Der Hauptunterschied zwischen den spektroskopischen und den Beugungsergeb-nissen rührt daher, daß bei den letzteren über das thermische Gleichgewicht gemittelt
Theorie: re
Experiment:
Rotations- harmon. anharmon.Spektroskopie >o » rz » re
Korrektur l Korrektur
Elektronen- harmon. | anharmon.beugung ra » ^ > >*e
Korrektur Korrektur
Abb. 1-2. Beziehung zwischen verschiedenen Abstandsarten. Die Symbole werden in Tab. 1-2erläutert. Die bei quantenchemischen Rechnungen resultierenden Gleichgewichtsabstände re erhältman aus experimentellen Daten erst nach Korrekturen, die auch die Anharmonizität der Molekül-schwingungen berücksichtigen.

1.2 Mikrowellenspektroskopie 9
wird, während bei der Mikrowellenspektroskopie ein bestimmter Schwingungszu-stand, üblicherweise der Grundzustand, betrachtet wird. Die dabei gewonnenen r0-oder rs-Werte sind von den Beugungswerten signifikant verschieden. Für eine C—C-Einfachbindung ist r0 um etwa 0.6 pm kürzer als ra. Die für den Schwingungsgrund-zustand ermittelten Bindungslängen aus Elektronenbeugung (rjj) und Mikrowellen-spektroskopie (rz) sollten innerhalb der experimentellen Fehlergrenzen überein-stimmen.
Bei genauer Kenntnis des molekularen Kraftfeldes läßt sich auch der Einfluß derAnharmonizität der Molekülschwingungen auf die Bindungslängen bestimmen, undman gelangt nach entsprechenden Korrekturen zu den re-Werten. In Abb. 1-2 ist derZusammenhang zwischen den verschiedenen Abstandsarten schematisch dargestellt.
Die Gleichgewichtsstruktur, re, ist zweifellos die am besten definierte Geometrie.Wegen der Schwierigkeit, die anharmonischen Korrekturen zu ermitteln, wird siejedoch nur selten bestimmt.
Die Bedeutung der vier wichtigsten experimentellen Methoden läßt sich in etwaan der Anzahl der bislang durchgeführten Strukturanalysen ablesen: Ca. 40000Röntgenuntersuchungen organischer Verbindungen stehen etwa 1500 Elektronen-beugungs- und 1200 Mikrowellen- sowie ungefähr 400 Neutronenbeugungsarbeitengegenüber.
l .2 Mikrowellenspektroskopie
1.2.1 Einleitung
Die innere Energie eines Moleküls setzt sich aus seiner elektronischen EQ, Schwin-gungs- Ev und Rotationsenergie Er zusammen, für die nur bestimmte (diskrete)Werte in Frage kommen. Der Übergang zwischen zwei verschiedenen Zuständendes Moleküls ist deshalb mit der Aufnahme oder Abgabe eines Energiequants ver-bunden, das nach dem Planckschen Gesetz [Gl. (1-2)] eine bestimmte Frequenz vbesitzt.
AE=hv (1-2)
Die Differenz zwischen zwei verschiedenen elektronischen Zuständen liegt in derGrößenordnung von 500 kJ/mol, von Schwingungszuständen bei etwa 20 kJ/molund von Rotationszuständen bei etwa 0.05 kJ/mol. Da auf jeden Freiheitsgrad desMoleküls die kinetische Energie l/2 £7"entfällt (bei 300 K sind das für l mol Molekülel .2 kJ), befinden sich die Moleküle nach dem Boltzmannschen Gesetz bei Raumtem-peratur normalerweise im elektronischen und vibronischen Grundzustand, besitzenaber etliche Quanten Rotationsenergie (Näheres s. Band 3 dieser Reihe).
Rotationsübergänge können von Strahlung angeregt werden, die aus dem Mikro-wellen-Gebiet des elektromagnetischen Spektrums stammt. Damit eine Absorption

10 l Methoden der Strukturanalyse
erfolgt, muß die MW-Strahlung Frequenzwerte besitzen, die genau mit den Rota-tionsfrequenzen des Moleküls übereinstimmen [Gl. (1-3)], und die Moleküle müssenein permanentes elektrisches Dipolmoment besitzen. Da die Rotationsenergie ge-quantelt ist, ergibt sich ein Linienspektrum. Das Aussehen des MW-Spektrums einesMoleküls wird durch die für Rotationen wichtigen Trägheitsmomente (Drehimpulse)bezüglich der Hauptachsen des Moleküls bestimmt. Diese Trägheitsmomente kön-nen aus dem Spektrum ermittelt werden. Sie sind durch die molekulare Geometrieund durch die Atommassen bestimmt. Die aus verschiedenen Isotopen aufgebautenMoleküle besitzen also unterschiedliche MW-Spektren.
Er = hvr (1-3)
Aufgrund der hohen Empfindlichkeit und des großen Auflösungsvermögens derMW-Spektroskopie kann diese Methode äußerst präzise Strukturparameter, Dipol-momente, Torsions- und Inversionsbarrieren u.a. liefern. Durch die Beschränkungauf polare Gase und den hohen Aufwand bei größeren Molekülen mit konformativerBeweglichkeit und niedriger Symmetrie sind der MW-Spektroskopie jedoch Grenzenfür ihre Anwendung gesetzt. Moleküle mit einer relativen Molekülmasse über 300wurden offenbar noch nicht untersucht.
Da die Rotationslinien äußerst genau registriert werden können, eignet sich dieMW-Spektroskopie auch zur Analyse von Gasgemischen. Mit Hilfe großer Radio-teleskope gelang häufig der Nachweis molekularer Teilchen bis zur Größe des Cyano-tetraacetylens, C9HN, im interstellaren Raum vor ihrer Synthese auf der Erde (Win-newisser, 1984).
1.2.2 Meßtechnik
Die Aufnahme von MW-Spektren erfolgt an freien Molekülen, d.h. in der Gas-phase bei möglichst niedrigem Druck (ca. 0.1 Pa oder 10~3 mbar). Wegen der sehrkleinen Absorptionskoeffizienten, die dem Quadrat des Dipolmoments proportionalsind, muß die Schichtdicke einige m betragen. Damit sich die Moleküle in möglichstniedrigen Rotationszuständen befinden, soll die Temperatur niedrig sein (üblicher-weise zwischen —80 und +20°C). Der am häufigsten untersuchte Spektralbereichliegt bei Wellenlängen von 0.75 bis 3.75 cm bzw. bei Frequenzen zwischen 8 und40 GHz (l GHz= 109Hz).
Als Strahlungsquelle dienen monochromatisch arbeitende HöchstfrequenzrÖhren,die bei der Aufnahme des Spektrums elektronisch durchgestimmt werden. Bis Endeder 60er Jahre verwendete man hauptsächlich Reflex- und Doppelhohlraum-Kly-strons. Heute arbeitet man mit Rückwärtswellenoszülatoren oder mit Festkörperoszil-latoren. Die MW-Strahlung wird in Hohlleitern mit zumeist rechteckigem Quer-schnitt geführt. In der Absorptionszelle befindet sich eine Elektrode, über die eineStark-Modulation der Absorptionssignale erfolgt. Diese lassen sich dann mit selekti-ven und äußerst empfindlichen Hochfrequenzverstärkern registrieren. Über Einzel-heiten informiert z. B. das am Ende dieses Kapitels aufgeführte Buch von Gordyund Cook (1984).

1.2 Mikrowellenspektroskopie 1 1
1.2.3 r - und r-Strukture
1.2.3.1 Energieterme und Rotationsspektrum
Bei der Interpretation der Rotationsspektren von Molekülen geht man üblicher-weise vom sog. starren Rotator aus, d.h. man läßt zunächst die Wechselwirkungenzwischen Rotation und Schwingungen außer acht. Die Rotation eines freien Mole-küls wird in die drei Komponenten bezüglich der Hauptträgheitsachsen a, b und czerlegt. Diese erhält man, in dem man den Trägheitstensor für ein beliebiges molekül-festes kartesisches Koordinatensystem diagonalisiert. Der Ausdruck für die Rota-tionsenergie lautet dann:
£ r=V2(/a^ + /X + /X) (1-4)
coa, cob und coc sind die Winkelgeschwindigkeiten und Pa, Pb und Pc die Kompo-nenten des Drehimpulsvektors P [Gl. (1-6)] bezüglich der drei Achsen. Die Wertevon 7a, 7b und 7C lassen sich nach Gl. (1-7) aus den Atommassen m{ und derGeometrie des Moleküls berechnen. r{ gibt den Abstand des Atoms / von derDrehachse an. Die Achsen a, b und c sind so gewählt, daß gilt 7a < 7b < /c .
Pa = /acoa (1-6)
Pb = 4«bP, = Icwc
/=Z»vf (1-7)i
Nach den Gesetzen der Wellenmechanik ist die Rotationsenergie gequantelt, d. h.auf ganz bestimmte Werte beschränkt. Der Gesamtdrehimpuls kann nur die nachGl. (1-8) durch die Rotationsquantenzahl J festgelegten Werte besitzen. Besitzt dasMolekül eine Figurenachse z (Symmetrieachse), so ist auch die Komponente desDrehimpulses in Richtung dieser Achse gequantelt [Gl. (1-9)]. Die zugehörige Quan-tenzahl K ist auf ganzzahlige Werte zwischen -/ und +/ beschränkt.
Pj = (h/2n) ]/J(J+V) .7 = 0, 1 ,2 . . . (1-8)
Pz = (h/2n)K Ä>0, + 1,±2, . . . ±J (1-9)
Die quantenmechanische Behandlung der Rotation hängt von der Symmetrie desjeweiligen Moleküls ab.
Für die Analyse der MW-Spektren ist es zweckmäßig, die nach Gl. (1-10) definier-ten Rotationskonstanten A, B und C einzuführen. Deren Werte sind dem MW-Spektrum zu entnehmen und aus ihnen über Gl. (1-7) die Strukturparameter zuermitteln.
A=F/Ia mit F= A/8712 (1-10)B = F/Ib
C = F/IC
12 l Methoden der Strukturanalyse
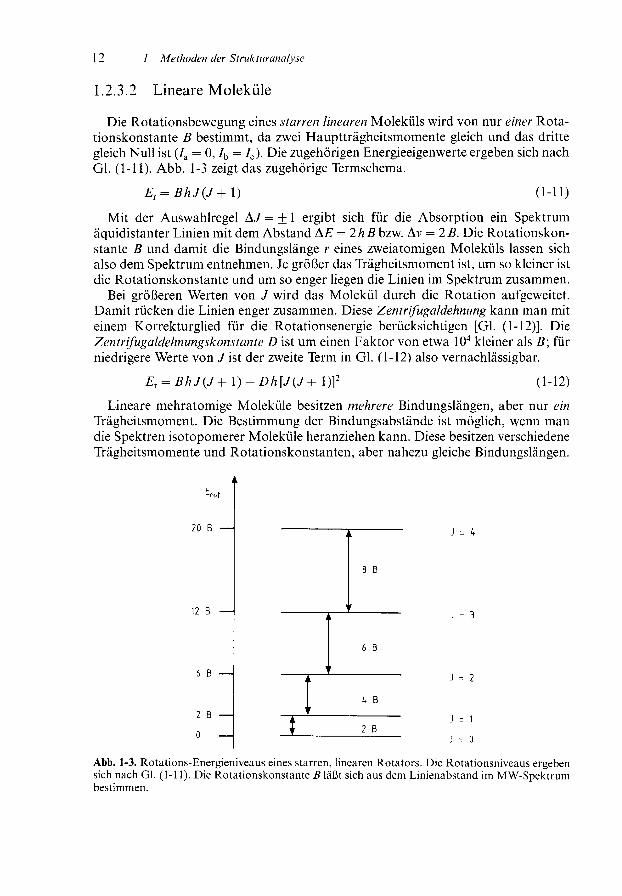
1.2.3.2 Lineare Moleküle
Die Rotationsbewegung eines starren linearen Moleküls wird von nur einer Rota-tionskonstante B bestimmt, da zwei Hauptträgheitsmomente gleich und das drittegleich Null ist (7a = 0, 7b = 7C). Die zugehörigen Energieeigenwerte ergeben sich nachGl. (1-11). Abb. 1-3 zeigt das zugehörige Termschema.
Mit der Auswahlregel A J = ± l ergibt sich für die Absorption ein Spektrumäquidistanter Linien mit dem Abstand A£ = 2h B bzw. Av = 2 B. Die Rotationskon-stante B und damit die Bindungslänge r eines zweiatomigen Moleküls lassen sichalso dem Spektrum entnehmen. Je größer das Trägheitsmoment ist, um so kleiner istdie Rotationskonstante und um so enger liegen die Linien im Spektrum zusammen.
Bei größeren Werten von J wird das Molekül durch die Rotation aufgeweitet.Damit rücken die Linien enger zusammen. Diese Zentrifugaldehnung kann man miteinem Korrekturglied für die Rotationsenergie berücksichtigen [Gl. (1-12)]. DieZentrifugaldehnungskonstante D ist um einen Faktor von etwa l O4 kleiner als B\ fürniedrigere Werte von /ist der zweite Term in Gl. (1-12) also vernachlässigbar.
r = BhJ(J+ l)-Dh[J(J+ l)]2 (1-12)
Lineare mehratomige Moleküle besitzen mehrere Bindungslängen, aber nur einTrägheitsmoment. Die Bestimmung der Bindungsabstände ist möglich, wenn mandie Spektren isotopomerer Moleküle heranziehen kann. Diese besitzen verschiedeneTrägheitsmomente und Rotationskonstanten, aber nahezu gleiche Bindungslängen.
20
12 B —
J =
6 B
J = 3
J = 2
J = 1
J = 0
Abb. 1-3. Rotations-Energieniveaus eines starren, linearen Rotators. Die Rotationsniveaus ergebensich nach Gl. (1-11). Die Rotationskonstante B läßt sich aus dem Linienabstand im MW-Spektrumbestimmen.

