induktion von matrix-metalloproteinase-9 (mmp-9) durch ... · inhaltsverzeichnis d. ergebnisse 37...
TRANSCRIPT

Klinik für Orthopädie und Sportorthopädie der Technischen Universität
München
Klinikum rechts der Isar
(Direktor: Univ.-Prof. Dr. R. Gradinger)
Induktion von Matrix-Metalloproteinase-9 (MMP-9) durch Thrombin in der Osteosarkomzellinie U2-OS
Alfred Eichbichler
Vollständiger Abdruck der von der Fakultät für Medizin der Technischen
Universität München zur Erlangung des akademischen Grades eines
Doktors der Medizin (Dr. med.)
genehmigten Dissertation.
Vorsitzender: Univ.-Prof. Dr. D. Neumeier
Prüfer der Dissertation:
1. Privatdozent Dr. Dr. J. Rechl
2. Univ.-Prof. Dr. R. Gradinger
Die Dissertation wurde am 12.06.2001 bei der Technischen Universität
München eingereicht und durch die Fakultät für Medizin am 14.11.2001
angenommen.
1

Meinen Eltern
2

Inhaltsverzeichnis
Inhaltsverzeichnis A. Einleitung 5
A.1 Maligne Transformation einer Zelle 5 A.2 Primäre Knochentumore 5 A.3 Transformation einer normalen Zelle in eine Tumorzelle 9 A.4 Autonome Versorgung von Wachstumssignalen 9 A.5 Tumorzellen sind gegen wachstumshemmende Signale resistent 10 A.6 Tumorzellen durchlaufen keine Apoptose 11 A.7 Grenzenloses replikatives Potential von Tumorzellen 12 A.8 Neoangiogenese bei Tumoren 13 A.9 Invasion und Metastasierung von Tumorzellen 14 A.10 Proteolyse der extrazellulären Matrix 15 A.11 Bedeutung der Typ-IV Kollagenasen bei der Metastasierung 16 A.12 Regulation des MMP-9-Gens 18 A.13 Intrazelluläre Signaltransduktionswege 18 A.14 Störungen der Hämostase bei Tumorpatienten 20 A.15 Wechselwirkung zwischen Tumorzelle und Blutgerinnung 21 A.16 Struktur und Funktion von Thrombin und dem spezifischen
Thrombinrezeptor 23 A.17 Thrombin und der spezifische Thrombinrezeptor in der Tumorbiologie 25 B. Fragestellung 27
C. Material und Methoden 28
C.1 Zellinien und Materialien 28 C.2 Material 28 C.3 Zellkultur 29 C.4 Zymographie 29 C.5 Northern Blot 30 C.6 Transfektionen: CAT, Luciferase 32 C.7 RT-PCR aus Gewebeproben 35 C.8 Invasionsassay 35
3

Inhaltsverzeichnis
D. Ergebnisse 37
D.1 Thrombin erhöht die Aktivität von pro-MMP-9 37 D.2 Untersuchung des Signaltransduktionsweges nach Stimulation mit
Thrombin 38 D.3 Thrombin stimuliert konzentrationsabhängig die RNA-Expression
von MMP-9 40 D.4 Zeitabhängige Veränderung der Aktivität von pro-MMP-9 nach
Stimulation mit Thrombin 41 D.5 Zeitabhängige Veränderung der Transktription von MMP-9 nach
Stimulation mit Thrombin 42 D.6 Thrombin aktiviert den MMP-9-Promotor auf Transkriptionsebene 43 D.7 Die MMP-9 Transkription wird über einen PI3-Kinase-abhängigen
Signaltransduktionsweg reguliert 46 D.8 Identifizierung des MMP-9-Promotorabschnittes, der für die
Stimulation durch Thrombin verantwortlich ist 47 D.9 Thrombin steigert die Invasivität von Tumorzellen 49 D.10 Nachweis des Thrombinrezeptors in Osteosarkomgewebe 50 E. Diskussion 52
E.1 Der Einfluss von Thrombin auf die Aktivität und die Expression
von Typ-IV Kollagenasen 52 E.2 Die Steigerung der Aktivität von MMP-9 durch Thrombin wird
durch Aktitivierung des Thrombinrezeptors vermittelt 55 E.3 Die Bedeutung des PI3-Kinase-Weges bei der Signalübermittlung 56 E.4 Identifizierung des regulierenden Abschnittes des MMP-9-Promotors
nach Stimulation durch Thrombin 57 E.5 Thrombin steigert die Invasion von Tumorzellen 58 E.6 Ausblick 59 F. Zusammenfassung 61
G. Literaturverzeichnis 63
H. Abkürzungsverzeichnis 79
I. Danksagung 81
4

A. Einleitung
A.1 Maligne Transformation einer Zelle Die ersten Beobachtungen bezüglich der Entstehung von Krebs durch exogene
Auslöser wurden im 18. Jahrhundert gemacht. Die Kausalität zwischen äußeren Faktoren und dem Auftreten von Malignomen wurde von John Hill bei Nasenpolypen durch Genuß von Schnupftabak und von Pervical Pott zwischen Hodenkarzinomen und der Exposition von Russpartikeln bei Schornsteinfegern erkannt. Ende des 19. Jahrhunderts berichtete L. Rehn über das Auftreten von Blasenkarzinomen bei der Exposition von aromatischen Aminen. Die ersten Grundzüge der viralen Karzinogenese wurden Anfang des letzten Jahrhunderts erkannt. Ellermann beschrieb die Übertragung einer Leukämie bei Hühnern (Ellermann et Bang, 1908). 1911 beschrieb Rous ein unbekanntes Agens, welches Tumore bei Hühnern induzieren konnte. Aus einem Sarkom von Hühnern wurde ein zellfreier Extrakt hergestellt und gesunden Hühnern injiziert. Nach wenigen Wochen traten Tumore mit dem gleichen Phänotyp auf (Rous, 1911). Peyton Rous erhielt 1966 für die Entdeckung eines tumorinduzierenden Virus den Nobelpreis für Medizin. Im zurückliegenden 20. Jahrhundert konnte mit Hilfe biochemischer und molekularbiologischer Techniken die Karzinogenese auf zellulärer Ebene untersucht werden.
A.2 Primäre Knochentumore Knochentumore unterteilt man in primäre, im Knochen entstandene Tumore, und in
sekundäre Tumore, die Metastasen eines anderen Primärtumors sind. Primäre Knochentumore werden nach den Ursprungszellen benannt und entstehen zumeist aus Osteozyten, Chondrozyten und Fibroblasten. Der Ursprung des Ewingsarkoms ist nicht völlig geklärt, wahrscheinlich entsteht es aus neuroektodermalem Gewebe (Tab. 1).
Osteozyt: Benigne: Osteoidosteom Osteoblastom Intermediate grade: Osteoklastom Maligne: Osteosarkom Chondrozyt: Benigne: Osteochondrom Enchondrom Chondrom Chondroblastom Chondromyxoidfibrom Maligne: Chondrosarkom
5

A. Einleitung
Fibrozyt: Benigne: Benignes fibröses Histiozytom Desmoplastisches Fibrom Maligne: Fibrosarkom Malignes fibröses Histiozytom Neuroektodermal: Maligne: Ewingsarkom Tabelle 1: Überblick über die häufigsten primären Knochentumore
Als Beispiele für benigne Knochentumore seien für Osteozyten das Osteoidosteom, für Chondrozyten das Osteochondrom und für Fibrozyten das Fibromyxom genannt. Der Hauptvertreter der bösartigen Geschwülste aus den Osteozyten ist das Osteosarkom, welches am häufigsten zwischen dem 15. und 20 Lebensjahr vorkommt. Das Chondrosarkom ist der zweithäufigste maligne primäre Knochentumor und betrifft vorwiegend das Erwachsenenalter. Das Ewingsarkom ist der dritthäufigste maligne Knochentumor, betroffen sind vor allem Kinder bis zum 15. Lebensjahr. Zwei Drittel der primären Knochentumore befallen Jugendliche, besonders zur Pubertät. Dabei scheint die Wachstumspotenz der Metaphyse eine Rolle zu spielen, da vor allem die wachstumsintensiven Metaphysen der langen Röhrenknochen die Hauptlokalisation sind. Bezogen auf das Osteosarkom ist zu einem hohen Prozentsatz die Kniegelenksregion, also die distale Femur- und proximale Tibiametaphyse betroffen (Mirra et Gold, 1987).
Primäre, maligne Knochentumore wachsen rasch invasiv, infiltrativ und es findet frühzeitig eine Metastasierung statt. Das Osteosarkom ist mit einer Inzidenz von 2-3 Fällen/106 Einwohner pro Jahr der häufigste solide maligne Knochentumor (Campanacci, 1990). Die Altersprädilektion dieses Tumors liegt in der zweiten Lebensdekade, nach den letzten Daten ist eine Tendenz zu einem höheren Erkrankungsalter zu erkennen (Stark et al., 1990; Hefti et Jundt, 1995). Es liegt eine geringe Knabenwendigkeit mit 1,5:1 vor.
Als mögliche Ursachen für die Entstehung eines Osteosarkoms sind bisher genetische Faktoren wie Veränderungen des Retinoblastom-Gens und des Tumorsuppressor-Gens p53 bekannt (Araki et al., 1991; Hansen et al., 1991; Nishida et al., 1994).
Die ersten Symptome sind in der Regel unspezifische, anfangs belastungsabhängige Schmerzen. In der Folgezeit treten Ruheschmerzen auf, und abhängig von der Lokalisation und Weichteildeckung ist eine Vorwölbung zu tasten. Nach 4-5 Wochen nehmen die Schmerzen deutlich zu. Der Zeitraum von 6 Wochen zwischen ersten Symptomen und der Diagnosestellung ist bei Osteosarkom-Patienten im Vergleich zu Patienten mit anderen Knochentumoren deutlich kürzer (Grimer et Sneath, 1990). Klinisch findet man häufig eine schmerzhafte Schwellung oder pergamentartig verdünnte Haut im Bereich der langen
6

A. Einleitung
Röhrenknochen. Bei der Labordiagnostik ist eine Erhöhung der alkalischen Phosphatase auffällig.
Das Nativröntgenbild in zwei Ebenen zeigt das typische Bild eines hochmalignen Tumors mit unscharfer Auflösung der Knochenstruktur, einer Unterbrechung der Kortikalis und einer Abhebung des Periostes. Aufgrund der inhomogenen Tumorstruktur lösen sich osteolytische und sklerotische Anteile ab. Durch das rasche Wachstum kommt es zu einer zwiebelschalenartigen Abhebung des Periostes und bei einer Unterbrechung zu sogenannten Codman-Dreiecken.
Abbildung 1: Seitliches Röntgenbild eines Kniegelenkes. Osteosarkom des distalen Femurs. Unscharfe Begrenzung des Tumors, permeative Destruktion, Unterbrechung der Kompakta/Kortikalis, keine Sklerosierung um den Tumor erkennbar, Codman-Dreieck dorsalseitig im proximalen Tumorbereich.
Zur Diagnostik wird neben der konventionellen Röntgenaufnahme die
Computertomographie (CT) und die Magnetresonanztomographie (MRT) eingesetzt. Mit der CT kann die Integrität der Kortikalis beurteilt werden. Mit Hilfe der MRT kann das Größenausmaß und die Infiltration in die umliegenden Weichteilstrukturen abgeschätzt werden. Entscheidend für die spätere operative Versorgung ist die Lagebeziehung des Tumors zu Nerven, Gefäßen und Muskelkompartimenten. Die Probeentnahme von repräsentativem und vitalem Tumorgewebe und die histologische Charakterisierung sind für das therapeutische Konzept entscheidend.
7