1.2 Mikrowellenspektroskopie 13
Beispiel 1-3 Kohlenstoffoxysulfid, O=C=S:
Aus den Spektren von zwei isotopen Spezies des Kohlenstoffoxysulfids ergaben sich folgendeWerte (Townes et al., 1948):
B D IMHz kHz 10-40gcm2
i 6 O = i 2 C = 3 2 S 6 0 8 1 . 4 8 0 16ÖÖ 137.974i6o=12C=34S 5932.843 1400 141.431
Unter der Annahme gleicher Bindungslängen in den Molekülen erhält man rc=0 =116.47 pm und rc=s = 155.76 pm.
Die Kombination verschiedener Isotopomerer liefert geringe Unterschiede in den Atomab-ständen:
16Q 12^ 32C vind ^O ^2f 34c
16Q 12£ 32C viri(J ^O ^3(^ 32c
16Q ^C\ 32C vrrjrl \%C) 12/"1 32C
u>O=uC=34$ und 16O=13C=34SMittelwert:
C=Opm
116.47116.29115.52116.25116.13
pm
155.76155.91156.53155.94156.04
Obwohl für verschiedene Isotopomere die Unterschiede in den Bindungslängenkleiner als l pm sind, übersteigen sie die Meßgenauigkeit bei weitem. Die Haupt-ursache ist darin zu suchen, daß nicht re-, sondern r0-Werte, also die mittlerenAbstände im Schwingungsgrundzustand, bestimmt wurden*). Da isotopomere Mo-leküle unterschiedliche Schwingungsfrequenzen besitzen, bewirkt die Anharmonizi-tät der Schwingungen, daß sich ihre r0-Werte unterscheiden. Aus der Gestalt derSchwingungspotentialkurve folgt, daß r0 stets etwas größer ist als re (vgl. Abb. 1-1).Wenn diese Potentialkurve bekannt ist, läßt sich auch re bestimmen.
Be = Bv + *(v + l/2) (1-13)
Gl. (1-13) verbindet für ein zweiatomiges Molekül die nicht direkt meßbareRotationskonstante #e für den Gleichgewichtsabstand re mit den Rotationskonstan-ten Bv angeregter Moleküle über die Rotations-Schwingungs-Wechselwirkungskon-stante a. Wenn B für zwei verschiedene Schwingungszustände bestimmt wurde, kanna ermittelt werden und damit dann Be und re. Für Kohlenmonoxid ergibt sich z. B.:
BQ a r0 re
MHz MHz pm pm12C16O 57897.75 524.16 113.079 112.82713C 16C 55345.10 488.48 113.072 112.827
Die so ermittelten Geometrieparameter bilden die sog. rQ-Struktur des Moleküls.Diese Methode läßt sich jedoch nur auf relativ einfache Moleküle anwenden, da dieRotations-Schwingungs-Wechselwirkungskonstanten für sämtliche Schwingungen
! Tatsächlich wird über r2 gemittelt, da r2 das Trägheitsmoment bestimmt [Gl. (1-7)].

14 l Methoden der Strukturanalyse
jedes isotopomeren Moleküls bekannt sein müssen. Für die Bindungslängen desKohlenstoffoxysulfids wurden folgende re-Werte gefunden (Watson, 1973): C=O115.87 pm, C=S 155.93 pm.
1.2.3.3 Symmetrische Kreiselmoleküle
Bei einem symmetrischen Kreisel besitzt das Molekül drei von Null verschiedeneTrägheitsmomente, von denen zwei gleich sind. Die Rotationsbewegung wird folglichdurch zwei Trägheitsmomente bestimmt. In diese Gruppen fallen Moleküle miteiner drei- oder höherzähligen Symmetrieachse, wie z. B. Chlormethan H3C—Cl mit7a < 7b = 7C (Abb. 1-4). Kugelkreisel mit Ia = Ib = 7C sind dipollos und besitzen keinMW-Spektrum. Hierzu gehören Moleküle mit Tetraeder- und Octaeder-SymmetriewieCH4, CC14, SF6.
Abb. 1-4. Hauptträgheitsachsen von Chlormethan. Die Achse a mit dem kleinsten Hauptträgheits-moment, 7a, fällt mit der C—Cl-Bindung zusammen. Die Achsen a, b und c schneiden sich imSchwerpunkt des Moleküls.
Die Eigenwerte des starren symmetrischen Kreisels werden gemäß Gl. (1-14) durchdie beiden Quantenzahlen 7 und K definiert.
E, = BhJ(J+ 1) + (A - B)hK2 (1-14)
Da die Figurenachse a des Moleküls mit der Dipolachse zusammenfällt, folgt fürK die Auswahlregel AÄ: = 0. Damit resultiert mit A J = + l für symmetrische Kreiselwie bei den linearen Molekülen ein MW-Spektrum äquidistanter Linien mit einemFrequenzabstand A v = 2 B, und die weitere Behandlung erfolgt analog [Näheres s.z. B. bei Gordy und Cook (1984)].

1.2 Mikrowellenspektroskopie 15
l. 2.3 A Unsymmetrische Kreiselmoleküle
Ein unsymmetrischer Kreisel besitzt drei verschiedene Hauptträgheitsmomente(7a 7* 7b 7^ /c)>
und das MW-Spektrum hängt von drei Rotationskonstanten [Gl. (1-5)]ab. Eine Besonderheit liegt beiplanaren Molekülen vor: Hier ist das größte Haupt-trägheitsmoment gleich der Summe der beiden anderen:
/c = 4 + 4 (1-15)
Da diese Beziehung - abgesehen von einem kleinen Term, dem sog. Trägheitsdefekt- exakt gilt, läßt sich mit ihrer Hilfe leicht entscheiden, ob ein Molekül planar istoder nicht.
Die Energieniveaus eines unsymmetrischen Kreisels können nicht wie im Fallelinearer Moleküle oder symmetrischer Kreisel in einer Gleichung explizit angegebenwerden. Nützlich ist der Asymmetrieparameter x [Gl. (1-16)], der Werte zwischen — l(B = C, verlängerter symm. Kreisel) und -h l (A = B, abgeplatteter symm. Kreisel)annehmen kann. Für die Rotationsenergie des unsymmetrischen Kreisels läßt sichdie allgemeine Gleichung (1-17) angeben. Hier bedeutet E(K) die reduzierte Energiefür einen fiktiven Kreisel mit A = l, B = x und C = — l, die numerisch zu finden(tabelliert) ist.
x = (2B - A - C)/(A - C) (1-16)
C)hJ(J+ 1) + (l/2)(A - C)hE(x) (1-17)
Das Schema der Rotationsenergieterme gemäß Gl. (l . 1 7) besitzt keine augenfälligeRegelmäßigkeit. Das MW-Spektrum eines unsymmetrischen Kreiselmoleküls be-steht dementsprechend aus unregelmäßigen Liniengruppen.
1.2.4 rs-Struktur
Nach der Ermittlung der Rotationskonstanten A, B und C aus dem MW-Spek-trum können über Gl. (1-5) und (1-7) die Geometrieparameter des Moleküls berech-net werden, sofern eine hinreichende Anzahl isotopomerer Moleküle herangezogenwird. Wie bereits am Beispiel des COS gezeigt wurde, schwanken die r0-Werte, jenachdem welche Isotopen-Kombination gewählt wird. Die Unsicherheit liegt beietwa l pm.
Für die re-Struktur benötigt man die Rotations-Schwingungs-Wechselwirkungs-konstanten a [Gl. (1-13)]. Und zwar muß a für jede Normalschwingung des Molekülsbekannt sein. Für die selektive Anregung einer jeden Normalschwingung ist also dasRotationsspektrum zu finden und zuzuordnen, was für größere Moleküle praktischunmöglich ist.
Die von Costain (1958) entwickelte Methode zur Bestimmung der sog. rs-Struktur(s für Substitution) liefert um eine Größenordnung verläßlichere Strukturparameterals die r0-Werte und ist bei vertretbarem Aufwand auch auf größere Moleküle wiez. B. Anilin anwendbar. Diese Methode beinhaltet, daß die Differenz der Trägheits-

16 l Methoden der Strukturanalyse
momente von zwei Isotopomeren für den Schwingungsgrundzustand und für dieGleichgewichtsgeometrie in guter Näherung übereinstimmt [Gl. (1-18)].
A/o^A/e (1-18)
Von Kraitchman (1953) stammen Gleichungen der Form (1-19) für die Koordina-ten eines Atoms im Hauptträgheitsachsensystem des Moleküls. Diese Gleichungenverlangen, daß jedes Atom, dessen Koordinaten gesucht werden, einmal gesondertsubstituiert wird. Wegen des hohen Auflösungsvermögens der Mikrowellenspektro-meter lassen sich die durch Isotopensubstitution verschobenen Spektrallinien ohneSchwierigkeiten voneinander trennen und häufig (z. B. bei 12C/13C, 32S/34S, 35C1/37C1)im natürlichen Vorkommen nachweisen. Präparative Isotopensubstitution ist jedocherforderlich, wenn das natürliche Vorkommen sehr gering ist (z. B. 1H/2H, 14N/15N).
rs(0=/(A/ f l,A/,,A/c) (1-19)
Die rs-Struktur stellt einen Kompromiß zwischen der zu ungenauen r0- und dereigentlich erwünschten re-Struktur dar. Die nach diesem Verfahren bestimmtenStrukturparameter haben eine Genauigkeit von einigen Zehntel pm bei Bindungslän-gen und etwa 0.5° bei Bindungswinkeln.
Beispiel 1-3 Benzonitril:
Die MW-Spektren von zehn Isotopomeren des Benzonitrils, C6H5CN, mit 2H, 13C oder 15N,lieferten die in Tab. 1-3 aufgeführte rs-Struktur.
Tab. 1-3. rs-Struktur von Benzonitril (Bak et al., 1962)
C7NC'C1
C'C2
C2C3
C3C4
C2HC3HC4H
1 15.9 ± 0.2 pm145.5 + 0.7139.1 +0.7139.3 ±0.9140.0 + 0.7106.9 ±0.8108.2 ±0.6108.1 ±0.2
C6C1C2
OC2C3
C2C3C4
C3C4C5
C HC4C3H
N
122.5 + 0.6°118.5 + 0.6120.3+0.4120.0 + 0.2121.8 + 0.7119.9 + 0.3
Bemerkenswert ist vor allem die Verformung des Benzolringes gegenüber dem regelmäßigenSechseck.
1.2.5 Qualitative Anwendungen, Konformationsanalyse
Häufig gestattet bereits das MW-Spektrum einer isotopen Spezies eine Antwortauf eine wichtige Frage bezüglich der Struktur eines Moleküls. So interessiert z.B.
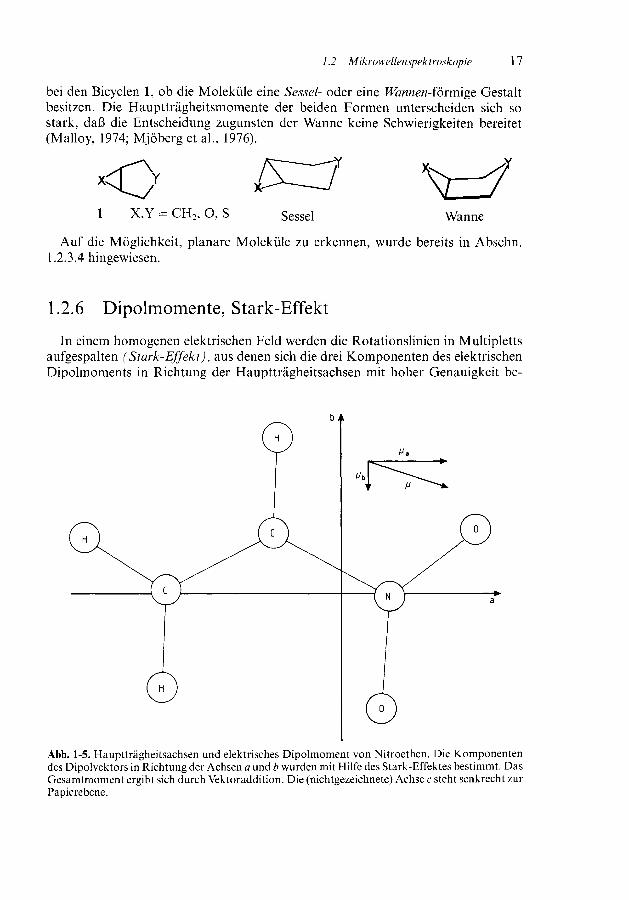
1.2 Mikrowellenspektroskopie 17
bei den Bicyclen l, ob die Moleküle eine Sessel- oder eine Wannen-förmige Gestaltbesitzen. Die Hauptträgheitsmomente der beiden Formen unterscheiden sich sostark, daß die Entscheidung zugunsten der Wanne keine Schwierigkeiten bereitet(Malloy, 1974; Mjöberg et al, 1976).
X,Y = CH2, O, S Sessel Wanne
Auf die Möglichkeit, planare Moleküle zu erkennen, wurde bereits in Abschn.1.2.3.4 hingewiesen.
1.2.6 Dipolmomente, Stark-Effekt
In einem homogenen elektrischen Feld werden die Rotationslinien in Multiplettsaufgespalten (Stark-Effekt), aus denen sich die drei Komponenten des elektrischenDipolmoments in Richtung der Hauptträgheitsachsen mit hoher Genauigkeit be-
Abb. 1-5. Hauptträgheitsachsen und elektrisches Dipolmoment von Nitroethen. Die Komponentendes Dipolvektors in Richtung der Achsen a und b wurden mit Hilfe des Stark-Effektes bestimmt. DasGesamtmoment ergibt sich durch Vektoraddition. Die (nichtgezeichnete) Achse c steht senkrecht zurPapierebene.

18 l Methoden der Strukturanalyse
stimmen lassen. Die MW-Spektroskopie gestattet also nicht nur eine Bestimmungdes Gesamtdipolmomentes, wie z. B. dielektrische Messungen (s. Abschn. 4.2), son-dern auch seiner Lage im Molekül. Allerdings ist keine Absolutbestimmung desDipolmomentes möglich, sondern die Messung erfolgt relativ zu einer Vergleichsver-bindung. Dazu verwendet man in der Regel Kohlenstoffoxysulfid (COS, ju =0.712 D).
Symmetrische Kreiselmoleküle zeigen einen linearen Stark-Effekt, d.h. die Fre-quenzverschiebung ist der angelegten Feldstärke proportional. Lineare und asym-metrische Kreiselmoleküle weisen dagegen einen quadratischen Stark-Effekt auf.Die Frequenzverschiebung erfolgt also mit dem Quadrat der Feldstärke.
Beispiel 1-4 Nitroethen:
Abb. 1-5 zeigt die Lage der Hauptträgheitsachsen im Molekül. Mit Hilfe des Stark-Effektes wurdendie Komponenten des elektrischen Dipolmomentes in Richtung der Achsen a und b bestimmt. Siebetragen jua = 3.51 ± 0.02 D und //b = 1.16 ± 0.08 D. Da das Molekül planar ist, ist /j,c = 0. DieVektoraddition liefert ein Gesamtmoment ju = 3.70 + 0.03 D (Hess et al., 1967). Eine dielektrischeBestimmung ergab für eine benzolische Lösung ein etwas kleineres Dipolmoment von 3.41 D.
1.2.7 Rotationsfeinstruktur von Schwingungs-und Elektronenspektren
In der Gasphase überlagern sich den Vibrationsenergien eines Moleküls die Rota-tionsenergien, und ein Übergang in einen anderen Schwingungszustand ist in derRegel auch mit einer Änderung des Rotationszustandes verbunden. Dies äußert sichin einer charakteristischen Rotationsfeinstruktur der IR- und Raman-Banden.
Wegen der Gültigkeit entsprechender Auswahlregeln muß ein Molekül nicht unbe-dingt ein permanentes Dipolmoment haben, um ein Rotationsschwingungsspektrum(IR oder Raman) zu besitzen. Im Gegensatz zur MW-Spektroskopie können alsodie Trägheitsmomente bzw. Rotationskonstanten [Gl. (1-7) bzw. (1-10)] auch vondipollosen Molekülen bestimmt werden. Die Einbeziehung isotopomerer Moleküleermöglicht dann eine Strukturanalyse auch von Molekülen, die mit der MW-Spek-troskopie nicht untersucht werden können. Daß das Verfahren wegen der Wechsel-wirkung von Schwingung und Rotation noch komplizierter ist als bei der Auswer-tung der reinen Rotationsspektren, bedarf keiner weiteren Erläuterung. Insbeson-dere die Analyse der Rotationsstruktur stark asymmetrischer Kreisel ist so schwierig,daß bis zum Einsatz elektronischer Rechenanlagen nur wenige Moleküle bis zurBestimmung der drei Trägheitsmomente durchgerechnet werden konnten. Anderer-seits bereitet die Strukturanalyse bei kleineren hochsymmetrischen Molekülen (Ku-gelkreisel und symmetrische Kreisel) keine besonderen Probleme. Bezüglich experi-menteller und methodischer Einzelheiten muß auf die einschlägige Literatur verwie-sen werden (s. z.B. Hollas, 1982).
Abschließend sei noch erwähnt, daß auch die Elektronenspektren (UV, sichtbar)gasförmiger Moleküle eine Feinstruktur besitzen, die auf Schwingungs- und Rota-tionsübergänge zurückgeht. Die Analyse der Rotationsfeinstruktur gestattet dieStrukturanalyse elektronisch angeregter Moleküle (vgl. Abschn. 9.6).

1.3 Elektronenbeugung 19
l. 3 Elektronenbeugung
1.3.1 Einleitung
Die de Broglie-Beziehung (1-20) zeigt, daß Elektronen, die durch ein elektrischesPotential V von einigen hundert Volt auf die kinetische Energie eV [Gl. (1-21)]beschleunigt werden, eine Wellenlänge /l [Gl. (1-22)] von etwa 100 pm besitzen.Solche langsamen Elektronen werden sowohl von den Atomkernen als auch von denElektronen in den Bindungen eines Moleküls gestreut, und es hat sich als unmöglicherwiesen, ein solches Beugungsdiagramm auszuwerten. Schnelle Elektronen werdendagegen nur an den Kernen gebeugt, und die Atomstreufaktoren lassen sich berech-nen. Deshalb werden gewöhnlich 40-kV-Elektronen verwendet, deren Wellenlängeetwa 6 pm beträgt. Da Elektronen viel stärker als Röntgenstrahlen gestreut werden,sind Belichtungszeiten von einigen Sekunden ausreichend, wogegen Röntgenbeu-gungsexperimente an Gasen gewöhnlich mehrere Stunden dauern.
rae = Elektronenmasse (1-20)v = Geschwindigkeit der Elektronen
e = Elementarladung (1-21)V = Beschleunigungsspannung
(1-22)
1.3.2 Meßprinzip und Strukturanalyse
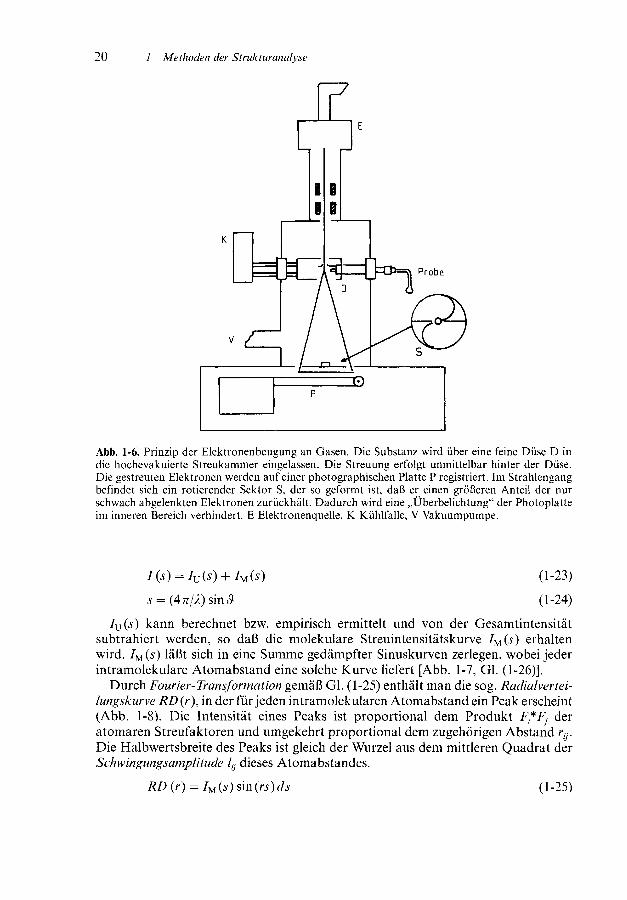
Elektronenbeugungs-untersuchungen werden nahezu ausschließlich an Gasendurchgeführt, aber auch Feststoffe können untersucht werden, letztere wegen desgeringen Durchdringungsvermögens der Elektronen allerdings nur in Folien oderdünnen Schichten. Das Meßprinzip ist in Abb. 1-6 schematisch dargestellt (Detailss. die am Ende des Kapitels angegebene Literatur).
Die Moleküle des Gases sind im Vergleich zur Wellenlänge der verwendetenElektronen sehr weit voneinander entfernt, so daß keine intermolekularen Beugungs-effekte auftreten. Die relative Anordnung der Atome innerhalb der Moleküle erzeugtähnlich wie bei einem Kristallpulver mit völlig regellos angeordneten Einzelkristallencharakteristische, ringförmige Interferenzmuster.
Die Intensität I (s) der an Gasen gestreuten Elektronen läßt sich gemäß Gl. (1-23)als Summe der Untergrundstreuung /u C$0, die nur die atomare und die unelastische,inkohärente Streuung, aber keine Information über die Molekülstruktur enthält,und der molekularen Streuung IM (s) auffassen. Anstelle des Streuwinkels 9 verwendetman den durch Gl. (1-24) definierten Streuparameter s als von der Energie derElektronen unabhängigen Variable.
20 l Methoden der Strukturanalyse
Abb. 1-6. Prinzip der Elektronenbeugung an Gasen. Die Substanz wird über eine feine Düse D indie hochevakuierte Streukammer eingelassen. Die Streuung erfolgt unmittelbar hinter der Düse.Die gestreuten Elektronen werden auf einer photographischen Platte P registriert. Im Strahlengangbefindet sich ein rotierender Sektor S, der so geformt ist, daß er einen größeren Anteil der nurschwach abgelenkten Elektronen zurückhält. Dadurch wird eine „Überbelichtung" der Photoplatteim inneren Bereich verhindert. E Elektronenquelle, K Kühlfalle, V Vakuumpumpe.
i(s) (1-23)
(1-24)
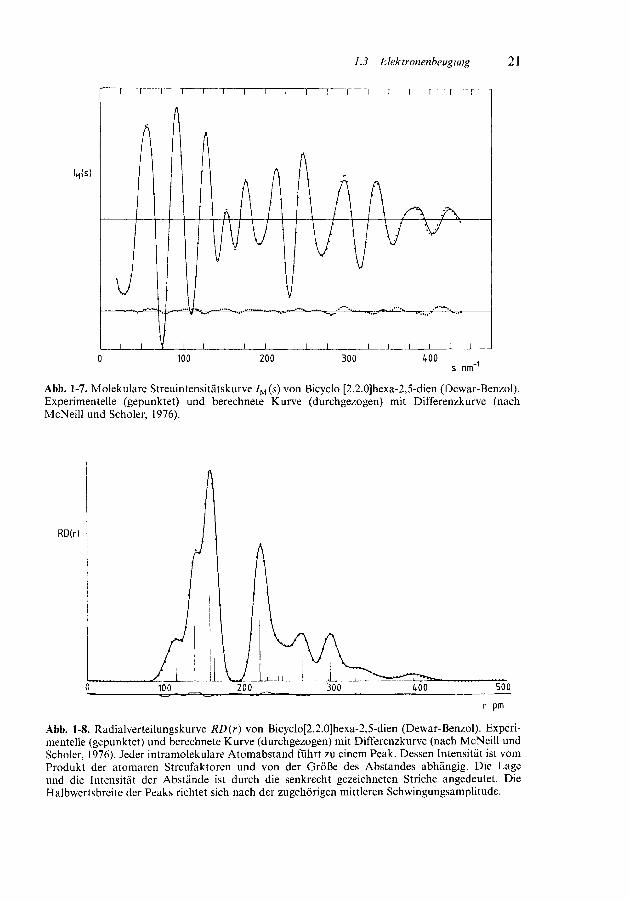
/u (s) kann berechnet bzw. empirisch ermittelt und von der Gesamtintensitätsubtrahiert werden, so daß die molekulare Streuintensitätskurve /M(^) erhaltenwird. /M (s) läßt sich in eine Summe gedämpfter Sinuskurven zerlegen, wobei jederintramolekulare Atomabstand eine solche Kurve liefert [Abb. 1-7, Gl. (1-26)].
Durch Fourier-Transformation gemäß Gl. (1-25) enthält man die sog. Radialvertei-lungskurve RD (r), in der für jeden intramolekularen Atomabstand ein Peak erscheint(Abb. 1-8). Die Intensität eines Peaks ist proportional dem Produkt FfFj deratomaren Streufaktoren und umgekehrt proportional dem zugehörigen Abstand rtj.Die Halbwertsbreite des Peaks ist gleich der Wurzel aus dem mittleren Quadrat derSchwingungsamplitude ltj dieses Atomabstandes.
n(rs)ds (1-25)
1.3 Elektronenbeugung 21
Abb. 1-7. Molekulare Streuintensitätskurve 7M (s) von Bicyclo [2.2.0]hexa-2,5-dien (Dewar-Benzol).Experimentelle (gepunktet) und berechnete Kurve (durchgezogen) mit Differenzkurve (nachMcNeill und Scholer, 1976).
RD(r)
r pro
Abb. 1-8. Radialverteilungskurve RD(r) von Bicyclo[2.2.0]hexa-2,5-dien (Dewar-Benzol). Experi-mentelle (gepunktet) und berechnete Kurve (durchgezogen) mit Differenzkurve (nach McNeill undScholer, 1976). Jeder intramolekulare Atomabstand führt zu einem Peak. Dessen Intensität ist vomProdukt der atomaren Streufaktoren und von der Größe des Abstandes abhängig. Die Lageund die Intensität der Abstände ist durch die senkrecht gezeichneten Striche angedeutet. DieHalbwertsbreite der Peaks richtet sich nach der zugehörigen mittleren Schwingungsamplitude.