A. Einleitung
Beim Osteosarkom handelt es sich um eine frühzeitige systemische Erkrankung. Zum Zeitpunkt der Diagnose kann bei 20 % der Patienten eine überwiegend pulmonale Metastasierung nachgewiesen werden. Mikrometastasen müssen bei 80 % der Patienten angenommen werden (Saeter et al., 1995).
Nach der histopathologischen Beurteilung, dem Staging und der Einteilung nach Enneking (Enneking et al., 1980) wird ein interdisziplinäres Therapiekonzept erstellt. Ein bewährtes Konzept ist das COSS-Protokoll (Cooperative Osteosarcoma Study) (Winkler et al., 1993). Im COSS-Protokoll von 1996 wird die Chemotherapie an entsprechende Risikogruppen adaptiert. Zunächst wird gemäß dem COSS-96-Schema eine neoadjuvante Chemotherapie über 3 Monate mit Adriamycin (90 mg/m2), Methotrexat (12g/m2, bei Alter > 25 (8g/m2) und Ifosfamid (3g/m2) oder Cisplatin (120 mg/m2) durchgeführt. Bei Hochrisikopatienten wird zusätzlich Etoposid (150mg/m2) postoperativ eingesetzt. Präoperativ werden 3 Zyklen und postoperativ 9 Zyklen durchgeführt. Das gesamte Therapiekonzept umfasst einen Zeitraum von 12 Monaten. Zeigt während der Chemotherapie die Verlaufskontrolle mittels MRT ein weiteres Wachstum, muß die systemische Therapie abgebrochen und die operative Therapie durchgeführt werden.
Die operative Therapie wurde in den letzten 30 Jahren deutlich verändert. In der Anfangszeit war die Amputation das operative Standardverfahren für maligne Knochentumore. Mit dem Einsatz der modernen bildgebenden Verfahren und der neoadjuvanten Therapieverfahren konnte das operative Vorgehen deutlich verbessert werden. Die operative Therapie bei malignen Knochentumoren wird im Sinne einer weiten bzw. einer radikalen Resektion durchgeführt. Die radikale Resektion mit der Entfernung des betroffenen Kompartimentes ist bei Befall von Extremitäten meist nur als Amputation möglich. Mit der Entwicklung der neoadjuvanten Therapie konnte in der Folgezeit nachgewiesen werden, daß bei extremitätenerhaltendem Vorgehen die Überlebenswahrscheinlichkeit nicht verschlechtert wird (Gebhardt et al., 1989). Damit wurden zunehmend rekonstruktive Maßnahmen mit der Verwendung von Tumorspezialprothesen zur Extremitätenerhaltung favorisiert. Die zytostatische Therapie wird mit Abschluß der Wundheilung nach dem COSS-96-Protokoll für weitere 9 Monate fortgesetzt.
Zu Beginn der neuen Therapieansätze mit dem COSS-77-Protokoll betrug die Überlebenschance nach 6 Jahren für das Osteosarkom 14 %. Mit dem von europäischen Tumorzentren entwickelten Therapiestandard ist der Prozentsatz auf 72 % gestiegen (Goorin et al., 1991; Marsden et al., 1991).
Die modernen Therapiekonzepte erzielen zunehmend eine gute Tumorkontrolle und Heilungsrate. Beim Einsatz von Chemotherapeutika muß jedoch vor allem bei Kindern und Jugendlichen auf die langfristigen Folgestörungen geachtet werden. So sind die kumulative Kardiotoxizität von Anthrazenen, die pulmonale Toxizität von Bleomycin und die renale
8

A. Einleitung
Toxizität von Cisplatin oft für die Nebenwirkungen einer Chemotherapie verantwortlich (Neglia et Nesbit, 1993; Morgan et Haugen, 1997).
A.3 Transformation einer normalen Zelle in eine Tumorzelle Die Entstehung einer Tumorzelle ist eine schrittweise Entwicklung, und diese
Schritte beinhalten genetische Veränderungen, die eine fortschreitende Transformation einer normalen in eine hoch maligne Zelle bewirken. Die Tumorzellen weisen verschiedene Veränderungen in den regulativen Kreisläufen auf, die Zellproliferation und Homöostase regulieren. Im folgenden werden die einzelnen Schritte aufgeführt.
A.4 Autonome Versorgung von Wachstumssignalen Normale Zellen benötigen für einen Wechsel von einem Ruhezustand in einen
proliferativen Zustand Wachstumssignale. Die Signale bestehen aus löslichen Wachstumsfaktoren, aus Kontakten zu extrazellulären Matrixkomponenten oder aus Zell-zu-Zell-Kontakten und werden über transmembrane Rezeptoren in die Zelle übermittelt (Weiss et Schlessinger, 1998; Giancotti et Ruoslahti, 1999; Christofori et Semb, 1999). Bei Tumorzellen findet man Veränderungen in diesem Regulationsmechanismus. Tumorzellen produzieren selbst Wachstumssignale und sind damit im Vergleich zu normalen Zellen von exogenen Signalen unabhängig. Diese Veränderungen können auf der Ebene der Wachstumsfaktoren, der Rezeptoren und der intrazellulären Signaltransduktionswege stattfinden.
Die meisten löslichen Wachstumsfaktoren werden von einem Zelltyp produziert, um einen anderen Zelltyp zu stimulieren. Tumorzellen erlangen die Fähigkeit, benötigte Wachstumsfaktoren selbst zu synthetisieren und damit eine autokrine Stimulation zu erreichen. Als Beispiele seien die Produktion von PDGF bei Glioblastomen und von TGFα bei Sarkomen genannt (Fedi et al., 1997). Durch Veränderungen auf der Ebene der Zelloberflächenrezeptoren kann die Proliferation der Tumorzellen ebenfalls gesteigert werden. Durch eine höhere Anzahl der jeweiligen Rezeptoren wird die Tumorzelle schon durch geringe Konzentrationen von Wachstumsfaktoren stimuliert. In den Gewebeproben von Tumoren des Magens und des Gehirns wurde eine Überexpression des EGF-Rezeptors, bei Magen- und Brustdrüsenkarzinomen eine Überexpression des HER-2-Rezeptors nachgewiesen (Yarden et Ullrich, 1988; Slamon et al., 1987). Bei Osteosarkompatienten konnte eine Korrelation zwischen der Expression von HER-2 und dem Ansprechen auf die Chemotherapie bzw. der Überlebensrate festgestellt werden (Gorlick et al., 1999). Ebenfalls in Osteosarkomgewebe konnte eine erhöhte Anzahl des Met-Rezeptors, einer Rezeptortyrosinkinase, nachgewiesen werden (Ferracini et al., 1995; Scotlandi et al., 1998).
Neben den bisher genannten Rezeptortyrosinkinasen haben auch die Integrine als extrazelluläre Matrixrezeptoren Einfluß auf die Profileration, die Zellform und die Mobilität
9

A. Einleitung
einer Zelle. Integrine, eine Familie von heterodimerischen Membranproteinen, binden an extrazelluläre Matrixproteine (Ruoslahti et Öbrink, 1996; Giancotti et Ruoslahti, 1999). So wurde gezeigt, daß durch die Bindung von Fibronektin, einem nichtkollagenen Matrixprotein, an den α5β1 Integrinrezeptor die Differenzierung von Osteoblasten und die Mineralisierung von Kollagen gesteuert wird (Moursi et al., 1997). Verlieren normale Zellen den Kontakt zur umgebenden Matrix, gehen sie in die Apoptose, was die Absiedelung einer normalen Zelle verhindert. Tumorzellen haben diese Kontrollfunktion der Integrinrezeptoren ausgeschaltet, sie können ohne Kontakt zu der ursprünglichen extrazellulären Matrix proliferieren, womit sie die Primärlokalisation verlassen können und Metastasen bilden (Frisch et Francis, 1994, Plath et al., 2000).
Nach Aktivierung der Wachstumsrezeptoren und der Integrinrezeptoren wird das Signal über intrazelluläre Signaltransduktionswege bis in den Zellkern weitergeleitet (Cobb et al., 1994; Schaeffer et Weber, 1999; Renshaw et al., 1997). Punktmutationen von Bestandteilen der Signaltransduktionswege können konstitutiv aktive Formen erzeugen und eine erworbene Autonomie von Wachstumsfaktoren bewirken. In einer Vielzahl von Tumoren und Tumorzellinien wird eine Mutation von Ras beschrieben. Ras aktiviert als Schlüsselprotein eine Vielzahl von nachfolgenden Signaltransduktionswegen und verursacht ein gesteigertes Tumorwachstum (Webb et al., 1998). In nahezu der Hälfte aller Proben von Kolonkarzinomen wurde eine Ras-Mutation nachgewiesen (Kinzler et Vogelstein, 1996).
Neben der bisher beschriebenen autonomen Stimulation können Tumorzellen die umgebenden Stromazellen zur Synthese von Wachstumsfaktoren induzieren (Skobe et Fusenig, 1998; Olumi et al., 1999). Zusätzlich wird in den letzten Jahren eine Unterstützung durch Entzündungszellen beobachtet (Cordon-Cardo et Prives, 1999; Coussens et al., 1999).
Durch die autonome oder heterotope Stimulation der Tumorzellen entfällt ein wichtiger Kontrollmechanismus, der bei normalen Zellen das Wachstum bei verschiedenen Zelltypen in einem Gewebe reguliert.
A.5 Tumorzellen sind gegen wachstumshemmende Signale resistent Die Zellzahl in normalem Gewebe wird neben dem Einfluß von
Wachstumshormonen auch durch inhibitorische Signale gesteuert. Dabei kann mit zwei verschiedenen Mechanismen die Proliferation gestoppt werden. Die Zelle wird von einem aktiven Zellzyklus in den G1/0-Status versetzt oder die Zelle verliert ihr proliferatives Potential und differenziert zu einer speziellen Gewebszelle. Tumorzellen müssen für ein ungebremstes Wachstum diese Inhibition ausschalten.
Die Kontrolle des Zellzyklus durch inhibitorische Signale wird vor allem während der G1-Phase ausgeführt. Auf molekularer Ebene münden inhibitorische Signale am RB-Protein (Retinoblastoma), der den E2F-Transkriptionsfaktoren bindet. Dadurch wird die Expression einer Vielzahl von Genen blockiert, die für den Zellzyklus wichtig sind (Weinberg, 1995). Durch Phosphorylierung des RB-Proteins wird E2F freigegeben, es
10

A. Einleitung
kommt zu einer Proliferation der Zelle. Die Phosphorylierung des RB-Proteins wird durch TGFß verhindert. Die Fähigkeit von TGFß, die Proliferation von malignen epithelialen Tumorzellinien zu inhibieren, wurde zunächst als mögliche Therapie angesehen (Sporn et Roberts, 1989). Es zeigte sich jedoch auch in normalen epithelialen Zellen der Brustdrüse, der Haut und verschiedener innerer Organe eine Inhibition der Proliferation durch TGFß (Lyons et Moses, 1990). Die Funktion des RB-Proteins kann bei Tumorzellen auf verschiedenen Ebenen herabgesetzt werden. Im Vergleich zu den normalen Retinazellen fehlen bei den Zellen eines Retinablastoms die Rezeptoren für TGFß. Durch diesen Mangel kann eine fehlende Regulation des Zellzyklus durch TGFß erklärt werden (Kimichi et al., 1988). Veränderungen der Signalkaskade von dem TGF-Rezeptor bis zu dem Rb-Protein können ebenfalls zu einer Phosphorylierung des RB-Proteins führen. Durch eine chronische Hyperphosphorylierung des RB-Proteins beobachtet man eine Konversion von normalen, wachstumsfaktor-abhängigen Melanozyten zu unkontrolliert wachsenden Melanomazellen (Halaban, 1999).
Die Proliferation eines Zellverbandes kann neben der Regulation des Zellzyklus auch durch die Ausbildung von postmitotischen, differenzierten Zellen kontrolliert werden. Während einer normalen Zellentwicklung wird der Transkriptionsfaktor Myc, welcher eine wachstumsstimulierende Wirkung besitzt, durch die hemmenden Transkriptionsfaktoren Mad verdrängt. Es resultiert eine Differenzierung der Zelle (Foley et Eisenmann, 1999). Tumorzellen benutzen verschiedene Strategien, diese terminale Differenzierung zu vermeiden. Das c-myc-Onkogen hebt durch die Überexpression von Myc die Wirkung von Mad auf, die Differenzierung wird geschwächt und das Wachstum der Zellen wird gefördert (Kraehn et al., 2001).
A.6 Tumorzellen durchlaufen keine Apoptose Das Wachstum eines Tumors wird neben der Proliferation auch durch den
fortlaufenden Untergang von Zellen bestimmt. Für diese Reduktion der Zellzahl ist die Apoptose, der programmierte Zelltod, die Hauptursache. Resistenz gegen die Apoptose wurde experimentell in Zellinien und in Biopsien von Tumoren nachgewiesen.
Der programmierte Zelltod kann mit Sensoren und Effektoren in zwei Regelkreise unterteilt werden. Sensoren überwachen die extrazelluläre Umgebung und intrazelluläre Abläufe wie DNA-Synthese, die nach Aktivierung ein Signal an die Effektor-Komponenten weiterleiten. Die Aktivierung der Effektoren führt über determinierte Schritte zum Tod der Zelle. Die Sensoren bestehen zum einen aus Oberflächenrezeptoren, an die Überlebens- oder Apoptosesignale anbinden. Als Beispiel für Überlebensfaktoren seien die IGF-Rezeptoren genannt. Durch Zugabe von IGF-1 wurde bei hämatopoetischen Zellen eine Inhibition der Apoptose beobachtet (Rodriguez-Tarduchy et al., 1992; Butt et al., 1999). Ein Apoptosesignal wird zum Beispiel durch Anbindung von TNFα an TNF-R1 übermittelt (Ashkenazi et Dixit, 1999). Intrazelluläre Sensoren überwachen Abläufe auf Zellebene und
11