22 l Methoden der Strukturanalyse
Die Streufaktoren F hängen mit den entsprechenden Werten / der Röntgenbeu-gung gemäß Gl. (1-26) zusammen. Z ist die Ordnungszahl des Atoms und C eineKonstante.
F(s) = C(Z-f(s))/s* (1-26)
Gl. (1-26) berücksichtigt, daß die Elektronen an den Atomrümpfen und nicht wiedie Röntgenstrahlen an den äußeren Elektronen gebeugt werden.
Beispiel 1-5 Dewar-Benzol:Den intensiveren Peaks der Radialverteilungskurve (Abb. 1-8) entsprechen folgende Atomab-
stände r und Schwingungsamplituden / (pm):
C1— H7
C3— H9
C1— C2
C2— C3
C3— C6
c!---c3c^-c5c -c4
112.4113.4134.5152.4157.4210.7259.5292.3
9.29.24.45.74.55.87.47.0
Nur in einfachen Fällen können sämtliche für die Geometrie des untersuchtenMoleküls relevanten Abstände der Radialverteilungskurve direkt entnommen wer-den. Im allgemeinen enthält ein Molekül mehrere etwa gleich lange Abstände, sodaß deren exakte Werte der Gesamtkurve nicht zu entnehmen sind (Abb. 1-8). Ausdiesem Grunde wird üblicherweise die Streukurve für ein bestimmtes Molekülmodellmit Gl. (1-27) berechnet. Die Strukturparameter rtj und ltj können jetzt - z. B. nachder Methode der kleinsten Fehlerquadrate - so weit verfeinert werden, daß sicheine optimale Übereinstimmung mit der experimentellen Streukurve ergibt. Fürdie Schwingungsamplituden ltj kann man auch spektroskopisch bestimmte Werteverwenden.
C = Konstante
Wie in Abschn. 1.1 erwähnt, handelt es sich bei den durch Elektronenbeugungermittelten Bindungslängen um ra- Werte. Durch eine Korrektur gemäß Gl. (1-28)erhält man die r^- Werte, die sich mit den Ergebnissen anderer Methoden vergleichenlassen. Im allgemeinen beträgt die Genauigkeit der so ermittelten Parameter beiBindungslängen einige Zehntel pm.
r« = ra + l2/ra (1-28)

1.3 Elektronenbeugung 23
1.3.3 Berücksichtigung spektroskopischer Daten:r -Strukturav
Wie bereits in Abschn. l. l erwähnt, können bei EB-Untersuchungen spektrosko-pische Daten mitverwendet werden. Dies betrifft einmal das Molekülmodell, dessenSymmetrie durch spektroskopische Untersuchungen ermittelt werden kann. Für dieStrukturanalyse lassen sich insbesondere auch die z.B. aus dem MW-Spektrumerhaltenen Rotationskonstanten des Moleküls verwenden, die ihrerseits mit derMolekülgeometrie [vgl. Gl. (1-7)] zusammenhängen.
Bei komplizierten Molekülen ist u. U. aus den EB-Daten eine Strukturanalyse nurunter bestimmten Annahmen möglich. So muß man evtl. einige Bindungslängenoder -winkel als gleich annehmen, um die Anzahl der zu optimierenden Parameterzu verringern.
Kann man nun die Verfeinerung des Strukturmodells gleichzeitig an EB- undspektroskopischen Daten durchführen, so sind solche Einschränkungen evtl. nichtmehr erforderlich, und das Ergebnis besitzt in jedem Fall höhere Zuverlässigkeit.Die aus EB- und MW-Daten ermittelten Strukturparameter werden als rav-Wertebezeichnet.
Beispiel 1-6 1,3-Butadien:
Das Molekül besitzt C2h-Symmetrie; zur Beschreibung seiner Geometrie sind neun Parametererforderlich. Aus den EB-Daten läßt sich die rJ-Struktur nur gewinnen, wenn sämtliche C—H-Bindungslängen und die C—C—H-Bindungswinkel als gleich angenommen werden. Unter Einbezie-hung der Rotationskonstanten von C4H6, C4H2D4 und C4D6, die aus dem Rotationsschwingungs-
Tab. 1-4. Strukturparameter von l ,3-Butadien. Die ^-Struktur wurdeallein aus Elektronenbeugungsdaten erhalten. Bei der rav-Struktur wur-den die Rotationskonstanten von C4H6, C4H2D4 und C4D6 mitverwen-det (Kuchitsu et al, 1968).
rl '„
C— Cc=cC TTa)— n '
c=c—cH1— C=CH2— C=CH3— C=C
146.3 + 0.3 pm134.2 + 0.2109.3 + 0.9123.6 + 0.3°
•)> 120.9+1.2J
146.3 + 0.3 pm134.1+0.2109.0 + 0.4123.3 + 0.3°122.4 + 2.2120.4 + 2.4123.0 + 3.2
a Mittelwert; die C—H-Bindungen wurden als gleich lang angenommen

24 l Methoden der Strukturanalyse
Spektrum ermittelt wurden, konnten sämtliche C—C—H-Winkel bestimmt werden. Die auf diesemWege erhaltene rav-Struktur unterscheidet sich hinsichtlich der übrigen Parameter nicht signifikantvon der r°-Struktur (Tab. 1-4). Dies muß aber keineswegs immer der Fall sein (Kuchitsu et al., 1968).
l .4 Röntgenstrukturanalyse
l .4. l Einleitung
Die Röntgenstrukturanalyse ist - gemessen an der Anzahl der durchgeführtenStrukturbestimmungen - die bei weitem wichtigste experimentelle Methode zurErmittlung von Strukturdaten. Die Genauigkeit der Strukturparameter ist zwaretwas schlechter als bei der Elektronenbeugung und der Mikrowellenspektroskopie(ca. l pm bei Bindungslängen und 1° bei Bindungswinkeln), und Wasserstoff-Atomelassen sich wegen ihrer geringen und nicht kugelsymmetrischen Elektronendichteschlecht lokalisieren. Da diese Methode aber nicht wie die beiden vorgenannten aufGase beschränkt ist, können im Prinzip sämtliche kristallisierbaren Verbindungenuntersucht werden. Dank weitgehend automatisierter Aufnahmetechniken und lei-stungsfähiger EDV-Auswertung ist die Feinstrukturanalyse einer organischen Ver-bindung heute innerhalb einiger Tage möglich. Bezüglich experimenteller und metho-discher Details sei auf die am Ende dieses Kapitels angegebene Literatur verwiesen.
Bei der Röntgenstrukturanalyse verwendet man monochromatische, harte Rönt-genstrahlung, z. B. die Ä^-Linie von Kupfer oder Molybdän mit einer Wellenlängevon 154.2 bzw. 71.1 pm. Die Wellenlänge liegt damit in der Größenordnung vonAtomen, Ionen oder Molekülen bzw. dem Abstand dieser Teilchen in einem Kristall-gitter. Deshalb ist es möglich, Beugungserscheinungen von Röntgenstrahlen anKristallen zu beobachten. Dieses Phänomen wurde 1912 von M. v. Laue und seinenMitarbeitern Friedrich und Knipping entdeckt, die damit gleichzeitig die Wellen-natur der Röntgenstrahlung nachwiesen. Bei der Beugung werden die Phasen dereinzelnen Röntgenstrahlen zueinander verändert, und man erhält Interferenzerschei-nungen, die mit dem Aufbau des Kristallgitters und damit der Struktur der Verbin-dung in unmittelbarem Zusammenhang stehen.
Die Elektronen der Verbindung werden zu Schwingungen mit der Frequenz dereinfallenden Röntgenstrahlung angeregt. Dadurch werden sie ihrerseits zu Quellenelektromagnetischer Strahlung mit der gleichen Wellenlänge wie die Primärstrah-lung. Eine Auswertung der Interferenzerscheinungen der Sekundärstrahlung liefertdie Dichte Verteilung der Elektronen innerhalb des Kristalls. In der Nähe der Atom-kerne besitzt die Elektronendichte Maxima und ist annähernd kugelsymmetrisch.Zwischen den Atomen ist sie niedrig, da sie nur von den Bindungselektronen her-rührt. Bei der Röntgen-Feinstrukturanalyse müssen die bei der Beugung auftreten-den Phasenverschiebungen der Streuwellen in die Auswertung einbezogen werden.

1.4 Röntgenstrukturanalyse 25
1.4.2 Aufbau kristalliner Stoffe
Kristalline Stoffe sind durch einen dreidimensional periodischen Aufbau gekenn-zeichnet. Im Prinzip läßt sich der ganze Kristall aus der Elementarzelle aufbauen.Eine solche regelmäßige Anordnung von Atomen, Ionen oder Molekülen entsprichtim allgemeinen ihrer dichtesten Packung und einem Minimum an potentieller Ener-gie. Ein makroskopischer Kristall besteht normalerweise aus einer großen Anzahlvon gegeneinander versetzten Kristalliten, die ihrerseits echte Einkristalle mit einemstreng regelmäßigen Aufbau darstellen. Röntgen-Feinstrukturanalysen können nurmit Einkristallen durchgeführt werden.
Jede (ideale) Kristallstruktur kann dadurch beschrieben werden, daß man dieKonstanten der Elementarzelle und die Atomanordnung in ihr angibt. Zur Charak-terisierung der Elementarzelle sind ihre Symmetrie, die drei Kantenlängen a, b undc sowie drei Winkel a, ß und y erforderlich (Abb. 1-9).
Abb. 1-9. Punktgitter, Elementarzelle und Gitterkonstanten. Die Punkte geben die Lage von Atomenin einem Kristall an. Jeder Gitterposition entspricht ein äquivalenter Punkt in der Elementarzelle,mit dem er durch Translation längs der Gitterachsen a, b und c zur Deckung gebracht werden kann.Die Elementarzelle wird durch die Gitterkonstanten charakterisiert. Das sind die Kantenlängen a,b und c sowie die Winkel oe, ß und y.
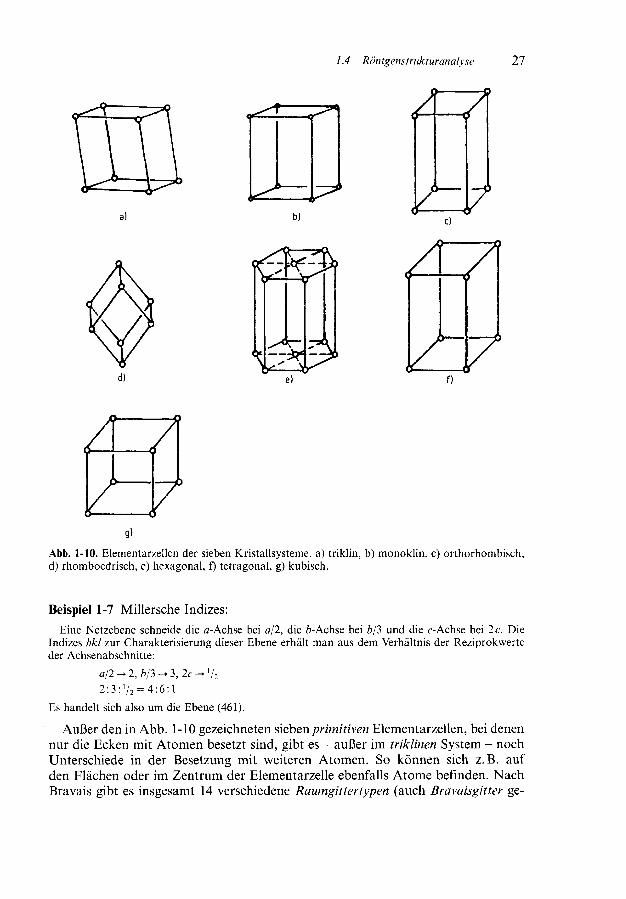
Mit diesen Gitterkonstanten lassen sich sieben verschiedene Gittertypen der32 Kristallklassen unterscheiden (Tab. 1-5, Abb. 1-10). Insgesamt sind bei Kristallen230 Raumgruppen mit unterschiedlicher Symmetrie möglich. Die 32 kristallographi-schen Punktgruppen sind den sieben Kristallsystemen in eindeutiger Weise zugeord-net. Zur Symmetrie s. Band l dieser Reihe, Kapitel 3.
Neben der Unterteilung des Kristallraumes in Elementarzellen ist die Aufgliede-rung durch Netzebenenscharen wichtig. Netzebenen sind Ebenen durch die Punktedes Kristallgitters. Sie treten als Scharen paralleler, äquidistanter Ebenen auf. DerAbstand zweier benachbarter Ebenen wird als d-Wert bezeichnet. Die Orientierungeiner Netzebenenschar in bezug auf die Kanten der Elementarzelle wird durch dieMillerschen Indizes, hkl, angegeben. Man findet sie, indem man die Schnittpunkte

26 l Methoden der Strukturanalyse
Tab. 1-5. Kristallsysteme und Kristallklassen.
Punktgruppen-symbola)
S
QQC2
QQhD2D2h
Qv
C3£>3C3iD3d
Qv
QQhC6h£)6
£)3h
D6h
QvC4
QhD4
Ah£)2d
QvroThohTd
H-M
11
2m2/m
222mmmmm2
33233m3m
666/m62262m6/mmm6mm
44/m4224/mmm442m4mm
23432m3m3m43m
System Elementarzelle Minimum vorhandenerSymmetrieelemente
triklin a ^ ß ^ J ¥= 90° keinea^b^c
monoklin a = ß = 90° eine zweizählige Achsey =£ 90° oder eine Spiegelebenefl/6/C
orthorhombisch a = ß = y = 90° beliebige Kombination vona ^ b c drei senkrecht aufeinander
stehenden zweizähligenAchsen oder Spiegelebenen
rhomboedrisch a = ß = y 90° eine dreizählige Achsea = b = c oder eine dreizählige Dreh-
inversionsachse
hexagonal a = ß = 90° eine sechszählige Achse7 = 120° oder eine sechszählige Dreh-a = b c inversionsachse
tetragonal a = ß = y = 90° eine vierzählige Achsea = b c oder eine vierzählige Dreh-
inversionsachse
kubisch a = ß = y = 90° vier dreizählige Achsen ima = b = c Winkel von 109° 28X
S = Bezeichnung nach SchoenfliesH-M = Bezeichnung nach Hermann u. Mauguin
der dem Ursprung der Elementarzelle benachbarten Netzebene mit den Elementar-zellenkanten bestimmt. Die Achsenabschnitte gibt man in Vielfachen der drei Gitter-konstanten an, also oa, pb und qc. Die Reziprokwerte I/o, l /p und l/q stehen ineinem ganzzahligen Verhältnis zueinander: I/o: l/p: l/q = h: k: L
1.4 Röntgenstrukturanalyse 27
-A
b)
g)Abb. 1-10. Elementarzellen der sieben Kristallsysteme, a) triklin, b) monoklin, c) orthorhombisch,d) rhomboedrisch, e) hexagonal, f) tetragonal, g) kubisch.
Beispiel 1-7 Millersche Indizes:
Eine Netzebene schneide die a-Achse bei a/2, die Z?-Achse bei b/3 und die c-Achse bei 2c. DieIndizes hkl zur Charakterisierung dieser Ebene erhält man aus dem Verhältnis der Reziprokwerteder Achsenabschnitte:
a/2-+2,b/3-*3,2c-+l/2
2:3:V 2 = 4:6:1
Es handelt sich also um die Ebene (461).
Außer den in Abb. 1-10 gezeichneten sieben primitiven Elementarzellen, bei denennur die Ecken mit Atomen besetzt sind, gibt es - außer im triklinen System - nochUnterschiede in der Besetzung mit weiteren Atomen. So können sich z.B. aufden Flächen oder im Zentrum der Elementarzelle ebenfalls Atome befinden. NachBravais gibt es insgesamt 14 verschiedene Raumgitter typen (auch Bravaisgitter ge-

28 l Methoden der Strukturanalyse
nannt). Man kennzeichnet den Gittertyp mit einem zusätzlichen Symbol: P (primi-tives Gitter, nur die Ecken der Elementarzelle sind besetzt); A, B oder C (einseitigflächenzentriertes Gitter; A -flächenzentriert bedeutet, daß das Zentrum der Fläche(100) besetzt ist; analoge Bedeutung haben die Symbole B und C); /(innenzentriertesGitter, das Zentrum der Elementarzelle ist besetzt). Näheres s. Lehrbücher derKristallographie.
Die Elementarzelle enthält alle Arten der in der Verbindung vorkommendenAtome in dem durch die Bruttoformel angegebenen Verhältnis. Sie kann bei Mole-külkristallen ein oder mehrere Formeleinheiten enthalten, wobei die einzelnen Mole-küle entsprechend ihrer Symmetrie unterteilt werden.
Beispiel 1-8 Naphthalin:
Naphthalin kristallisiert in einem monoklinen Gitter mit den Konstanten a = 823.5, b = 600.3 undc = 865.8 pm sowie a = y = 90° und ß = 122.9°. Aus der Dichte der Kristalle und dem Volumen derElementarzelle folgt, daß sich in ihr zwei Formeleinheiten befinden müssen. In Abb. 1-11 erkenntman insgesamt zehn Naphthalin-Moleküle. Die an den Ecken befindlichen liegen zu jeweils einemAchtel und die beiden mittleren Moleküle je zur Hälfte innerhalb der Elementarzelle. Aus Symme-triegründen wird zur Beschreibung des Aufbaus der Elementarzelle nur ein halbes Molekül alsasymmetrische Einheit benötigt. Naphthalin besitzt daher die molekulare Symmetrie D^.
Die Struktur der Molekülkristalle ist durch die Gestalt der Einzelmoleküle be-stimmt. Eine räumlich möglichst dichte Anordnung mit möglichst vielen zwischen-molekularen Wechselwirkungen (z. B. Wasserstoffbrücken) wird bevorzugt. Der zwi-
Abb. 1-11. Anordnung der Moleküle in kristallinem Naphthalin (nach Abrahams et al., 1949).Gezeichnet sind insgesamt zehn Moleküle, die aber nur teilweise innerhalb der Elementarzelle liegen.Insgesamt befinden sich zwei Formeleinheiten in der Elementarzelle.

l A Röntgenstrukturanalyse 29
schenmolekulare Abstand wird durch den van-der-Waals-Radius der Atome be-stimmt (vgl. Abschn. 3.5.3).
1.4.3 Beugung von Röntgenstrahlen und Bestimmung derGitterkonstanten von Einkristallen
Wir betrachten (Abb. 1-12) eine mit Gitterpunkten belegte Netzebenenschar, aufdie unter dem Winkel 9 Röntgenstrahlen fallen. Ein Teil der Strahlung wird an denNetzebenen reflektiert. Die von den Streuzentren auf verschiedenen Netzebenenausgehenden Beugungsstrahlen besitzen maximale Intensität, sofern ihr Gangunter-schied 2dhk!sm9 ein ganzzahliges Vielfaches der Wellenlänge ist (BraggscheGleichung):
= Wellenlänge der Strahlung (1-29)
dhM kennzeichnet den Abstand der Netzebenen. Der Reflexionswinkel S wird auchals Glanzwinkel, die Zahl n als Ordnung der Reflexion bezeichnet. Die BraggscheGleichung bildet die Grundlage für die geometrische Auswertung von Röntgenauf-nahmen. Großen Netzebenenabständen entsprechen kleine Winkel 9 und um-gekehrt.
Ein Reflex tritt nur auf, wenn für den einfallenden Strahl eine bestimmte Netz-ebenenschar die Braggsche Gleichung erfüllt. Durch eine Messung der Reflexions-winkel 9 lassen sich die Netzebenenabstände dhki und damit bei richtiger Zuordnung(Indizierung) der Reflexe zu Netzebenenscharen die Gitterkonstanten bestimmen.In jedem Kristallsystem besteht ein mathematischer Zusammenhang zwischen denGitterkonstanten, den Millerschen Indizes einer Netzebene und dem zugehörigenNetzebenenabstand. Bei hochsymmetrischen Gittern (kubisch, tetragonal, hexago-nal, trigonal) kann man mit Hilfe einer Debye-Scherrer-Pulveraufnahme die Gitter-
einfallenderRöntgenstrahl
gebeugterRöntgenstrahl
Netzebenen-schar
Abb. 1-12. Beugung von Röntgenstrahlen an einer Netzebenenschar. Maximale Intensität dergebeugten Strahlen infolge konstruktiver Interferenz ist nur bei ganz bestimmten Werten des WinkelsS möglich, die über das Braggsche Gesetz, Gl. (1-29), mit dem Netzebenenabstand d zusammen-hängen.