A. Einleitung
initiieren bei Veränderungen die Apoptose. p53 wird als intrazellulärer Sensor durch Schäden der DNA aktiviert, es tritt der programmierte Zelltod ein. Wird die Osteosarkomzellinie U-2 OS mit UV-Licht bestrahlt, wird durch DNA-Schäden eine p53 induzierte Apoptose beobachtet (Yonish-Rouach et al., 1993; Allan et Fried, 1999). Neben den bisher erwähnten Sensoren wird die Lebensdauer auch durch Adhäsionsmoleküle wie Integrine beeinflußt. Verliert eine Zelle den Kontakt zur umgebenden extrazellulären Matrix, wird bei normalen Zellen ebenfalls die Apoptose eingeleitet (Frisch et Francis, 1994).
Die proapoptotischen Signale aktivieren Effektoren, eine Freisetzung von Cytochrom C von den Mitochondrien wird bewirkt (Green and Reed, 1998). Durch Cytochrom C werden die Kaspasen-8 und -9 aktiviert. Durch die Aktivierung von Kaspase-8 und –9 werden kaskadenartig weitere Kaspasen aktiviert. Kaspasen sind Proteasen, die im letzten Schritt durch selektive Zerstörung von subzellulären Strukturen und des Genoms den Zelltod bewirken (Thornberry and Lazebnik, 1998).
Tumorzellen können durch unterschiedliche Strategien Apoptose umgehen. Apoptose kann durch eine Überexpression von antiapoptotischen Signalen oder durch einen Funktionsverlust von regulativen Bereichen gehemmt werden. Die Synthese eines Apoptosehemmers konnte bei bestimmten Leukämieformen nachgewiesen werden. Bei einem hohen Prozentsatz der follikulären Lymphome wird in den malignen Zellen eine chromosomale Translokation nachgewiesen. Durch diese Translokation wird das Onkogen bcl-2 hochreguliert und es wird vermehrt das bcl-2 Protein gebildet, welches Apoptose von Leukämiezellen hemmt (Tsujimoto et al., 1985; Vaux et al., 1988). In der Folgezeit wurde gezeigt, daß durch die Expression des bcl-2-Gens in Anwesenheit von dem Onkogen myc bei transgenen Mäusen ein Lymphom entsteht. Kennzeichen der Lymphomzellen ist eine verlängerte Überlebenszeit, die proliferierende Wirkung von myc wird nicht beobachtet (Strasser et al., 1990).
A.7 Grenzenloses replikatives Potential von Tumorzellen Mit den bisher beschriebenen Strategien der Autostimulation, der Umgehung von
Wachstumshemmung und Apoptose können Tumorzellen im Vergleich zu normalen Zellen schneller wachsen. Bei der Analyse der Chromosomen vor und nach der Zellteilung konnte gezeigt werden, daß die Enden der Chromosomen nach jeder Teilung verkürzt sind. Die Enden der Chromosomen werden von Telomeren, bestehend aus tausenden von Wiederholungen einer 6-Basen-Sequenz, gebildet. Bei der Verdoppelung der Chromosomen wird die DNA von der Polymerase repliziert. Da die Polymerase an den Enden der Chromosomen befestigt ist, kann dieser Abschnitt nicht kopiert werden. Es gehen die letzten 50-100 bp der Chromosomen verloren. Mit dem Verlust der Telomeren entstehen End-zu-End-Fusionen und der Zelltod tritt ein (Counter et al., 1992). Bei maligne entarteten Zellen ist ein Abbau der Telomere nicht zu erkennen, der Verbrauch der Telomeresequenzen wird
12

A. Einleitung
ersetzt. Das Enzym Telomerase, welches an die Telomere die Hexanukleotide-Wiederholungen TTAGGG anfügt, wird bei nahezu 90% aller Tumorzellenarten gesteigert synthetisiert (Bryan et Cech, 1999). Die Telomerase ermöglicht damit eine unbegrenzte Zellteilung. Die Bedeutung der Telomerase konnte durch die Expression in normalen Zellen bestätigt werden. Die telomerase-positiven Zellen weisen eine verlängerte Lebenszeit auf. Bei länger passagierten Zellinien können die durch den Telomereabbau eintretenden Veränderungen aufgehoben werden (Bodnar et al., 1998; Halvorsen et al., 1999).
Neben der Synthese der Telomerase wurde ein weiterer Mechanismus, die Telomerestruktur zu erhalten, beschrieben. Der genaue Mechanismus konnte jedoch noch nicht genauer dargestellt werden. Bei einem Bruchteil der Tumorzellen, die telomerase-negativ sind, werden lange, stabile Telomere nachgewiesen (Bryan et al., 1997; Perrem et al., 1999). Durch die Erhaltung der Telomerstruktur durch die Telomerase erhalten Tumorzellen eine wichtige Fähigkeit zur grenzenlosen Replikation.
A.8 Neoangiogenese bei Tumoren Zellen benötigen für den Stoffwechsel Sauerstoff und Nährstoffe, die über das
Blutgefäßsystem transportiert werden. Für eine ausreichende Versorgung beträgt die maximale Distanz zwischen einer Zelle und einem Kapillargefäß etwa 100-150 µm, bei einer abnehmenden Sauerstoffkonzentration beobachtet man eine Reduzierung der Zellproliferation (Tannock, 1968; Bedford et Mitchell, 1974; Bouck et al., 1996). Das Wachstum eines Zellverbandes setzt damit eine Neubildung des Gefäßsystems voraus. Bereits vor 50 Jahren wurde eine solche Gefäßneubildung bei Tumoren beobachtet (Ide et al., 1939; Algire et al., 1945). In einem Tierexperiment konnte die Bedeutung der Neoangiogenese bei dem Wachstum eines Tumors bewiesen werden. Tumore, die im Glaskörper eines Kaninchenauges wachsen, haben für 100 Tage eine unveränderte Grösse von 0,5 mm. Erreicht ein Tumor jedoch die Oberfläche der Retina, entsteht eine Neovaskularisation und der Tumor vergrössert sich innerhalb von 14 Tagen im Vergleich zu der Ausgangsgrösse um das 19 000 fache (Brem et al., 1976). Ein direkter Beweis für die Hypothese, daß eine Neovaskularisation für das Tumorwachstum notwendig ist, konnte durch die Suppression des vascular endothelial growth factor (VEGF) gezeigt werden. Mäusen wurde subkutan eine Tumorzellinie eingesetzt, die VEGF als einzigen Mediator für die Neoangiogenese produziert. Mit der Blockade von VEGF durch Antikörper wurde das Tumorwachstum um mehr als 90 Prozent reduziert (Kim et al., 1993).
Die Angiogenese wird durch verschiedene Wachstumsfaktoren, deren Oberflächenrezeptoren oder durch Adhäsionsmoleküle reguliert. VEGF, als löslicher Wachstumsfaktor, bindet an den von Endothelzellen präsentierten spezifischen Rezeptor, eine transmembrane Tyrosinkinase, und stimuliert die Angiogenese. Neben VEGF wird auch durch die Anbindung des fibroblast growth factor (FGF) an den FGF-Rezeptor eine gesteigerte Angiogenese beobachtet (Compagni et al., 2000; Saaristo, 2000). Als Beispiele
13

A. Einleitung
für Inhibitoren seien Thrombospondin-1 und Angiostatin genannt. Durch die Anbindung von Thrombospondin-1 an den transmembranen CD36-Rezeptor wird die Proliferation von Endothelzellen inhibiert (Lawler, 2000). Angiostatin wird durch Aufspaltung von Plasmin freigesetzt und hemmt ebenfalls proliferierende Endothelzellen. Die Hemmung der Neoangiogenese durch Angiostatin bewirkt im Tiermodell eine Hemmung des Tumorwachstums (Bergers et al., 1999).
A.9 Invasion und Metastasierung von Tumorzellen Patienten mit einem Osteosarkom sterben in der Regel nicht an ihrem Primärtumor,
sondern an den hämatogen verbreiteten Metastasen in der Lunge. Betrachtet man das gesamte Krankengut der Tumorpatienten, ist die Metastasenbildung die Ursache für 90 % aller Todesfälle (Sporn, 1996). Der Übergang von lokalem Tumorwachstum zu einer metastatischen Erkrankung ist durch die Fähigkeit der Tumorzellen definiert, am Primärort Gewebsgrenzen zu überwinden und in das umgebende Gewebe einzudringen
Abbildung 2: (Quigley and Armstrong, 1998) Neben der veränderten Adhäsion ist die Fähigkeit, umliegende Strukturen aufzulösen
der zweite bestimmende Faktor für eine erfolgreiche Invasion und Metastasierung.
14

A. Einleitung
A.10 Proteolyse der extrazellulären Matrix Das infiltrative Wachstum und die Überwindung von Organgrenzen korreliert mit
der Agressivität eines Tumors. Durch zyklische Anbindung und Ablösung von der extrazellulären Matrix und durch Migration dringt ein Tumor in anliegende Kompartimente ein. Mit dem Einsatz von proteolytischen Enzymen kann eine Tumorzelle zielgerichtet Strukturen der extrazellulären Matrix auflösen. Das Auflösen von extrazellulären Strukturen ist auch bei physiologischen Umbauvorgängen zu beobachten. Die Fortbewegung von Entzündungszellen zu einem Infektionsherd hin, die Implantation eines Trophoblasten in die Uterusschleimhaut und die Ausbildung von Blutgefäßen benötigt eine zielgerichtete Proteolyse.
Mit der Synthese und der Präsentation von proteolytischen Enzymen lösen Tumorzellen die umgebende Matrix auf und infiltrieren in anliegende Strukturen. Die Bestandteile der extrazellulären Matrix sind verschiedene Kollagentypen wie Fibrin, Laminin, Fibronektin und Vitronektin (Basbaum et Werb, 1996). Die Proteasen werden in 5 unterschiedliche Gruppen unterteilt:
• Aspartylproteasen: Kathepsin D und Pepsin • Caspasen: Caspase-8, -9 als Schlüsselenzyme der Apoptose • Cysteinproteasen: Kathepsin B, H und L sowie Caspasen und Papain • Metalloproteasen: Matrixmetalloproteasen • Serinproteasen: uPA, tPA, Plasmin, Thrombin und Trypsin
Es wurde für vier Klassen der Proteasen, nämlich die Serin- (Reuning et al., 1998),
Cystein- (Thomsson et al., 1995), Aspartyl- (Rochefort et al.,1990) und Metalloproteasen (Duffy et McCarthy, 1998) eine Korrelation zwischen der Aggressivität des Tumors und der Konzentration der Proteasen nachgewiesen.
Die Familie der Matrixmetalloproteinasen (MMP) spielt durch die hohe Substratspezifität insbesondere bei der Auflösung von extrazellulären Proteinen und damit bei der Tumorinvasion eine wichtige Rolle (Duffy et McCarthy, 1998). Die Erstbeschreibung einer Metalloprotease erfolgte 1962. Bei dem Umbau der Flossen von Kaulquappen konnte ein Enzym nachgewiesen werden, welches das Kollagen der extrazellulären Substanz auflöst (Gross et Lapiere, 1962). Die meisten Matrixmetalloproteasen wurden erstmals von Tumorzellinien geklont (Coussens et Werb, 1996). Liotta identifizierte bei Untersuchungen über das Invasionsverhalten eine Typ IV-Kollagenase, welche die extrazelluläre Matrix auflöst (Liotta et al., 1980). MMP haben alle eine gleiche Domänenstruktur mit einer katalytischen Region, welche eine zinkbindende Stelle enthält.
15

A. Einleitung
Kollagenasen: Interstitielle Kollagenase MMP-1 Neutrophile Kollagenase MMP-8 Kollagenase-3 MMP-13 Xenopus Kollagenase MMP-18 Gelatinasen: Gelatinase A MMP-2 Gelatinase B MMP-9 Stromelysine: Stromelysin-1 MMP-3 Stromelysin-2 MMP-10 Matrylisin MMP-7 Stromelysin-3 MMP-11 Elastase: Metalloelastase MMP-12 Membran Typ-MMP: MT1-MMP MMP-14 MT2-MMP MMP-15 MT3-MMP MMP-16 MT4-MMP MMP-17 MT5-MMP MMP-21 Unklassifiziert: Enamelysin MMP-20 MMP-19 MMP-23 MMP-24 Tabelle 2: Unterteilung der Matrixmetalloproteinasen
A.11 Bedeutung der Typ-IV Kollagenasen bei der Metastasierung Die lokale Destruktion der Basalmembran ist eine Voraussetzung für das infiltrative
Wachstum bösartiger Tumorzellen. Viele Primärtumore sind von einer intakten Basalmembran umgeben. Die Basalmembran besteht aus kollagenen und nichtkollagenen Glykoproteinbestandteilen. Die wesentliche strukturelle Komponente der kollagenen Bestandteile ist das Typ-IV Kollagen, welches mit einem Anteil von 50 % das Grundgerüst der Basalmembran bildet (Yurchenco et al.,1992).
Bei Osteosarkomen beobachtet man eine frühzeitige Überwindung der umliegenden Organgrenzen wie des Periostes und anliegender Muskelfaszien. Damit können die Tumorzellen infiltrativ in angrenzende Muskelgruppen wachsen.
16