30 l Methoden der Strukturanalyse
konstanten aus den Netzebenenabständen dhkl bestimmen. Für die Gitterkon-stante a eines kubischen Gitters gilt z. B. die Beziehung
a = dhkl \/h2 + k2 + l2 (1_30)
Die Kristallsymmetrie kann nach dem Laue-Verfahren an einem feststehendenEinkristall ermittelt werden. Die Gitterkonstanten werden nach der Drehkristall-methode bestimmt, bei der der Einkristall so justiert wird, daß er um eine Gitter-achse gedreht werden kann. Diese Technik findet auch bei den modernen Einkristall-Diffraktometern Verwendung, in denen die Intensitäten aller nachweisbaren Einzel-reflexe mit Hilfe von Zählrohren gemessen werden.
Wichtig für die Bestimmung der Kristallsymmetrie ist es, daß für bestimmte Wertevon h, k und / keine Reflexe auftreten können, da für jede Raumgruppe ganzbestimmte, durch die Symmetrie bedingte, Auslöschungsgesetze gelten. Diese Ge-setze sowie die Symmetrien innerhalb der Intensitäten geben Anhaltspunkte für dieErmittlung der Raumgruppe.
l .4.4 Strukturaufklärung
l .4.4. l Strukturfaktoren
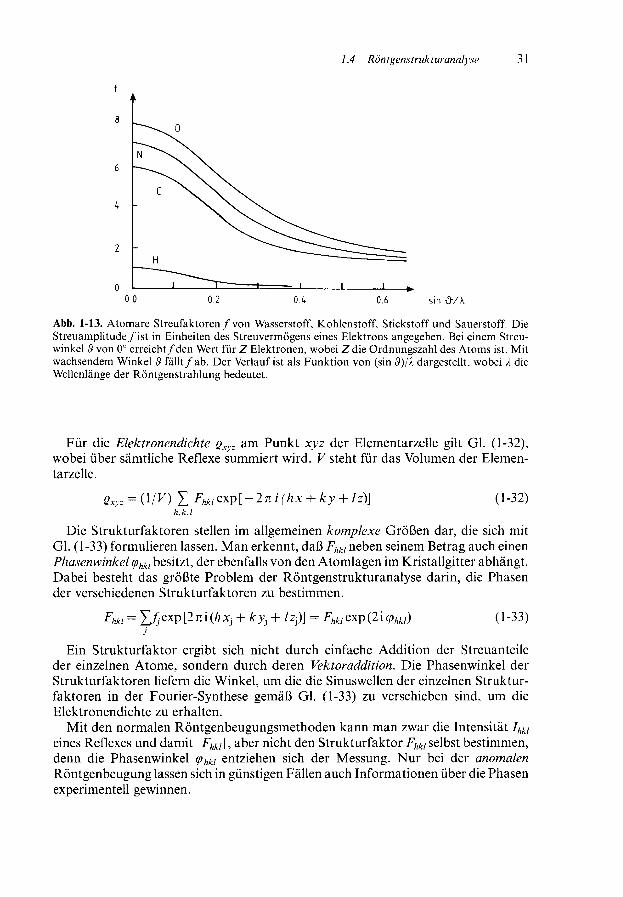
Wie bereits erwähnt, ist es zur Bestimmung der Strukturparameter erforderlich,die Intensitäten Ihkl der zu den Netzebenen hkl gehörigen Reflexe zu bestimmen. DieIntensität hängt von der Anzahl und der Art der Atome ab, die auf der jeweiligenNetzebene liegen. Sie ist von der Anzahl der Elektronen und damit von der Ord-nungszahl und auch vom Streuwinkel 3 abhängig. Die atomaren Streufaktoren f sindbei einem Streuwinkel von 0° gleich der Ordnungszahl des Elements oder bei einemIon gleich der Anzahl seiner Elektronen. Bei einem Streuwinkel ungleich 0° streuendie Elektronen die Röntgenstrahlen nicht mehr in Phase, sondern es kommt zuInterferenzen zwischen verschiedenen Regionen der Elektronenwolke. Deshalb ver-mindert sich die Gesamtstreuung (Abb. 1-13).
Abgesehen von einem berechenbaren geometrischen Faktor ist die Intensität Im
eines Reflexes gleich dem Quadrat des sogenannten Strukturfaktors Fhki. Ein solcherStrukturfaktor läßt sich berechnen, da er in eindeutiger Weise von der Lage derAtome in der Elementarzelle abhängt. Wenn z.B. aufgrund der Symmetrie derElementarzelle und anderer Befunde bei einem einfach gebauten Molekül recht guteVorstellungen über die Atomlagen in der Elementarzelle bestehen, läßt sich anhandberechneter Strukturfaktoren die Güte des Modells überprüfen. Der R- Wert [Resi-dualwert, Gl. (1-31)] dient als Anhaltspunkt, wie weit man wahrscheinlich an dierichtige Struktur herangekommen ist. Üblicherweise wird man jedoch unbekannteAtomlagen aus den gemessenen Strukturfaktoren zu ermitteln haben.
mit (1-31)^ — Whkl \ beob. ~~ \Ffikl\ber.
1.4 Röntgenstrukturanalyse 31
0.4 0.6
Abb. 1-13. Atomare Streufaktoren / von Wasserstoff, Kohlenstoff, Stickstoff und Sauerstoff. DieStreuamplitude/ist in Einheiten des Streuvermögens eines Elektrons angegeben. Bei einem Streu-winkel B von 0° erreicht/den Wert für Z Elektronen, wobei Z die Ordnungszahl des Atoms ist. Mitwachsendem Winkel «9 fällt/ab. Der Verlauf ist als Funktion von (sin $)/A dargestellt, wobei A dieWellenlänge der Röntgenstrahlung bedeutet.
Für die Elektronendichte Qxyz am Punkt xyz der Elementarzelle gilt Gl. (1-32),wobei über sämtliche Reflexe summiert wird. V steht für das Volumen der Elemen-tarzelle.
-27i i(hx /z)] (1-32)h, k, l
Die Strukturfaktoren stellen im allgemeinen komplexe Größen dar, die sich mitGl. (1-33) formulieren lassen. Man erkennt, daß Fm neben seinem Betrag auch einenPhasenwinkel (phki besitzt, der ebenfalls von den Atomlagen im Kristallgitter abhängt.Dabei besteht das größte Problem der Röntgenstrukturanalyse darin, die Phasender verschiedenen Strukturfaktoren zu bestimmen.
(1-33)i = L//exP P ni (A *j + kyj + /Zj)] = Fm exp (2 i cphkl)
Ein Strukturfaktor ergibt sich nicht durch einfache Addition der Streuanteileder einzelnen Atome, sondern durch deren Vektoraddition. Die Phasenwinkel derStrukturfaktoren liefern die Winkel, um die die Sinuswellen der einzelnen Struktur-faktoren in der Fourier-Synthese gemäß Gl. (1-33) zu verschieben sind, um dieElektronendichte zu erhalten.
Mit den normalen Röntgenbeugungsmethoden kann man zwar die Intensität Im
eines Reflexes und damit | Fhkl\, aber nicht den Strukturfaktor Fm selbst bestimmen,denn die Phasenwinkel q>hkl entziehen sich der Messung. Nur bei der anomalenRöntgenbeugung lassen sich in günstigen Fällen auch Informationen über die Phasenexperimentell gewinnen.

32 l Methoden der Strukturanalyse
l .4.4.2 Schweratom-Methode
Dieses von A. L. Patterson entwickelte Verfahren beruht auf der Patterson-Funk-tion [Gl. (1-34)], in der im Gegensatz zu Gl. (1-33) nur die Absolutwerte der Fhkh
nicht aber die Phasenwinkel erforderlich sind. P(x,y,z) besitzt Maxima für dieAtomabstände rtj innerhalb der Elementarzelle, deren Intensität dem Produkt Zt • Zjder Ordnungszahlen der beteiligten Atome proportional ist. Jedes beliebige Atom-paar in der Elementarzelle des untersuchten Kristalls „erzeugt" also ein Maximumin der Patterson-Funktion, und die Aufgabe besteht nun darin, die zugehörigenAtomlagen zu ermitteln.
P(x,y,z) = (l/V) X \Fhkl\2exp[2ni(hx + ky + lz)] (1-34)
h, k, l
Die Schwierigkeit dieses Unterfangens wächst mit steigender Anzahl der Atome inder Elementarzelle, denn bei n Atomen gibt es n2 -n interatomare Abstandsvektoren.Z.B. gehören zu den 36 Atomen in der Elementarzelle des Naphthalins (Beispiel1-8) 1260 Maxima der Patterson-Funktion. Wenn einige der Atome besondersschwer sind, z. B. Brom- Atome in einem organischen Molekül, dann verursachendie Vektoren zwischen diesen Atomen die bei weitem größten Peaks. Mit diesenVektoren werden jetzt die Schweratome lokalisierbar. Anhand der Positionen derSchweratome können nun für weitere Atome ungefähre Lagen abgeschätzt werden,und damit werden den meisten Strukturfaktoren die richtigen Phasen zugeordnet.Jetzt läßt sich mit einer Fourier-Synthese nach Gl. (1-33) die Elektronendichte-Verteilung berechnen. Da die Phasen nur die Lagen der Schweratome berücksichti-gen, zeigt die erste Fourier-Synthese nicht die ganze Struktur des Moleküls, sondernnur Teile davon, was aber zumeist ausreicht, um bessere Phasen für die nächsteFourier-Synthese zu erhalten. Auf diese Weise sollte das Strukturproblem nacheinigen solcher sukzessiven Fourier-Synthesen gelöst sein.
1.4.4.3 Direkte Phasenbestimmung
Zwischen den Phasen der stärkeren Reflexe gibt es systematische Beziehungen, diesich mittels Wahrscheinlichkeits-theoretischer Methoden dazu verwenden lassen, diePhasenwinkel direkt, d. h. ohne jede Annahme über die Natur oder die Lagen derAtome in der Elementarzelle, mit kalkulierbarer Wahrscheinlichkeit zu bestimmen.Ob ein auf diese Weise ermittelter Phasensatz der richtige ist, läßt sich damit überprü-fen, ob die resultierende Elektronendichteverteilung chemisch sinnvoll ist.
Nach der Sayre-Gleichung [Gl. (1-35)] besteht eine Beziehung zwischen einemStrukturfaktor Fhkl und den Faktorenpaaren, deren Indizes sich zu hkl addieren.Die Summation in Gl. (1-35) wird durch die Produkte großer F- Werte dominiert.Die Sayre-Gleichung ermöglicht also Wahrscheinlichkeitsberechnungen für die Pha-senwinkel.
Fhki = K Z Fhfk'ir'Fh-hr,k-k'j-ir (1-35)v, v, r
K = Konstante

1.4 Röntgenstrukturanalyse 33
Bei zentrosymmetrischen Kristallen reduziert sich das Phasenproblem auf dieFrage nach dem Vorzeichen. Für die Vorzeichen von drei zu intensiven Reflexengehörigen Strukturfaktoren kann man nach Gl. (1-35) erwarten, daß Gl. (1-36) gilt.Für nichtzentrosymmetrische Kristalle läßt sich analog (Gl. 1-37) annehmen.
Sign(F^) = Sign^^O-Sign^,^^^^,) (1-36)
<Phki = Vh'k'r + 9h-h>,k-k'j-r (1-37)
Die Entwicklung leistungsfähiger EDV-Programme für die Bewältigung des ho-hen Rechenaufwandes hat seit Beginn der 70er Jahre zu einem stürmischen Anstiegder durch direkte Methoden gelösten Strukturen geführt.
l .4.4.4 Strukturverfeinerung
Wie bereits in Abschn. 1.3 dargestellt, wird man sämtliche Parameter, die zurvollständigen Beschreibung des Strukturmodells erforderlich sind, z.B. nach derMethode der kleinsten Fehlerquadrate, so verfeinern, daß sich eine optimale Anpas-sung der berechneten an die beobachteten Strukturfaktoren ergibt. Der R-Wert[Gl. (1-31)] soll also einen Minimalwert erreichen. Üblicherweise gelangt man zu R-Werten < 0.10. Bei R = 0.02 kann man von einer ausgezeichneten Übereinstimmungsprechen.
Um einen derart guten R-Wert zu erhalten, genügt es allerdings nicht, nur dieGeometrie der Atomanordnung zu optimieren. Vielmehr muß auch das Schwin-gungsverhalten der Atome mit berücksichtigt werden. Die thermische Bewegung der
C2 C1 C7 N9
(b)
Abb.1-14. Darstellung der Molekülstruktur von 1,4-Dicyanobenzol mit thermischen Ellipsoiden.Die Größe der Ellipsoide ist so gewählt, daß sie 50% der Aufenthaltswahrscheinlichkeit der Atomewiedergeben, a) Projektion in die Ebene des Benzolringes, b) Projektion in die Ebene senkrecht zumBenzolring parallel zur Längsachse des Moleküls. Die Wasserstoff-Atome wurden der Übersichtlich-keit halber weggelassen. (Mit freundlicher Genehmigung aus Colpapietro et al., 1984.)

34 l Methoden der Strukturanalyse
Atome beschreibt man mit Schwingungs- oder Temperaturkoeffizienten. Bei isotropenTemperaturkoeffizienten geht man von einer sphärischen Schwingungsbewegungaus und optimiert für jedes Atom einen Parameter. Für die anisotrope Temperatur-schwingung sind sechs Parameter pro Atom erforderlich: drei für die räumlicheOrientierung und drei weitere für die Hauptachsen des Schwingungsellipsoids. Ineiner graphischen Darstellung wählt man die Größe eines Ellipsoids zumeist so, daßes 50% der Aufenthaltswahrscheinlichkeit des schwingenden Atoms wiedergibt.
Zur Ermittlung der Standardabweichungen s. Abschn. 1.1.3.
Beispiel 1-9 1,4-Dicyanobenzol (Terephthalsäuredinitril):Das Diagramm in Abb. 1-14 zeigt die Anisotropie der thermischen Bewegung der Atome im
Kristall.
1.4.5 Meßtechnik und zukünftige Entwicklung
Zur Messung werden heute zumeist rechnergesteuerte Einkristalldiffraktometereingesetzt. Abb. 1-15 zeigt das Schema einer Versuchsanordnung. Ein solches Systembesteht aus einer Röntgenröhre, einem Szintillationszähler als Detektor, einer Mecha-nik für die Positionierung von Kristall und Meßsonde und einem Prozeßrechner, derauch zur Lösung des Phasen- und Strukturproblems verwendet werden kann. Jenach Größe des Moleküls werden zwischen 1000 und 100000 Reflexe ausgemessen.
Besondere Probleme und Schwierigkeiten können die Absorption der Röntgen-strahlung, die auch mit einer Zersetzung der Probe verbunden sein kann, Fehlanord-nungen im Kristall, Zwillingsbildung von Einkristallen u. ä. verursachen.
Die Einführung sog. Flächendetektoren, deren Entwicklung kurz vor dem Ab-schluß steht, läßt eine drastische Verkleinerung der Aufnahmezeiten und damit dieEliminierung etwaiger durch die zeitliche Instabilität der Meßanordnung bedingteFehlerquellen erwarten.
Eine andere Entwicklung erfolgt mit Hilfe der Synchrotronstrahlung. Diese fälltals „Nebenprodukt" in den Speicherringen für Elementarteilchen (Elektronen oderPositronen) ab und reicht lückenlos vom Mikrowellenbereich bis zu den hartenRöntgenstrahlen. In Deutschland sind Strukturuntersuchungen im HamburgerSynchrotronstrahlungslabor (HASYLAB) möglich. Die Synchrotronstrahlung be-sitzt den Vorteil hoher Monochromasie und gestattet auch eine Messung schwacherReflexe.
Durch Messungen bei verschiedenen Wellenlängen innerhalb und außerhalb deranomalen Dispersion ergibt sich eine wesentliche Erleichterung bei der Bewältigungdes Phasenproblems. Die Messung bietet zudem den Vorteil, daß Mikrokristalle mitca. l jim Länge ausreichen - bei der „normalen" Röntgenstrukturanalyse benötigtman Einkristalle, die mindestens einige Zehntel mm lang sind. Damit sind auch Bio-und Makromoleküle, von denen man keine größeren Kristalle erhält, der Struktur-analyse zugänglich. Als Beispiele seien hier Proteine, Enzyme und Viren erwähnt.
Wegen der Kürze der Meßdauer eignet sich die Synchrotronstrahlung auch fürdas Studium zeitabhängiger Phänomene wie z. B. Festkörperreaktionen.

1.4 Röntgenstrukturanalyse 35
Abb. 1-15. Vierkreisdiffraktometer. Der Einkristall A wird über die drei Eulerschen Winkel /, (p, CDso ausgerichtet, daß ein gewünschter Reflex in der horizontalen Ebene erscheint. Der Detektor Cist nur in dieser Ebene verstellbar und empfängt den Reflex bei einem Ablenkungswinkel 2 <9 zumdirekten Strahl. B ist die Röntgenröhre.
1.4.6 Absolutbestimmung der Konfiguration chiralerVerbindungen durch anomale Röntgenbeugung
Die Konfiguration chiraler Moleküle gibt sich nur einem chiralen Medium zuerkennen. Deshalb kann man Enantiomere zwar mit polarisiertem Licht, nicht abermit Röntgenstrahlen unterscheiden. Daß die Röntgenstrukturanalyse sich dennochzur Absolutbestimmung der Konfiguration eignet, konnten Bijvoet, Peerdeman undvan Bommel 1951 am Beispiel des L-( + )-Natrium-rubidium-tartrats, NaRbC4H4O4,zeigen. Das Prinzip der sog. anomalen Röntgenbeugung beruht auf einer Verletzungdes Friedeischen Gesetzes, nach dem die Reflexe für entgegengesetzte Gitterebenen,hkl und Tikl, gleiche Intensitäten besitzen müssen. Wird nun eines der Atome, imobigen Falle handelte es sich um das Rubidium-Ion, durch die Röntgenstrahlungelektronisch angeregt, so unterscheidet sich die Intensität der Reflexpaare in signifi-kanter Weise je nachdem, welche Konfiguration vorliegt. Auf diese Weise konntedie von E. Fischer 1891 willkürlich getroffene Zuordnung von Enantiomeren zur L-bzw. D-Reihe bestätigt werden. Auf die Möglichkeit zur experimentellen Phasenbe-stimmung wurde bereits in Abschn. 1.4.4.3 hingewiesen.
36 l Methoden der Strukturanalyse
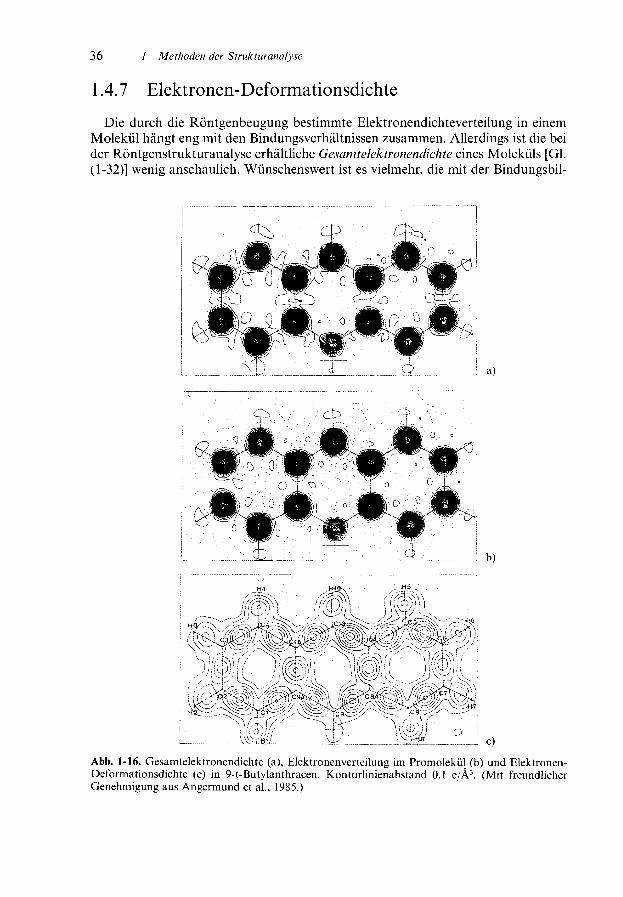
l .4.7 Elektronen-Deformationsdichte
Die durch die Röntgenbeugung bestimmte Elektronendichteverteilung in einemMolekül hängt eng mit den Bindungsverhältnissen zusammen. Allerdings ist die beider Röntgenstrukturanalyse erhältliche Gesamtelektronendichte eines Moleküls [Gl.(1-32)] wenig anschaulich. Wünschenswert ist es vielmehr, die mit der Bindungsbil-
a)
Abb. 1-16. Gesamtelektronendichte (a), Elektronenverteilung im Promolekül (b) und Elektronen-Deformationsdichte (c) in 9-t-Butylanthracen. Konturlinienabstand 0.1 e/Ä3. (Mit freundlicherGenehmigung aus Angermund et al., 1985.)

1.5 Neutronenbeugung 37
düng einhergehenden Änderungen in der Elektronenverteilung zu ermitteln undgraphisch darzustellen. Hierzu wurde der Begriff Elektronen-DeformationsdichteEDD eingeführt. Dabei handelt es sich um die Elektronendichte, die sich durchDeformation der kugelsymmetrischen Ladungsverteilung als Folge der Ausbildungchemischer Bindungen zwischen ursprünglich freien Atomen ergibt.
Die kugelsysmmetrische Elektronenverteilung freier Atome läßt sich rechnerischgenau ermitteln. Nimmt man nun an, daß sich bei der Bildung von Molekülen freieAtome ohne Änderung ihrer Elektronenverteilung auf die Abstände im Molekülnähern, so ergibt sich ein idealisiertes Molekülmodell, das als Promolekül bezeichnetwird.
Aus der Differenz zwischen der gefundenen Elektronen Verteilung des realen Mole-küls und der berechneten des Promoleküls erhält man die EDD. Eine positiveDeformationsdichte an einer Stelle des Moleküls bedeutet, daß bei der Ausbildungder Bindungen eine Ladungserhöhung eingetreten ist; negative Deformationsdichtenfindet man analog an Stellen vermindeter Elektronendichte.
Abb. 1-16 zeigt für das 9-t-Butylanthracen einen Vergleich zwischen der Gesamt-elektronendichte, der Elektronenverteilung im Promolekül und der EDD. Manerkennt, daß zwischen allen gebundenen Atomen positive EDD-Werte gefundenwerden.
Aus zahlreichen, sowohl gemessenen als auch berechneten, EDDs unterschiedli-cher Verbindungen ist zu entnehmen, daß je nach Elementen und Bindungstypensowohl positive als auch negative Deformationsdichten zwischen gebundenen Ato-men auftreten.
Wegen der Schwierigkeiten mit der genauen Lokalisierung von Wasserstoff-Ato-men bei der Röntgen-Strukturanalyse führt man EDD-Bestimmungen auch in Kom-bination mit der Neutronenbeugung durch (s. Abschn. 1.5.3).
l. 5 Neutronenbeugung
1.5.1 Einleitung
Wie die Röntgenstrukturanalyse wird die Neutronenbeugung vornehmlich zumStudium kristalliner Stoffe eingesetzt. Da man auf einen Kernreaktor als Neutronen-quelle angewiesen ist, wird man diese Methode in erster Linie auf Probleme anwen-den, die sonst nicht gelöst werden können. Dazu gehört z. B. die genaue Lokalisie-rung von Wasserstoff-Atomen in kristallinen Stoffen.
Da bei allen neutronenliefernden Kernprozessen schnelle Neutronen mit sehrkurzer Wellenlänge freigesetzt werden, müssen sie durch geeignete Moderatoren(z.B. Paraffin) auf thermische Energien abgebremst werden, denen nach derde Broglie-Gleichung [vgl. Gl. (1-20)] Wellenlängen in der Größenordnung von100 pm entsprechen. Die Monochromatisierung erfolgt durch Beugung an einemKristall, üblicherweise aus Blei oder Kupfer, mit dessen Hilfe aus dem polychromati-