A. Einleitung
Die Migration von Zellen beobachtet man bei physiologischen Prozessen wie z.B. bei dem Austritt von Entzündungszellen aus dem Gefäßsystem, bei der Entwicklung der Plazenta oder bei der Organentwicklung. Das infiltrative Wachstum eines malignen Tumors beruht auf der Migration einzelner Tumorzellen in das umgebende Stroma. Liotta stellte für die Interaktion zwischen einer Tumorzelle und der extrazellären Matrix eine 3-Schritt-Hypothese auf (Liotta et Stetler-Stevenson, 1990):
1. Adhäsion der Tumorzelle an die Matrix 2. Lokale Proteolyse der Matrixproteine durch tumorassoziierte Proteasen 3. Migration der Tumorzelle in die veränderte Matrix
Die Auflösung der Basalmembran und damit von Typ-IV Kollagen ermöglicht eine
Überwindung von Organgrenzen und Gefäßwänden. Unter den Matrixmetalloproteinasen lösen die Typ-IV Kollagenasen spezifisch Typ-IV Kollagen in der helikalen Region auf (Liotta et al., 1979). Sie spalten aber auch Typ-V, -VII, -IX und -X Kollagen sowie Laminin, Fibronektin und Gelatin. Mit der Expression von Typ-IV Kollagenasen wird die Basalmembran als Gewebebarriere aufgelöst und das metastatische Potential eines Tumors charakterisiert (Liotta et al., 1980; Nakajima et al., 1991).
Typ-IV Kollagenasen bestehen aus zwei Enzymen mit 72 kDa und 92 kDa Größe. In der MMP-Familie werden sie als MMP-2 und MMP-9 benannt (Liotta et al., 1981). MMP-2 und -9 werden als proteolytisch inaktive Zymogene sezerniert und durch Abspaltung von aminoterminalen Aminosäuren aktiviert. MMP-2 wird im Unterschied zu den anderen Metalloproteasen an der Zelloberfläche von membrane-type Matrixmetalloproteasen (MT-MMP) aktiviert (Morgunova et al., 1999; Nakamura et al., 1999). Die Aktivität der Typ-IV Kollagenasen wird durch physiologische Inhibitoren, tissue inhibitor of metalloproteinase (TIMP) geblockt. Durch Interaktion mit TIMP-1 wird MMP-9 inhibiert (Wilhelm et al.; 1989), TIMP-2 blockt MMP-2 (Howard et al., 1991). Im Unterschied zu den restlichen Metalloproteasen interagieren MMP-2/-9 bereits als Proenzyme mit TIMP-1/-2 (DeClerck et al., 1989).
Typ-IV Kollagenasen wurden in Gewebeproben und Zellinien von verschiedenen Tumorentitäten nachgewiesen (Mignatti et Rifkin, 1993; Tryggvason et al., 1993). In Mammakarzinom-Gewebe wurden Typ-IV Kollagenasen immunhistochemisch dargestellt, MMP-9 konnte in normalem Mammagewebe nicht detektiert werden (Barsky et al., 1983; Basset et al., 1990). In Gewebeschnitten von Carcinomen der Lunge, des Darmes und der Haut wurden sowohl MMP-2 als auch MMP-9 nachgewiesen (Canete-Solar et al., 1994; Pyke et al., 1992 et 1993). Experimentell konnte gezeigt werden, daß MMP-9 bei der Invasion in die Gefäßwand und damit bei der Intravasation und hämatogenen Metastasierung wichtig ist (Kim et al., 1998). Transfiziert man eine nicht-metastatische Zellinie stabil mit dem MMP-9-Gen, metastasiert sie (Bernhard et al., 1990 et 1994). Durch
17

A. Einleitung
die Inkubation einer Mammakarzinom-Zellinie mit Blutplättchen synthetisieren die Tumorzellen vermehrt MMP-9. Vergleicht man in einem Invasionsassay die stimulierten Zellen mit unstimulierten, zeigt sich, daß die stimulierten Zellen eine Matrigelschicht schneller überwinden (Belloc et al., 1995).
A 12 Regulation des MMP-9-Gens Die Aktivität der Metalloproteasen wird auf unterschiedlichen Ebenen reguliert, was
auf deren Bedeutung bei biologischen Funktionen hinweist. Diese Regulation kann zum einen durch die Expression der Gene und zum anderen in der Kontrolle der proteolytischen Aktivität der Proteinprodukte durch die Inhibierung mit TIMP-1 und –2 stattfinden.
Die transkriptionelle Regulation der Metalloproteasen wird durch Wachstumsfaktoren und Zytokine, durch Hormone und durch die Zellform beeinflußt (Birkedal-Hansen et al., 1993). Durch Zell-Matrix-Interaktionen und durch Bestandteile der extrazellulären Matrix wird die Produktion von Metalloproteasen ebenfalls verändert (Sweeney et al.,1991; Huhtala et al., 1995).
Analysiert man die Transkription von MMP-2 und MMP-9, zeigt sich eine geringe Beeinflussung von Zytokinen oder Wachstumsfaktoren auf MMP-2 (Brown et al., 1990). Die Aktivität von MMP-9 hingegen wird durch Zytokine, Wachstumsfaktoren und Interaktionen von Tumorzellen mit den umliegenden Zellen deutlich verändert. Unter dem Einfluß von TNFα findet sich bei einer Riesenzell-Zellinie eine konzentrationsabhängige Aktivierung von MMP-9 (Rao et al., 1999). Durch den Kontakt mit Stromazellen sezernieren Tumorzellen vermehrt MMP-9. Kultiviert man eine Karzinomzellinie mit einer Fibroblastenzellinie, wird die Promotoraktivität um das 4-5fache erhöht und damit die proteolytische Aktivität des Tumors an der Grenzfläche durch das umgebende Stroma gesteigert (Lengyel et al., 1995). Durch Infektion mit dem Epstein-Barr-Virus, assoziiert mit malignen Erkrankungen wie dem Burkitt- oder dem Hodgkin-Lymphom, wird MMP-9 vermehrt gebildet (Yoshizaki et al., 1998).
Die Transkription des MMP-9-Gens wird durch den Promotor reguliert. Die wichtigsten Transkriptionsstellen sind Bindungsstellen für AP-1, NF-κB und SP-1 (Sato et al., 1993; Gum et al., 1996; Bond et al., 1998; Yoshizaki et al., 1998). Im Unterschied zu den anderen Metalloproteasen, die nur eine AP-1 Bindungsstelle besitzen, sind in der Promotorregion von MMP-9 zwei AP-1-Bindungsstellen angelegt (Sato et Seiki, 1993). Verschiedene Studien haben die Bedeutung der AP-1-Bindungsstelle bei der Regulation von MMP-9 gezeigt (Benbow et Brinkerhoff, 1997).
A.13 Intrazelluläre Signaltransduktionswege Die Funktion von Zellen in einem multizellulären Organismus wird durch eine
Vielzahl von extrazellulären Signalen wie Wachstumsfaktoren, Neurotransmitter oder
18

A. Einleitung
Hormone reguliert. Diese Signale werden über verschiedene Klassen von Plasmamembranrezeptoren in das Zellinnere weitergeleitet. Die Mehrheit dieser Rezeptoren ist der Familie der G-proteingekoppelten Rezeptoren (G-protein coupled receptor: GPCR) zuzuordnen. Die Anbindung eines Liganden bewirkt eine Konformationsänderung des Rezeptorproteins, und das Signal wird in das Zellinnere an G-Proteine weitergeleitet, die an der zytoplasmatischen Oberfläche der Plasmamembran angebunden sind. G-Proteine sind aus einer α-, β- und γ-Einheit aufgebaut, die im Ruhezustand nonkovalent verbunden sind. In dieser Phase ist GDP an die α-Einheit angelagert. Mit der Aktivierung durch einen Membranrezeptor dissoziiert GDP von der α-Einheit und wird durch GTP ersetzt. Die Aktivierung des G-Proteins bewirkt eine Aufspaltung in eine α-Einheit und eine βγ-Einheit. Die dissoziierten Untereinheiten Gαq aktivieren downstream die MAP Kinase oder im Falle von Gßγ die PI 3-Kinase. Nach wenigen Sekunden hydrolisiert die α-Einheit GTP zu GDP und bindet wieder an die βγ-Einheit (Conklin and Bourne, 1993; Neer, 1995). Das Signal wird durch die aktivierten G-Proteine an intrazelluläre Signaltransduktionswege wie die MAPK-Familie oder die Phosphoinositol 3-Kinase (PI3K) übermittelt (Faure et al., 1994; Malarkey et al., 1995; Belisle et Abo, 2000). Durch spezifische Regulatoren des G-Protein Signals (RGS) findet eine Kontrolle der Signaltransduktion statt (Witherow et al., 2000).
Abbildung 3: Signaltransduktion
19

A. Einleitung
Nach Aktivierung durch Rezeptortyrosinkinasen oder G-Protein gekoppelte Rezeptoren übermittelt der MAPK-Signaltransduktionsweg über eine Kaskade von verschiedenen Proteinkinasen Signale bis auf die Promotorebene (Cobb et al., 1994). MAP Kinasen werden in drei Untergruppen aufgeteilt. ERK 1 und ERK 2 (extracellular signal-regulated kinases) werden durch Wachstumsfaktoren wie PDGF (Denhardt, 1996), Endothelin (Wang et al., 1992) oder Thrombin (Kyriakis et al.,1993) aktiviert. Nach Rezeptoraktivierung wird ein Adapterprotein (Grb2) an den zytoplasmatischen Abschnitt gebunden und anschließend mit einem sogenannten Austausch-Protein (Sos) verbunden (Egan et al., 1993). Dieser Komplex überführt Ras-GDP in die aktive GTP-Form. Aktives Ras rekrutiert Raf-Kinasen (Raf-1, A-Raf, B-Raf) (Zhang et al., 1993). Raf wiederum phosphoryliert MEK1 (Papin et al., 1995), welches mit einer hohen Substratspezifität ERK1 und ERK2 aktiviert. Die ERK-Proteine gelangen in den Zellkern und regen unterschiedliche Genpromotoren an (Gonzalez et al., 1993). Die zwei anderen MAPK-Kaskaden Jun-N-terminale Kinase und p38 Kinase werden durch osmotischen Stress beeinflusst (Ip et Davis, 1998, Lewis et al., 1998).
Es konnte nachgewiesen werden, daß Bestandteile des MAPK-Weges bei der Metastasierung von Bedeutung sind (Webb et al., 1998; Yoshioka et al., 1999). Mit der Aktivierung des MAPK-Weges wurde die Aktivität von MMP-9 und daraus resultierend die Migration erhöht (McCawley et al., 1999).
Neben der Aktivierung des MAPK-Signaltransduktionsweges wird die Phosphoinositol 3-Kinase (PI3K) durch GPCR aktiviert. Die Phosphoinositol 3-Kinase wurde erstmals 1985 beschrieben (Whitman et al., 1985). Die Aktivierung der PI3K bewirkt Veränderungen des Zellzyklus und der Zellform entsprechend einer malignen Transformation von Zellen (Klippel et al.1998). Drei Klassen von PI 3-Kinasen werden unterschieden (Domin et Waterfield, 1997). Typ I PI 3-Kinasen können durch Zelloberflächen-Rezeptoren aktiviert werden und phosphorylieren PI 4,5-Diphosphat zu PI 3,4,5-Triphosphat. Sie bestehen aus einer 110 kDa grossen katalytischen Untereinheit und aus einer 85 kDa grossen regulatorischen Untereinheit (Stephens et al., 1991). Die zweite Klasse der PI3K, die Typ IA PI3K, bestehen aus einer katalytischen α, β, oder δ p110 Untereinheit und aus einer regulatorischen p50, p55, oder p85 (α oder β) Untereinheit (Hiles et al., 1992; Pons et al., 1995). Typ IB PI 3-Kinase ist die dritte Klasse, bestehend aus einer katalytischen p110γ Untereinheit und einer regulatorischen p101 Untereinheit. Die Ib PI3-Kinasen werden von GPCR aktiviert, indem die βγ-Einheit an die p101-Untereinheit der PI 3-Kinase bindet (Krugmann et al., 1999).
A.14 Störungen der Hämostase bei Tumorpatienten Bereits 1865 beschrieb Trousseau Störungen der Blutgerinnung bei Tumorpatienten.
Er beobachtete das gehäufte Auftreten von Phlebitiden infolge von tiefen Venenthrombosen bei Patienten mit einem Pankreaskarzinom (Trousseau, 1864; Rickles et Edwards., 1983). In
20

A. Einleitung
der Folgezeit wurde die gesteigerte Bildung von Thrombosen vor allem bei Patienten mit einem Malignom der Lunge, des Pankreas oder des Gastrointestinaltraktes nachgewiesen (Morrison, 1932; Fisch et al., 1951; Dvorak, 1987). Die Ausbildung einer Lungenembolie als Folge einer tiefen Venenthrombose ist bei Tumorpatienten neben der Grundkrankheit die zweithäufigste Todesursache (Donati, 1994). Im Umkehrschluß sollte bei dem Vorliegen einer tiefen Venenthrombose nach Ausschluß einer Gerinnungsstörung eine Tumorerkrankung als Ursache bedacht werden. So konnte bei 12% der Patienten mit einer tiefen Venenthrombose in der Folgezeit ein Malignom nachgewiesen werden (Cornuz et al., 1996).
Analysiert man die Komponenten der Blutgerinnung bei Tumorpatienten, ist bei 95 % aller Patienten eine Veränderung der Blutgerinnung nachweisbar. Es wird eine deutliche Steigerung der extrinsischen Kaskade und damit der Generierung von Thrombin festgestellt (Rickles et al., 1992; Kakkar A. K. et al., 1995). Als Folge haben Tumorpatienten nach einer Operation im Vergleich zu Patienten ohne Malignom ein zweifach erhöhtes Risiko eine Thrombose zu entwickeln (Kakkar V. V. et al.,1970), und die gesteigerte Blutgerinnung bewirkt bei Tumorpatienten einen erhöhten Umsatz von Fibrinogen und Thrombozyten im Sinne einer Verbrauchskoagulopathie (Yodo et Abe, 1981; Wehmeirer et al., 1991). Fibrinablagerungen als Endprodukt der Blutgerinnung werden histologisch an der Tumor-/Stromagrenze und auf im Blut zirkulierenden Tumorzellen nachgewiesen (Dvorak et al., 1981; Long et al.,1960; Zacharski et al., 1983). Aufgrund der Hyperkoagulopathie sollte deshalb bei Tumorpatienten eine Thromboseprophylaxe mit niedermolekularem Heparin durchgeführt werden (Piccioli et al., 1996).
A.15 Wechselwirkung zwischen Tumorzelle und Blutgerinnung Bereits 1964 wurde das Modell der plasmatischen Gerinnungsfaktoren aufgestellt,
welche als inaktive Vorstufen im Blut vorliegen und deren schrittweise Aktivierung zur Bildung von Fibrin führt (Davie et Ratnoff, 1964; MacFarlane, 1964). Die Blutgerinnung wird in ein intrinsisches und ein extrinsisches System unterteilt. Der intrinsische Weg beginnt mit dem Kontakt von Faktor XII mit negativ geladenen Phospholipiden, wie zum Beispiel Phosphatidylserin, welche auf der Oberfläche von aktivierten Thrombozyten oder beschädigten Zellen zu finden sind. Aktivierter Faktor XII überführt zusammen mit Kininogen Präkallikrein in Kallikrein, welches in einer positiven Rückkoppelung verstärkt Faktor XIIa bildet. Als zweites Substrat aktiviert Faktor XIIa den Faktor XI, welcher nun seinerseits mit Kalzium den Vitamin K-abhängigen Faktor IX aktiviert. Im nächsten Reaktionsschritt müssen neben Faktor IXa noch Kalzium, Phospholipide und Faktor VIIIa vorhanden sein, um Faktor X in Faktor Xa zu überführen (Colman et al., 1987). Bei der Hämophilie A und B handelt es sich um plasmatische Gerinnungsstörungen, welche durch einen X-chromosomal rezessiv vererbten Mangel an Faktor VIII und IX verursacht sind und bereits im 19. Jahrhundert von Otto klinisch beschrieben wurden. Faktor Xa bildet mit
21
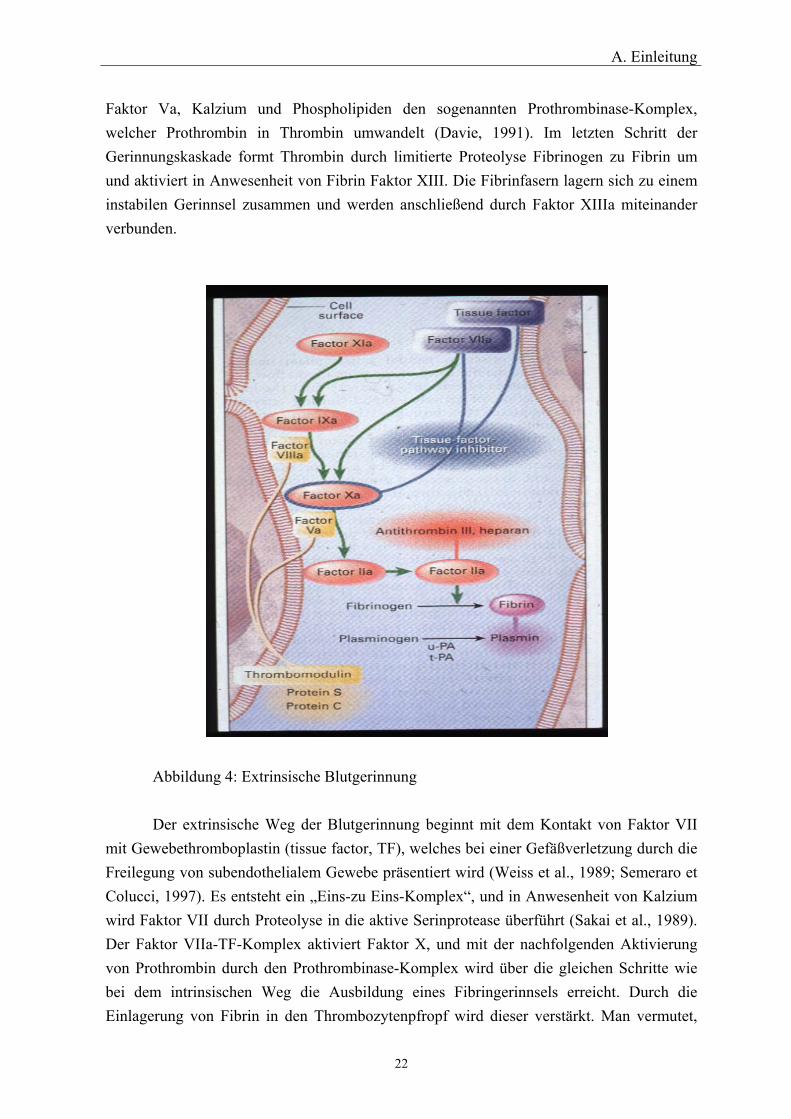
A. Einleitung
Faktor Va, Kalzium und Phospholipiden den sogenannten Prothrombinase-Komplex, welcher Prothrombin in Thrombin umwandelt (Davie, 1991). Im letzten Schritt der Gerinnungskaskade formt Thrombin durch limitierte Proteolyse Fibrinogen zu Fibrin um und aktiviert in Anwesenheit von Fibrin Faktor XIII. Die Fibrinfasern lagern sich zu einem instabilen Gerinnsel zusammen und werden anschließend durch Faktor XIIIa miteinander verbunden.
Abbildung 4: Extrinsische Blutgerinnung Der extrinsische Weg der Blutgerinnung beginnt mit dem Kontakt von Faktor VII
mit Gewebethromboplastin (tissue factor, TF), welches bei einer Gefäßverletzung durch die Freilegung von subendothelialem Gewebe präsentiert wird (Weiss et al., 1989; Semeraro et Colucci, 1997). Es entsteht ein „Eins-zu Eins-Komplex“, und in Anwesenheit von Kalzium wird Faktor VII durch Proteolyse in die aktive Serinprotease überführt (Sakai et al., 1989). Der Faktor VIIa-TF-Komplex aktiviert Faktor X, und mit der nachfolgenden Aktivierung von Prothrombin durch den Prothrombinase-Komplex wird über die gleichen Schritte wie bei dem intrinsischen Weg die Ausbildung eines Fibringerinnsels erreicht. Durch die Einlagerung von Fibrin in den Thrombozytenpfropf wird dieser verstärkt. Man vermutet,
22

A. Einleitung
daß der extrinsische Weg für den Beginn und die Instandhaltung der Gerinnungskaskade wichtig ist, wohingegen der intrinsische Weg für den weiteren Aufbau des Fibringerinnsels entscheidend ist (Davie et al., 1991).
Tumorzellen können die Blutgerinnung durch verschiedene Mechanismen aktivieren. Auf dem direkten Weg wird durch Präsentation von TF auf der Oberfläche von Tumorzellen ein Komplex mit Faktor VIIa gebildet und der extrinsische Weg aktiviert (Gordon, 1992; Rao, 1992; Mueller et al., 1992). Indirekt können Tumorzellen durch Sekretion von Zytokinen wie TNF-α oder IL-1ß Endothelzellen oder Monozyten anregen, TF zu produzieren oder Thrombozyten werden aktiviert, welche mit der Oberfläche die Aktivierung von Gerinnungsfaktoren fördern (Pearlstein et al., 1981; Lorenzet et al., 1983; Murray, 1991). Die Anzahl der Thrombozyten ist dabei ein sensitiver Indikator für die Aktivierung der intravaskulären Gerinnung. Werden Fibrosarkomzellen in die venöse Blutstrombahn von Mäusen injiziert, beobachtet man eine Reduktion der Thrombozyten um 50 %. Gleichzeitig sinkt die Aktivität von Faktor Xa und der Spiegel von Fibrinogen. Durch die Gabe von Heparin wird das Bild der disseminierten intravaskulären Gerinnung unterdrückt. Neben der Reduktion der intravaskulären Gerinnung hat die Gabe von Heparin aber auch einen Einfluß auf die Bildung von Lungentumoren durch die Fibrosarkomzellen. In der Gruppe der mit Heparin behandelten Tiere ist eine deutlich geringere Anzahl von Lungentumoren nachzuweisen (Amirkhosravi et al., 1993). In einer klinischen Studie konnte mit dem Einsatz von niedermolekularem Heparin bei Patientinnen mit einem gynäkologischen Malignom eine signifikante Verlängerung der Überlebenszeit festgestellt werden (Tempelhoff et al., 1999).
A.16 Struktur und Funktion von Thrombin und dem spezifischen Thrombinrezeptor
Thrombin entsteht aus der inaktiven Vorstufe Prothrombin, welche in der Leber als Vitamin K-abhängiger Blutgerinnungsfaktor synthetisiert wird (Shapiro et McCord, 1978). Nach Anlagerung an den sogenannten Prothrombinase-Komplex wird durch Spaltung von Prothrombin aktives Thrombin freigesetzt (Mann et al., 1990). Aktives Thrombin ist mit einem Molekulargewicht von 39 kDa annähernd halb so groß wie Prothrombin (71.9 kDa) und besteht aus einer A- und einer B-Kette.
Zusätzlich zu der Fähigkeit, Fibrinogen aufzuspalten weist Thrombin wichtige zellaktivierende Funktionen auf. Thrombin beeinflusst die Zellform von Blutplättchen (Davey et Luscher, 1967), die Proliferation und die Migration von Endothelzellen (Chen et Buchanan, 1975) und Monozyten (Bar-Shavit et al., 1983). 1991 wurde von zwei Arbeitsgruppen der spezifische Thrombinrezeptor kloniert und anschließend in weiteren Zellinien detektiert (Vu et al., 1991; Rasmussen et al., 1991; Zhong et al., 1992; Gerszten et al., 1994). In der Folgezeit konnte bewiesen werden, daß mit der Gabe von Thrombin die
23

A. Einleitung
zellulären Veränderungen durch die Aktivierung des Thrombinrezeptors erreicht werden (Hung et al., 1992a; Negrescu et al., 1995; McNamara et al., 1993).
Der Thrombinrezeptor besteht aus 425 Aminosäuren und weist eine Reihe von strukturellen Eigenschaften der G-Protein gekoppelten Rezeptoren auf. Der siebenspännige Rezeptor hat jeweils drei intra- und drei extrazelluläre Schleifen. Intrazellulär liegt ein C-terminales Ende und extrazellulär eine lange N-terminale Domäne vor (Vu et al., 1991a). Im Unterschied zu den meisten zellulären Wachstumsfaktor-Rezeptoren benötigt der Thrombinrezeptor für die Aktivierung keine Ausbildung eines Liganden-Rezeptor-Komplexes. Der Rezeptor dient als Substrat für eine proteolytische Abspaltung, wodurch eine irreversibel aktivierte Form des Oberflächenproteins entsteht. Thrombin bindet an den Rezeptor und spaltet das carboxyterminale Ende zwischen der Position 41 und 42. Das neue Rezeptorende bindet intramolekular an die zweite extrazelluläre Domäne des Rezeptors und bewirkt eine Konformationsänderung (Vu et al, 1991b, Chen et al., 1994). Deshalb wird der Thrombinrezeptor auch als Protease aktivierter Rezeptor (protease-activated receptor) oder PAR beschrieben.
Abbildung 5: Thrombin-Rezeptor (Grand et al., 1996) Das synthetische Peptid SFLLRN, welches aus den ersten sechs Aminosäuren des
neuen extrazellulären Endes des Rezeptors nach der Aktivierung besteht, kann als Agonist von Thrombin in der Zellkultur eingesetzt werden (Gerszten et al., 1994). Mit diesem sogenannten TRAP (Thrombin Rezeptor aktivierendes Peptid) wird der Einfluß von
24

A. Einleitung
Thrombin über den Thrombinrezeptor nachgewiesen. Durch den Einsatz von TRAP werden verschiedene Zelltypen wie Blutplättchen, Endothelzellen, Fibroblasten im gleichen Ausmaß wie durch Thrombin aktiviert (McNamara et al., 1993; Herbert et al., 1992; Vassallo et al., 1992).
Nach der Aktivierung leitet der Thrombinrezeptor als sogenannter G-Protein gekoppelter Rezeptor Signale über G-Proteine wie Gq oder Gi, Tyrosinkinasen und Ras-verbundene Signalwege in den Zellkern weiter (Hartwig et al., 1995; Trejo et al., 1996).
Die Zugabe von Thrombin hat bei verschiedenen Zelltypen unterschiedliche Wirkungen (Connolly et al., 1994, 1996). Aufgrund dieser Beobachtung und durch selektive Blockade wurden verschiedene Thrombinrezeptoren identifiziert. Neben PAR-1 (Andersen et al., 1999), konnten PAR-2 (Nystedt et al., 1994), PAR-3 (Ishihara et al., 1997) und PAR-4 (Kahn et al., 1998; Xu et al., 1998) nachgewiesen werden. Im Unterschied zu PAR-1,-3 und –4 wird PAR-2 nicht von Thrombin, sondern von Trypsin und Tryptase oder den Blutgerinnungsfaktoren VIIa und Xa aktiviert (Molino et al., 1997; Camerer et al., 2000). PAR-3 scheint als Kofaktor bei der Aktivierung von PAR-4 zu wirken, da eine alleinige Aktivierung von PAR-3 keine transmembrane Signalweiterleitung einleitet (Nakanishi-Matsui et al., 2000). Die Bedeutung von zwei verschiedenen Rezeptorvarianten auf einer Zelle konnte durch unterschiedliche Konzentrationen von Thrombin erklärt werden. Beim Menschen ist PAR 1 für die Antwort bei niedrigen Thrombinkonzentrationen notwendig, PAR 4 leitet das Signal bei hohen Konzentrationen in das Zellinnere weiter (Coughlin, 1999; Shapiro et al., 2000).
A.17 Thrombin und der spezifische Thrombinrezeptor in der Tumorbiologie Nach der Verletzung eines Gefäßes wird die Blutung durch die Aktivierung der
Gerinnungskaskade und durch die Ausbildung eines Thrombus gestoppt. Aktiviertes Thrombin bewirkt in dieser Phase neben der Polymerisierung von Fibrin eine Aktivierung von Blutplättchen, Aggregation und Degranulation der Thrombozyten sind die Folge (Davey et Luscher, 1967). Der Verschluß einer Gefäßverletzung stellt die sogenannte Akutphase der Wundheilung dar. In der nachfolgenden Entzündungsphase wandern Entzündungszellen, Fibroblasten und Endothelzellen in das Granulationsgewebe ein, es wird neues Bindegewebe gebildet. Unter dem Einfluß von Thrombin wird eine Steigerung der Adhäsion, Proliferation und Migration dieser Zellen beobachtet (Bar-Shavit et al., 1983; Chen et Buchanan, 1975; Herbert et al., 1992; McNamara et al., 1993). Dabei ist die enzymatische Aktivität von Thrombin für den Anstieg der Endothelpermeabilität entscheidend (Aschner et al, 1990). Damit spielt Thrombin neben der Blutgerinnung auch in der zweiten Phase der Wundheilung eine zentrale Rolle, es wirkt in diesem Abschnitt als Hormon und Wachstumsfaktor.
Durch die Klonierung des spezifischen Thrombinrezeptors konnte ein Modell für die direkte Wirkung von Thrombin auf Zellen entwickelt werden (Vu et al., 1991; Rasmussen et
25

A. Einleitung
al., 1991). In der Folgezeit wurde der Thrombinrezeptor in verschiedenen Zellinien nachgewiesen (Zhong et al., 1992; Gerszten et al., 1994). Durch Zugabe von TRAP wird selektiv der Thrombinrezeptor aktiviert, treten die gleichen Reaktionen wie nach Gabe von Thrombin auf, ist der Rezeptor nachgewiesen. Der Thrombinrezeptor konnte damit in verschiedenen Tumorzellinien nachgewiesen werden (Fischer et al., 1995; Nierodzik et al., 1995, 1996), wobei sich eine deutlich höhere Expression des Thrombinrezeptors bei den aggressivsten Zellinien zeigte (Even-Ram et al., 1998).
Durch die Präsentation von TF auf der Zelloberfläche kann eine Melanomzellinie die extrinsische Gerinnungskaskade initiieren. Mit der Zugabe der Gerinnungsfaktoren II (Prothrombin), V, VII und X beobachtet man eine gesteigerte Proliferation der Zellen. Diese Steigerung kann durch Zugabe von Hirudin, einem Thrombinhemmer, gehemmt werden (Fischer et al., 1995). Im Tiermodell kann durch die Gabe von Hirudin die Metastasierungsrate einer Melanomzellinie gesenkt werden (Esumi et al., 1991). In einem weiteren Experiment konnte der Einfluß von Thrombin auf die Proliferation einer Zellinie bestätigt werden. Nach Transfektion mit der cDNA des Thrombinrezeptors konnte durch die Zugabe von Thrombin ein deutlicher Anstieg der Proliferation festgestellt werden (Hung et al., 1992). Im Umkehrschluß wird mit Antisense-Sequenzen des Rezeptors in der Zellkultur von Gefäßzellen die mitogene Wirkung von Thrombin unterdrückt (Chaikof et al., 1995).
Das Metastasierungspotential eines Tumors wird neben der Proliferation auch durch Veränderungen der Adhäsion beeinflußt. Wird Thrombin in einer Konzentration, die eine Fibrinbildung ausbildet, zugegeben, werden vermehrt Integrine an der Zelloberfläche präsentiert, eine vermehrte Adhäsion ist zu beobachten (Chiang et Huang, 1996; Wojtukiewicz et al., 1992).
In der Zellkultur konnte in einem Invasionsassay bewiesen werden, daß Tumorzellen nach Inkubation mit Thrombin Gewebebarrieren schneller durchdringen. Wird eine hoch aggressive Brustkrebszellinie mit Thrombin stimuliert, so überwinden die Tumorzellen schneller eine Matrigel-Schicht, wohingegen das Invasionsverhalten einer weniger aggressiven Tumorzellinie durch Thrombin nicht beeinflußt wird (Henrikson et al., 1999).
Neben dem invasiven Wachstum eines Tumors ist die Metastasierungsrate für die Aggressivität bestimmend. Tumorzellen entwickeln nach Zugabe von Thrombin eine vermehrte Adhäsion und Proteolyse, damit wird das metastatische Potential eines Tumors erhöht. Im Tiermodell konnte nach Inkubation von Zellinien eines Kolonkarzinoms, eines Bronchialkarzinoms und eines Melanoms mit Thrombin eine Steigerung der pulmonalen Metastasierung zwischen dem 10-100fachen beobachtet werden (Nierodzik et al., 1992; Wojtukiewicz et al., 1993). Einen ähnlichen Effekt erreicht man, wenn Tumorzellen mit Blutplättchen oder mit von Willebrandfaktor inkubiert werden (Honn et al., 1988; Nierodzik et al., 1995; 1998).
26

B. Fragestellung Die Typ-IV Kollagenase löst die Hauptbestandteile der Basalmembran auf und
ermöglicht damit Tumorzellen infiltrativ und invasiv zu wachsen. Thrombin, welches bei Tumorpatienten vermehrt durch die Hyperkoagulopathie freigesetzt wird, erhöht die Aktivität von Matrixmetalloproteasen.
In diesem Zusammenhang ergaben sich folgende Fragestellungen: 1. Welchen Einfluß hat die Zugabe von Thrombin auf die Aktivität von Typ-IV
Kollagenase? Nachdem gezeigt werden konnte, daß Thrombin MMP-9 aktiviert, ergaben sich
folgende Fragen: 2. Über welchen Rezeptor erfolgt die Aktivierung? 3. Ist die Aktivierung konzentrationsabhängig und nach welcher Zeitdauer ist eine
Steigerung zu beobachten? 4. Wird durch Zugabe von Thrombin das Invasionsverhalten einer Tumorzellinie
verändert? 5. Welcher Signaltransduktionsweg ist für die intrazelluläre Weiterleitung
verantwortlich? 6. Ist der Thrombinrezeptor in Gewebeproben von Knochentumoren nachweisbar?
27

C. Material und Methoden
C.1 Zellinien und Materialien HT 1080 und NIH 3T3 freundlicherweise von Dr. D. Boyd (MD Anderson, Cancer Center Houston, USA) zur Verfügung gestellt. U-2 OS (Ponten J and Saksela E., 1967; ATCC-Nummer HTB-96) freundlicherweise von Dr. M. Atkinson (Institut für Pathologie des GSF-Forschungszentrums für Umwelt und Gesundheit, Neuherberg) zur Verfügung gestellt.
• Gibco BRL Life Technology, Karlsruhe: Fötales Kälberserum, HEPES, DMEM mit
Glutamax, PBS
• Falcon / Becton Dickinson, Plymouth, England: Gewebekulturplatten ∅ 10cm,
Gewebekulturflaschen T175, Gewebekulturflaschen T75, Gewebekulturflaschen
T25, Gewebekultur Sixwell, 96-well-Mikrotiterplatten
• Nunc, Wiesbaden: Gewebekulturplatten (24,5 x 24,5 cm)
• Seromed Biochrom KG, Berlin: BSA, EDTA (1%), Penicillin/Streptomycin, Trypsin
(2,5%)
• Sigma- Aldrich GmbH, Steinheim: L-Arginin, L-Asparagin
C.2 Material • Amersham Pharmacia Biotech, Bedford, USA: Acetyl- Coenzym A, ECL Reagenz,
[α-32P]-dCTP PB 10205 (3000Ci/mmol), MicroSpin G-25 Säulen • Amicon Millipore Corp., Bedford, USA: Centricon Filter • Amresco, Solon, USA: Ammoniumpersulfat (APS) • Becton Dickinson, Bedford, USA: BIOCOAT® Invasion Chamber MATRIGEL®
Basement Membrane Matrix (Lot. Number 902489, 909233), EGF • Biometra Göttingen: Epson GT-9900, ScanPack 3,0 Software • Boehringer Mannheim, Penzberg: Klenow Enzym, Random Primed Labeling Kit,
Trennsäule Nick Column • Calbiochem Bad Soden: LY 294002 (PI3-Kinase), AG 1478 (EGF-Rezeptor-
Tyrosinkinase), PD 98059 (MAP Kinase Kinase), SB 203580 (p38-Kinase), humanes Thrombin
• Gibco BRL Life Technology, Karlsruhe: Bench Mark Molekulargewichtmarker, DNAse 1, Kompetente Zellen DH 5α, Superscript II RNAse H Reverse Transkriptase, Trizol-Reagent
• ICN Biomedicals, Aurora, USA: Chloramphenicol D-threo-[1,2-14C], SDS • Merck, Darmstadt: 2-Propanol, Chloroform, DMSO, Coomassie blue, Ethanol
absolut z. A., Essigsäure, Gelatine, Natriumcarbonat, t-RNA • Molecular Dynamics, Sunnyvale, USA: Phosphor-Imager 445 SI
28

C. Material und Methoden
• Pierce Rockford, USA: BCA Protein Reagent Kit • Qiagen, Hilden: SuperFect Transfektion Reagent • Roth Karlsruhe: Glycerol, Methanol, Triton X-100, Tris • Schleicher & Schüll, Daßel: Nitrocellulose Optitran BA-S85 0,45µm • Sigma-Aldrich GmbH, Steinheim: p-Aminobenzamidin, Aprotinin, EDTA,
Formaldehyd, Gelatine, Glycin, Glutaraldehyd, Kalziumchlorid, MBP, ß-Mercaptoethanol, MTT, ONPG, PMSF, Proteinase K, Rnase freies Wasser, Sodiumorthovanadat
• Whatman Maidstone, England: Dünnschichtchromatographie (TLC) Platten
C.3 Zellkultur Die verwendeten Zellinien wurden in Dulbecco`s modified Eagle Medium (DMEM)
von Gibco mit 10 % FCS, 0,01 M HEPES, 40 U/ml Penicillin, 40 µg/ml Streptomycin, 272 mM Arginin und 550 mM Asparagin bei 37°C und 5 % CO2 kultiviert. Mediumwechsel erfolgte jeden zweiten Tag mit Absaugen des Kulturmediums, Waschen der Zellen mit sterilem PBS und Zugabe von vorgewärmtem Medium. Bei ca. 80 % Konfluenz wurden die Zellen mit Split-Lösung (0,5 % Trypsin; 0,02 % EDTA) abgelöst. Nach Zentrifugation und Resuspendierung des Zellpellets erfolgte die Aussaat im Verhältnis von 1:3 bis 1:6.
C.4 Zymographie Da MMP’s von Zellen in das Kulturmedium sezerniert werden, verwendet man zur
Analyse den Zellüberstand. Der Zellüberstand wird in einem gelatinhaltigen SDS-Polyacrylamid-Gel aufgetrennt, nach Auftrennung werden die Gelatinasen mit Ca2+ aktiviert und durch Anfärbung des Gels mit Coomasieblau wird das verbliebene Gelatin dargestellt. Ungefärbte Areale entsprechen der Gelatinase-Aktivität der jeweiligen Proben.
In Petrischalen mit 3 cm Durchmesser werden 2,0 x 105 Zellen in 2 ml Komplettmedium (DMEM mit Glutamax, 10 mM HEPES, 272 mM Asparagin, 550 mM Arginin, 1000 U Penicillin/Streptomycin, 10 % fötales Kälberserum) ausgesät und über 24 h bis zu einer Konfluenz von ca. 70 % kultiviert. Die Zellen werden 2x mit sterilem PBS gewaschen und für 24 h in serum- und antibiotikafreiem Medium gehalten. Um Einflüsse von autokrinen Wachstumsfaktoren zu vermeiden, wird vor Stimulation mit Thrombin das serumfreie Medium ausgetauscht und die Zellen in 1 ml kultiviert. Nach 24 h wird der Zellüberstand abgenommen und abzentrifugiert. Die Zellen werden gezählt und die Proben bis zur weiteren Analyse bei –80° C gelagert.
Zur Auftrennung der Proben wird ein 1,5 mm dickes SDS/Polyacrylamidgel mit Gelatin (Sammelgel: 19 ml ddH2O, 3 ml Acrylamid 40 %, 7,4 ml 0,5 M Tris pH 6,8, 300 µl 10 % SDS, 100 µl APS 10 %, 30 µl TEMED; Trenngel: 60 mg Gelatin, 63,8 ml ddH2O, 22,4 ml Acrylamid 40 %, 30 ml 1,5 M Tris pH 8,8, 4,1 ml 10 % SDS, 600 µl APS 10 %, 60
29

C. Material und Methoden
µl TEMED) verwendet (Woessner, 1995). Die Proben werden mit 3 x nicht-reduzierendem Lämmli-Puffer (187,5 mM Tris-HCl, pH 6,8; 6 % SDS; 30 % Glyzerin; 0,1 % Bromphenolblau) 5 min bei 95° C in einem Heizschüttelblock denaturiert und in dem Polyacrylamidgel mit einem SDS-haltigen Laufpuffer (0,1 % SDS; 192 mM Glycin; 25 mM Tris) in einer Gelapparatur von Biorad (Protean II grosse Kammer) elektrophoretisch aufgetrennt. Die Elektrophorese wird zum Einlaufen der Proben 30 min mit 50 V und weitere 3 h mit 180 V durchgeführt. Anschließend wird das Gel zur Entfernung des SDS in einer Triton X-100-Lösung (2,5 % Triton X-100; 50 mM Tris-HCl, pH 7,5; 0,05 % NaN3) für 2 h auf einem Schüttler bei RT inkubiert. Nach dreimaligem Waschen mit je 300 ml destilliertem Wasser erfolgt die Inkubation bei 37° C im Wasserschüttelbad mit einem kalziumhaltigen Puffer (10 mM CaCl2; 0,15 M NaCl; 50 mM Tris-HCl, pH 7,5; 0,05 % NaN3) für mindestens 16h. Durch Einbinden des Kalziums in das katalytische Zentrum werden die MMP`s aktiviert und das Gelatin wird aufgespalten. Anschließend wird das Gel für 1 h gefärbt (0,2 % Coomassie Blau; 10 % Essigsäure; 20 % Methanol) und für 1 h entfärbt (10 % Essigsäure; 20 % Methanol). Das Gel wird zwischen zwei Cellophanblätter gelegt und für 2 h in einem Heizofen getrocknet. Zur Dokumentation und Quantifizierung werden die Gele mit dem Software Programm Scan-Pack 3.0 eingelesen.
C.5 Northern Blot Nach der Entwicklung des Southern-Blotting als ein wichtiges Analyseverfahren von
DNA (Southern, 1975) wurde 2 Jahre später ein equivalentes Verfahren für RNA vorgestellt (Alwine et al., 1977). In der Beschreibung als Northern-Blotting, ursprünglich als humoristische Anlehnung an den Southern-Blot gedacht, ist es ein Standardverfahren in der Analyse der RNA-Expression.
Die RNA wird in einem formalinhaltigen Agarosegel aufgetrennt, anschließend erfolgt der Transfer auf eine Nylonmembran und zuletzt wird mit einer spezifischen, radioaktiv markierten cDNA-Probe die RNA-Expression dargestellt. 106 U-2 OS-Zellen werden in Petrischalen mit 10 cm Durchmesser in 10 ml Komplettmedium ausgesät. Nach 24 h wird bei ca. 70 % konfluenten Zellen das Medium abgesaugt und mit sterilem PBS gewaschen. Die Zellen werden 24 h in serum- und antibiotikafreiem Medium kultiviert. Vor Stimulation erfolgt ein Wechsel des serumfreien
Mediums. Die Isolierung der RNA erfolgt mit dem TRIZOL Reagent von Gibco BRL. Das Kulturmedium wird abgesaugt, die Zellen auf Eis mit 4° C gekühltem PBS gewaschen und mit 1 ml/10 cm2 Reagenz lysiert. Das Lysat wird in ein Reaktionsgefäß überführt, anschließend für 5 min bei RT inkubiert. Nach Zugabe von 0,2 ml Chloroform je ml Reagenz werden die Reaktionsgefäße 15 s geschüttelt und nach 2-3 min Inkubation in der Tischzentrifuge mit 12 000 rpm für 10 min bei 4° C abzentrifugiert. Die wässrige Phase wird in ein neues Reaktionsgefäß überführt und nach Zugabe von 0,5 ml Isopropanol je ml
30

C. Material und Methoden
eingesetzter TRIZOL Reagent wird RNA gefällt. Nach Zentrifugation mit 12 000 rpm für 10 min bei 4° C wird das RNA-Pellet mit 75 % Ethanol zweimal gewaschen und anschließend getrocknet. Die RNA wird in doppeldestilliertem H2O aufgenommen, ein Aliquot photometrisch bei 260 nm gemessen und nach der unten aufgeführten Formel die Konzentration bestimmt. Die RNA wird bei –20° C gelagert.
c = OD260nm x f x n [µg/ml] c: Konzentration OD: Optische Dichte f: Verdünnungsfaktor (n: 50 µg/ml für DNA, 40 µg/ml für RNA und 37 µg/ml für Oligonukleotide bei einer Schichtdicke der Küvette von 1 cm)
10 µg RNA werden in einem formaldehydhaltigen 1 % Agarosegel aufgetrennt. Die
RNA wird dazu in einem formamidhaltigen Ladepuffer (50 % Formamid; 5,8 % Formaldhyd; 1 x MOPS) aufgenommen, und als Laufpuffer für das Gel wird 1 x MOPS (20 mM MOPS; 8 mM NaAc; 1 mM EDTA) verwendet. Der Laufpuffer zirkuliert während der Auftrennung. Vor dem Auftragen wird die RNA 10 min bei 70 °C denaturiert und auf Eis abgekühlt. Nach der Elektrophorese wird das Agarosegel unter UV-Licht (λ= 260 nm) kontrolliert und photodokumentiert. Es wird damit eine mögliche Degradation der RNA beurteilt.
Der Transfer der RNA auf eine positiv geladene Nylonmembran (NY 04 von Boehringer, Mannheim) erfolgt mittels Kapillarblot. Die Membran wird entsprechend der Gelgröße zugeschnitten, für 3 min in dd H2O und für 3 min in SSC (20 x SSC 3 M NaCl; 0,3 M NaAc, pH 7,0) vorinkubiert. Der Transfer der RNA erfolgt über Nacht mit Hilfe von Kapillarkräften. Am nächsten Morgen wird der Transfer weitere 2 h durchgeführt. Die Transfereffizienz wird durch Beleuchtung des Gels und der Membran unter UV-Licht kontrolliert. Zur Fixierung der RNA wird die Membran für 2 h bei 80 °C gebacken.
Die Nylonmembran wird für 30 min in Church-Puffer (500 ml 1 M Na2HPO4; 10 ml 0,1 M EDTA; 70 g SDS; 490 ml ddH2O) mit 10 mg t-RNA bei 65 °C in einem Hybridisierungsröhrchen prähybridisiert.
Für die radioaktive Markierung der DNA-Fragmente wird der Random Primed Labeling-Kit von Boehringer-Mannheim verwendet und als Gensonde ein Abschnitt der cDNA von MMP-9 eingesetzt. 50 ng der zu markierenden Gensonde werden mit ddH2O auf ein Volumen von 9 µl gebracht. Dieser Ansatz wird 10 min bei 95°C denaturiert und danach auf Eis abgekühlt. Anschließend werden in einem 20 µl-Reaktionsansatz, mit 0,05 mM dNTPs, 2 µl Reaktionspuffer Nr. 6 (10x), 1 µl Klenow-Polymerase ( 2 U/µl ) und 50 µCi [α-32P]-dCTP zugegeben. Dieser Ansatz wird 30 min bei 37°C inkubiert und zuletzt die
31

C. Material und Methoden
Reaktion mit 1 µl 0,5 M EDTA-Lösung inaktiviert. Bis zur Abtrennung der freien Nukleotide wird der Ansatz auf Eis gelagert.
Vor Verwendung der markierten Gensonde werden die freien Nukleotide über eine Trennsäule (Boehringer) entfernt. Die Trennsäule wird mit 1 ml TE-Puffer ( 10 mM Tris HCl, ph 7,5; 1 mM EDTA) gewaschen und darf bis zur Verwendung nicht austrocknen. Der radioaktive Ansatz wird in 400 µl TE-Puffer aufgenommen, auf die Trennsäule gegeben und das Eluat in einem Reaktionsgefäß gesammelt. Anschließend werden weitere 400 µl TE-Puffer auf die Säule gegeben und das Eluat in einem neuen Reaktionsgefäß aufgenommen. Von beiden Proben wird in einem ß-Zähler die Radioaktivität der Proben bestimmt. Die spezifische Bindung ist in der II. Fraktion und die freien Nukleotide befinden sich in der Trennsäule. Nach dem Messen wird die Gensonde 10 min bei 95 °C denaturiert, abzentrifugiert und auf Eis gestellt. Die Hybridisierungslösung, auf 65°C vorgewärmt, wird mit 106 cpm/ml der radioaktiven Gensonde versetzt und in das Hybridisierungsröhrchen gegeben. Die Hybridisierung erfolgt bei 65 °C über Nacht. Anschließend wird die Nylonmembran zur Entfernung der unspezifischen Bindungen mit 25 ml einer Waschlösung (2 x SSC; 1 % SDS) bei 58 °C für zweimal 15 min und zweimal 30 min gewaschen. Die Northern-Blots werden für 24 h in Phosphor Screen-Kassetten exponiert und anschließend mit dem Phosphor-Imager von Molecular Dynamics quantitativ ausgewertet.
C.6 Transfektionen: CAT, Luciferase Die genomische Sequenz von MMP-9 umfaßt 7,7 Kilobasen (kb) und ist in 13 Exone
unterteilt, die in einen 2,5 kb großen mRNA-Abschnitt übersetzt werden (Huhtala et al., 1991). Die Transkription wird durch einen 2 kbp langen Abschnitt im 5`-Bereich vor dem Start der Transkription gesteuert. Die Expression von MMP-9 wird u. a. durch die Transkriptionsrate reguliert, und durch Promotorstudien kann damit der Einfluß von inhibierenden oder aktivierenden Stoffen untersucht werden.
Für Promotorstudien wird Plasmid-DNA mit der Promotorsequenz in die Zellinie eingeschleust. Wird die DNA als Plasmidform im Zellkern aufgenommen, ist eine transiente Transfektion durchgeführt worden. Bei einer stabilen Transfektion wird die DNA in das Genom der Zellen eingebaut.
Zum Nachweis der Aktivierung von MMP-9 auf der Promotor-Ebene werden transiente Transfektionen durchgeführt. Die Osteosarkomzellinie U-2 OS wird mit einem MMP-9-CAT-Reporter-Konstrukt und mit β-Galaktosidase, zur Normalisierung, kotransfiziert. Mit der Transkription des zelleigenen MMP-9-Genabschnittes wird das nachgeschaltete Reportergen abgelesen. Damit wird das Reporterenzym gebildet, und durch Messung der Enzymaktivität kann direkt auf die Transkriptionsrate des MMP-9-Gens und damit auf die Promotoraktivität geschlossen werden. Der eingesetzte MMP-9-Promotor umfaßt die Positionen von -670 bp bis 0 bp (bezogen auf den Transkriptionsbeginn des 5`-Endes). Die wichtigsten Bindungsstellen in diesem Bereich sind die beiden AP-1-
32

C. Material und Methoden
Bindungsstellen bei Position -533 und- 79 bp. In den aufgeführten Plasmidkonstrukten wird als Reporterenzym neben der Chloramphenicol-Acetyl-Transferase (CAT) auch die Luciferase eingesetzt. Zur Normalisierung der Transfektionseffizienz wird die ß-Galaktosidase verwendet.
Für die Transfektionen wird das Superfect Transfection Reagent® von Qiagen verwendet. Die hierbei eingesetzte Methodik ist eine von Qiagen weiterentwickelte Transfektionsart mittels Liposomen, die im Vergleich zu anderen Möglichkeiten des DNA-Transfers wie zum Beispiel der Kalzium-Phosphat-Methodik oder der Elektroporation eine höhere Transfektionseffizienz in der U-2 OS-Zellinie hat (Felgner et al., 1987). Neben den eingesetzten MMP-9-Konstrukten werden als Positiv-Kontrolle H-Ras, als Negativkontrolle der Leervektor pSV-2 Neo eingesetzt.
Bei dem Transfektionsansatz muß die DNA-Menge der verwendeten Konstrukte die gleiche Molarität besitzen. Deshalb wird bei dem MMP-9-CAT-Reporter-Konstrukt aufgrund der Größe insgesamt eine höhere DNA-Menge im Vergleich zum Leervektor eingesetzt. Für das MMP-9-Reporter-Konstrukt wird für Petrischalen mit 10 cm Durchmesser je 5 µg und für Petrischalen mit 3 cm Durchmesser 1 µg Konstrukt eingesetzt. Als Positivkontrolle wird H-Ras mit 2,5 µg bzw. 0,3 µg DNA verwendet, als Negativkontrolle pSV-2-Neo, der Leervektor, mit 1,5 µg bzw. 0,2 µg. Um die Effizienz der Transfektionen zu vergleichen, wird jeweils ein Expressionsplasmid mit ß-Galaktosidase kontransfiziert. Die eingesetzte Vektormenge beträgt 2,5 µg bzw. 0,3 µg.
In Petrischalen mit 10 cm Durchmesser werden 106 Zellen in 10 ml oder in Petrischalen mit 3 cm Durchmesser 2 x 105 Zellen in 2 ml Komplettmedium ausgesät. Am folgenden Tag werden die zu 60-70 % konfluenten Zellen mit sterilem PBS gewaschen. Für Petrischalen mit 10 cm Durchmesser wird in einem 4 ml Transfektionsröhrchen aus Polystyrol in 300 µl serumfreiem Medium die DNA und 60 µl SuperFect Transfektion Reagent pipettiert. Der Ansatz wird fünfmal durch Pipettieren gemischt. Nach 10 min Inkubation bei RT werden 3 ml serumfreies Medium zugegeben und die Lösung auf die mit PBS gewaschenen Zellen aufgetropft. Nach 2-3 h werden 6 ml Komplettmedium hinzugefügt und über Nacht belassen. Pro Petrischalen mit 3 cm Durchmesser werden 100 µl serumfreies Medium, 10 µl SuperFect Transfektion Reagent und 600 µl serumfreies Medium eingesetzt. Am nächsten Morgen werden die Zellen mit PBS gewaschen und in serumfreiem Medium kultiviert. Nach 24 h erneut Wechsel des serumfreien Mediums und Stimulation mit 0,5-2 U Thrombin /ml.
Das Abernten der Zellen erfolgt 36 h nach der Transfektion. Die Zellen werden mit kaltem PBS gewaschen und pro Platte 1 ml TEN-Puffer (40 mM Tris-HCl; 1 mM EDTA; 6 mM NaCl, pH 8) zugegeben. Nach Inkubation für 10 min bei 4°C werden die Zellen mit Zellschabern abgelöst, in ein Reaktionsgefäß überführt und in der Tischzentrifuge bei 14 000 rpm für 4 min bei 4°C abzentrifugiert. Der Überstand wird abgesaugt und das Zellpellet in 100 µl 0,25 M Tris, pH 7,8 resuspendiert. Die Zellen werden anschließend durch
33

C. Material und Methoden
viermaliges wechselndes Inkubieren auf Trockeneis und im Wasserbad bei 37 °C lysiert. Das Zellysat wird 30 min bei 4 °C mit 14 000 rpm abzentrifugiert und der Überstand in ein neues Reaktionsgefäß überführt. Bis zur weiteren Aufarbeitung können die Proben bei –80 °C gelagert werden.
Zur Bestimmung der Transfektionseffizienz wird in die Zellen neben dem Reporterplasmid das lacZ-Gen von E. coli, welches für das Enzym ß-Galaktosidase kodiert, kotransfiziert. Für die Normalisierung der unterschiedlichen Ansätze wird die kolorimetische Färbung des X-Gal gemessen (Alam et al., 1990). Zu 10 µl Zellysat (oder 10 µl 0,25 M Tris, pH 7,8 als Negativkontrolle) werden 75 µl ß-Gal-Puffer (6,5 mM NaPO4; 16,8 mM ß-Mercaptoethanol; 0,15 mM MgCl2) und 75 µl ONPG (0,0013 g/ml in 0,1 M NaPO4) zugegeben. Der Ansatz wird im Wasserbad bei 37 °C für 1-3 h, abhängig von der Gelbfärbung, inkubiert und die Farbreaktion mit 50 µl 1 M Na CO3 gestoppt. Zur Messung werden 150 µl der Lösung auf eine ELISA-Platte aufgetragen und bei 405 nm im ELISA-Meßgerät (Titertek Multiscan MCC/340 Lesegerät) gemessen.
Nach der Normalisierung anhand der X-Gal-Färbung werden die Proteine der Zellysate 10 min bei 72 °C denaturiert, anschließend 5 min bei RT mit 14 000 rpm abzentrifugiert und der Überstand in ein neues Reaktionsgefäß überführt. CAT ist im Vergleich zu ähnlich wirkenden Enzymen sehr hitzestabil, mit der Denaturierung wird sichergestellt, daß nur die Aktivität von CAT gemessen wird. Entsprechend der ß-Galaktosidase-Aktivität werden die Proben mit 0,25 M Tris, pH 7,8 auf ein Volumen von 90 µl normalisiert.
Mit 4 µM Chloramphenicol D-threo-[1,2-14C] (111 mCi/mmol) und 120 µg/ml Azetyl-Coenzym A (4 mg /ml in 0,25 M Tris, pH 7,8) wird im Wasserbad bei 37 °C über Nacht die CAT-Reaktion gestartet. Im Anschluß daran wird zu jeder Probe 800 µl Ethylazetat hinzugefügt und für 30 s mit dem Vortexer gemischt. Nach Zentrifugation für 6 min bei 14 000 rpm wird die organische Phase unter strikter Vermeidung der Verunreinigung durch Mitführen der wässrigen Phase in ein neues Reaktionsgefäß überführt. Nach Eindampfen über 45 min in einer Vakuumzentrifuge werden die Pellets in 20 µl Ethylazetat resuspendiert und fraktioniert auf eine Dünnschicht-Chromatographie-Platte aufgetragen. Mit einem organischem Laufpuffer (95 % Chloroform, 5 % Methanol) werden die Proben für 90 min aufgetrennt. Die Radioaktivität auf der Chromatographie-Platte wird über Nacht mit dem PhosphorImager 445 SI von Molecular Dynamics quantitativ gemessen.
Durch die Klonierung des Luciferase-Gens von Photinus pyralis (de Wet et al., 1987) ist ein alternatives Reportersystem entwickelt worden (Nordeen, 1988). Der Vorteil der Luciferase-Methode im Vergleich zur CAT-Methode ist die schnellere Auswertung der Transfektion und die nichtradioaktive Technik. Durch Zugabe von Luciferin als Substrat, ATP, Mg2+, Sauerstoff und Zellysat entsteht eine biolumineszente Reaktion. Die dabei enstehende Lichtreaktion wird in einem Luminometer quantitativ gemessen und entspricht
34

C. Material und Methoden
der Transkriptionsrate des MMP-9-Luc-Reporter-Konstruktes. Das Zellysat wird nach den gleichen Arbeitsschritten wie für den CAT-Assay hergestellt. Für die Lichtreaktion werden neben den Zellysaten die Reagenzien A und B der Firma Berthold verwendet. Bei der erstmaligen Verwendung eines Luciferase-Assays beginnt man mit geringen Mengen Zellysat, um die notwendige Lysatmenge zu titrieren. Bei den MMP-9-Konstrukten werden 80 µl Zellysat mit 100 µl Reagenz A gemischt, und nach Zugabe von 100 µl Reagenz B in dem Lumat-Gerät wird die Lichtreaktion gemessen. Die Werte werden gemäß der ß-Galaktosidase-Aktivität normalisiert.
C.7 RT-PCR aus Gewebeproben Von 2 Patienten (16. und 11. Lj.) mit einem histologisch gesicherten Osteosarkom
wird ein Bruchteil des Gewebes für die Aufarbeitung verwendet. Von dem Osteosarkomgewebe werden 50-100 mg homogenisiert, mit dem TRIZOL- Reagent lysiert und die RNA isoliert. Für die Homogenisierung des Gewebes wird ein Keramikmörser mit Seife und Alkohol gereinigt, um Verunreinigungen auszuschließen. Der Mörser wird mit flüssigem Stickstoff gekühlt, damit die Gewebeprobe während der Zerkleinerung nicht auftaut. Die in Stickstoff gelagerte Probe wird pulverisiert und in 1 ml TRIZOL Reagent aufgenommen. Das Lysat wird in ein Reaktionsgefäß überführt und die RNA nach den gleichen Arbeitsschritten, wie unter Kapitel X (Northern-Blot) beschrieben, extrahiert. Nach einem Verdau mit DNAse-I erfolgte die Umschreibung von RNA zu DNA mit Hilfe der reversen Transkription. Für die anschließende PCR wurden 2 µl der entstandenen cDNA-Lösung mit 2 U Expand high fidelity polymerase, 200 µM dNTP, 2 mM MgCl2 und 300mM Thrombinrezeptor-Primer eingesetzt. Die Primer waren für das 5`-Ende CCCTGCTCGAAGGCTACTAT und für das 3`-Ende CTACATGCTGAACGTATGGACG. Nach der PCR wurde das Produkt auf einem 1,5% Agarose-Gel aufgetrennt und unter UV-Licht dokumentiert.
C.8 Invasionsassay Matrigel® ist ein lösliches Basalmembran-Extrakt aus Engelberth-Holm-Swarm-
Sarkomen bei Mäusen. Es enthält u. a. Laminin, Kollagen Typ IV, Heparansulfatproteoglykan und Entaktin (Kleinmann et al., 1986). Durch Beschichtung einer Transwell-Membran mit Matrigel wird die Basalmembran im Modell nachgebaut und kann für Untersuchungen des Invasionsverhaltens von Zellen eingesetzt werden (Albini et al., 1987). Die Osteosarkomzellen werden in der oberen Kammer einer Invasionskammer kultiviert, und nach 72 h wird die Anzahl der nach unten gewanderten Zellen quantitativ erfaßt.
35

C. Material und Methoden
Z e l l k a m m e r e i n s a t z
S e r u m f r e i e s M e d i u m
M a t r i g e l s c h i c h tO s t e o s a r k o m z e l l e n
F i b r o b l a s t e n - K u l t u r m e d i u m
Abb. 6: Invasionsassay Als Chemotaxis wird das Kulturmedium einer NIH 3T3-Zellinie in die untere
Kammer gegeben. Die Fibroblastenzellinie wird bei einer Konfluenz von ca. 70 % mit PBS gewaschen und 24 h in serumfreiem Kulturmedium gehalten. Dieses Medium wird abgenommen, abzentrifugiert und bis zur Verwendung bei –20 °C gelagert. Für den Invasionassay werden BIOCOAT® Matrigel® Invasionskammern von Becton Dickinson mit einer Porengröße von 8 µm eingesetzt. Die Matrigel®-Membranen werden vor der Verwendung unter der Zellkulturwerkbank auf RT gewärmt und anschließend für 1 h im Inkubationsschrank mit serumfreiem Medium beschichtet. Es werden 3 x 105 U-2 OS-Zellen in 1,5 ml serumfreiem Medium in die obere Kammer des Einsatzes ausgesät. In die untere Kammer werden 2,5 ml des Fibroblastenmediums pipettiert. Nach 1 h, wenn die Zellen adhärent sind, erfolgt die Stimulation mit 1 U/ml Thrombin in die obere Kammer. Der Invasionsansatz wird für 72 h im Inkubationsschrank kultiviert. Für den Invasionsassay werden Dreifach-Ansätze verwendet.
Die Auswertung erfolgt durch Anfärbung der nach unten penetrierten Zellen mit MTT oder Kristallviolett (Mosmann et al., 1983). Nach Entfernen des Mediums wird mit Wattestäbchen die obere Matrigelschicht abgenommen. Damit werden nur die durch die Membran nach unten gewanderten Zellen angefärbt. Durch Inkubation für 3 h in Komplettmedium mit MTT (0,2 mg/ml) werden die MTT-Kristalle von den Mitochondrien der Zellen aufgenommen. Anschließend werden die Membranen getrocknet, mit einem Skalpell ausgeschnitten und die Membranen in 1 ml DMSO ausgewaschen. Die Farbe löst sich in DMSO, 150 µl der Lösung werden auf eine ELISA-Platte aufgetragen und die Absorption bei 570 nm gemessen. Die Intensität der Absorption entspricht der Anzahl der migrierten Zellen. Eine alternative Färbung kann mit Kristallviolett (0,2 % Kristallviolett) durchgeführt werden.
36

D. Ergebnisse Die Invasion eines bösartigen Tumors in benachbarte Strukturen wie
Muskelkompartimente oder Gefäße erfordert die Auflösung der Basalmembran. Die kollagene Komponente der Basalmembran besteht hauptsächlich aus Kollagen-Typ IV, welches durch die beiden Typ-IV Kollagenasen MMP-2 und –9 abgebaut wird (Nakajima et al., 1991).
Bei Tumorpatienten werden im Vergleich zu normalen Patienten vermehrt thromboembolische Komplikationen beobachtet. In klinischen Untersuchungen konnte man bei Malignompatienten eine Steigerung der Blutgerinnung nachweisen (Kakkar A. et al., 1995). In vivo konnte gezeigt werden, daß mit der Gabe von Heparin die