38 l Methoden der Strukturanalyse
sehen Strahl nach dem Braggschen Gesetz [Gl. (1-29)] eine bestimmte Wellenlängeausgeblendet wird.
Die Wechselwirkung der Neutronen mit den Atomen besteht überwiegend auselastischen Streuprozessen an den Atomkernen. Ein Vergleich mit den an der gleichenProbe durchgeführten Röntgenstreuexperimenten (Streuung an den Elektronen)ermöglicht demzufolge eine experimentelle Bestimmung der Elektronenverteilungin der Substanz (s. Abschn. 1.5.3).
Das Prinzip der Strukturanalyse mit Neutronen ist dem der Röntgenkristallogra-phie weitgehend analog. Der Nachweis der gestreuten Neutronen erfolgt mittelsspezieller Detektoren (BF3- oder 3He-Zählrohre). Das Streuvermögen eines Atomsfür Neutronen hängt vom jeweiligen Kernniveauschema ab. Mit Neutronen lassensich also einzelne Isotope unterscheiden. Im Gegensatz zur Röntgenbeugung sinddie Streufaktoren für Neutronen nicht vom Streuwinkel abhängig. Dies liegt daran,daß die Atomkerne als Beugungspunkte für die Neutronen wesentlich kleiner sindals ihre Elektronenwolke, an der die Röntgenstrahlen gebeugt werden. Während dieStreufaktoren für Röntgenstrahlen der Ordnungszahl proportional sind, variierensie für Neutronen nur um einen Faktor von etwa 3, d. h. alle Atome besitzen ungefährdas gleiche Streuvermögen. Deshalb ist es bei der Neutronenbeugung kein Problem,leichte Atome neben schweren zu finden, und demzufolge liegt das Hauptanwen-dungsgebiet dieser Methode bei der Strukturanalyse organischer Verbindungen inder Lokalisierung von Wasserstoff-Atomen. - Hinsichtlich näherer Einzelheiten seiauf die am Ende des Kapitels angegebene Literatur verwiesen.
1.5.2 Strukturanalyse
Wie bei der Röntgenstrukturanalyse können bei der Neutronenbeugung Pulver-und Einkristallmethoden verwendet werden. Da die Streufaktoren für Neutronennicht mit dem Streuwinkel abfallen, sind die Fehler bei einer Fourier-Synthese [Gl.(1-33)] aufgrund einer begrenzten Anzahl von Termen weitaus größer, da die Reihefür Neutronen weder endlich noch konvergent ist. Diese Schwierigkeit umgeht mandurch die Berechnung von Differenz-Synthesen.
Da Röntgenstrahlen an den Elektronen, die Neutronen aber an den Atomkernengebeugt werden, unterscheidet sich auch das Ergebnis beider Methoden: Bei derRöntgenstrukturanalyse kann nur aus der Elektronendichteverteilung auf die Kern-lagen geschlossen werden. Bei kovalenten Bindungen oder bei Atomen mit einsamenElektronenpaaren fällt der Schwerpunkt der Elektronenwolke zumeist nicht mitdem Atomkern zusammen, dessen Position bei der Neutronenbeugung direkt gemes-sen wird. Bei einer Kombination beider Methoden lassen sich dann Einzelheitenbezüglich der Elektronenverteilung in (polaren) kovalenten Bindungen und der„Gestalt" einsamer Elektronenpaare ermitteln.
Beispiel 1-10 Hexamethylentetramin:Das Molekül mit der Formel C6H12N4 besitzt kubische Symmetrie (rd). Die asymmetrische Einheit
beinhaltet nur drei Atome (C, H und N). Die Molekülstruktur wird durch die vier Atomkoordinatenu, v, x und z beschrieben (Tab. 1-6): Das Kohlenstoff-Atom liegt bei (0 0 u) und den äquivalentenPositionen, das Stickstoff-Atom bei (t; v v) und das Wasserstoff-Atom bei (x x z).

L 5 Neutronenbeugung 39
Tab. 1-6. Lageparameter für die Atome von Hexamethylentetramin inEinheiten der Gitterkonstante a = 701.9 pm.
u(C)»(N)*(H)z (H)
Neutronenbeugunga)
0.2381 ±0.00080.1221 ± 0.00050.0899 + 0.00110.3258 + 0.0012
Röntgenbeugungb)
0.2375 ± 0.00030.1237 ±0.00020.0802 ± 0.00200.3292 ± 0.0030
a Becka und Cruickshank, 1963b Duckworth et al., 1970
Tab. 1-6 entnimmt man, daß die Werte von u für den Kohlenstoff innerhalb der Standardabwei-chung der Neutronenbeugung übereinstimmen. Dies war zu erwarten, da der sp3-hybridisierteKohlenstoff eine symmetrische Elektronenwolke besitzt. Demgegenüber zeigt sich für die v-Wertedes Stickstoffs eine Differenz, die dreimal so groß ist wie die Standardabweichung der Neutronenbeu-gung. Demnach differiert das Zentrum der Elektronenwolke (Röntgen) um 1.8 pm von der Lagedes Atomkerns (Neutronen). Dies läßt sich mit dem einsamen Elektronenpaar erklären, das sich ineinem vom Atomkern wegweisenden Orbital befindet. Auch die Parameter x und z des Wasserstoff-Atoms in Tab. 1-6 weisen für beide Methoden signifikante Unterschiede auf, und zwar ist die C—H-Bindungslänge bei der Neutronenbeugung um 6 pm länger als bei der Röntgenmethode.
Neuere Untersuchungen zeigten, daß die Verfälschung von Atomlagen bei derRöntgenanalyse gemindert werden kann, wenn man Reflexe bei möglichst großenStreuwinkeln (sin$/A > 10~2 pm"1) in die Messung einbezieht. Generell findet manjedoch Unterschiede zwischen beiden Methoden von bis zu 20 pm für Wasserstoff-Bindungslängen (C—H, N—H, O—H usw.) und zumeist weniger als l pm beiBindungslängen mit Stickstoff- und Sauerstoff-Atomen (C—N, C—O usw.).
Proteine und andere biologisch wichtige Moleküle sind im allgemeinen zu großfür eine Röntgenstrukturanalyse unter Verwendung direkter Methoden zur Lösungdes Phasenproblems. Zumeist wird man dann auf die Schweratom-Methode zurück-greifen. Da solche Moleküle etwa zur Hälfte aus Wasserstoff-Atomen bestehen, vondenen einige Schlüsselrollen für die Struktur und die Funktion des Proteins spielen,ist der Einsatz der Neutronenbeugung bei der Strukturanalyse von Proteinen ver-ständlich. Bei der Lokalisierung bestimmter Wasserstoff-Atome macht man sich dasunterschiedliche Streuvermögen von Wasserstoff und Deuterium zunutze, indemman Wasserstoff-Atome gezielt durch Deuterium ersetzt.
1.5.3 X-N-Methode zur Bestimmung der Elektronen-Deformationsdichte
Wie bereits in Abschn. 1.4.7 erwähnt, ergeben sich bei der Röntgen-MethodeSchwierigkeiten, wenn leichte Atome wie Wasserstoff in die Elektronen-Deforma-tionsdichte EDD einbezogen werden sollen. Diese Probleme lassen sich lösen, wennman für das Promolekül die bei der Neutronenbeugung ermittelten Lageparameterverwendet, und dann wie üblich die berechnete Elektronenverteilung von der durchRöntgenbeugung bestimmten Gesamtelektronendichte QX [vgl. Gl. (1-32)] subtra-

40 l Methoden der Strukturanalyse
0OC H
Abb. 1-17. Nach der X-N-Methode gewonnene Elektronen-Deformationsdichte in Oxalsäure. (Mitfreundlicher Genehmigung aus Coppens, 1977.)
hiert. Die EDD zeigt dann die durch die chemische Bindung verursachten Änderun-gen in der Elektronenverteilung.
In Abb. 1-17 ist die nach der X-N-Methode gewonnene Elektronen-Differenz-dichte der Oxalsäure dargestellt. Die Erhöhung der Elektronendichte in den Bindun-gen auf Kosten der Atome sowie die einsamen Elektronenpaare an den Sauerstoff-Atomen sind deutlich zu erkennen.
Für Einfach-, Doppel- und Dreifachbindungen ergaben X-N-Analysen deutlicheUnterschiede in der Elektronenverteilung. Untersuchungen an Dreiringverbindun-gen ließen die nach dem Coulson-Moffitt-Modell erwartete hohe Elektronendichteaußerhalb des Ringes deutlich erkennen (vgl. Band l, Seite 266).
Wie in Abschn. 1.4.7 beschrieben, können ähnliche Ergebnisse auch bei aus-schließlicher Verwendung von Röntgenbeugungsmessungen (X-X-Methode) erhal-ten werden. Bei Problemen, an denen Wasserstoff-Atome beteiligt sind, ist jedocheine X-N- einer X-X-Analyse vorzuziehen. Die EDD kann auch mit Hilfe quantenche-mischer Rechnungen ermittelt werden.
Weiterführende und ergänzende Literatur zu Kapitel l
Allgemeines zu Molekülstruktur und StrukturparameternCoulson C.A. (1977): Geometrie und elektronische Struktur von Molekülen. Verlag Chemie,Weinheim.Lide D. R. und Paul M. A. (1974): Critical Evaluation of Chemical and Physical Structural Informa-tion. National Academy of Sciences, Washington.

Weiterführende und ergänzende Literatur zu Kapitel l 41
Natta G. und Farina M. (1976): Struktur und Verhalten von Molekülen im Raum. Verlag Chemie,Weinheim.Robiette A. G. (1973), „The Interplay Between Spectroscopy and Electron Diffraction", in Sinn G. A.und Sutton L. E. (Hrsg.): Molecular Structure by Diffraction Methods. Specialist Periodical Report,Bd, l, S, 160, The Chemical Society, London.Schröder B. und Rudolph J. (Hrsg.) (1985): Physikalische Methoden in der Chemie. VCH Verlags-gesellschaft, Weinheim.Stuart H.A., Funck E. und Müller-Warmuth W. (1967): Molekülstruktur. Physikalische Methodenzur Bestimmung der Struktur von Molekülen und ihre wichtigsten Ergebnisse. Springer-Verlag, Berlin.
MikrowellenspektroskopieChantry G. W. (Hrsg.) (1979): Modern Aspects ofMicrowave Spectroscopy. Academic Press, London.Gordy W. und Cook R. L. (1984): Microwave Molecular Spectra. 3. Aufl., Wiley, New York.Kroto H.W. (1975): Molecular Rotation Spectra. Wiley, London.Zeil W. (1973), „Mikrowellenspektroskopie", in Körte F. (Hrsg.), Methodicum Chimicum l/l, 418.
ElektronenbeugungAlmenningen A., Bastiansen O., Haaland A. und Seip H. M. (1965), „Strukturbestimmung freierMoleküle durch Elektronenbeugung", Angew. Chem. 77, 877.Davis M.I. (1971): Electron Diffraction in Gases. Marcel Decker, New York.Goodman P. (Hrsg.) (1981): Fifty Years of Electron Diffraction. Reidel Publishing Company,Dordrecht/Holland.Haase J. (1973), „Elektronenbeugungsanalyse", in Körte F. (Hrsg.), Methodicum Chimicum l/l, 596.Rymer T.B. (1970): Electron Diffraction. Methuen, London.
Kristallstruktur und RöntgenstrukturanalyseBuerger M. J. (1977): Kristallographie. Eine Einführung in die geometrische und röntgenographischeKristallkunde. De Gruyter, Berlin.Kitaigorodsky A. I. (1973): Molecular Crystals and Molecules. Academic Press, New York undLondon.Dunitz J.D. (1979): X-Ray Analysis and the Structure of Organic Molecules. Cornell UniversityPress, Ithaca.Haussühl S. (1979): Kristallstrukturbestimmung. Verlag Chemie, Weinheim.Keller E. (1982), „Röntgenstrukturanalyse von Molekülen", Chemie Unserer Zeit 16, Teil l S.71,Teil 2 S. 116.Paulus E.F. (1980), „Strukturanalyse durch Beugung an Kristallen", in Ullmanns Encyklopädie dertechnischen Chemie, 4. Aufl., Band 5, S. 235, Verlag Chemie, Weinheim.Schultze-Rhondorf E. (1973), „Röntgenstrukturanalyse", in Körte F. (Hrsg.), Methodicum Chimi-cum, l/l, 532.
NeutronenbeugungBacon G.E. (1975): Neutron Diffraction. 3.Aufl., Clarendon Press, Oxford.Bacon G.E. (1977): Neutron Scattering in Chemistry. Newnes-Butterworths, Sevenoaks.Dachs H. (Hrsg.) (1978): Neutron Diffraction. Topics in Current Physics, Bd. 6, Springer-Verlag,Berlin.Fuess H. (1979): The Application of Neutron Diffraction to Chemistry. Mod. Phys. Chem. 2, AcademicPress, London.Will G. (1973), „Neutronenbeugung", in Körte F. (Hrsg.), Methodicum Chimicum, l/l, 570.
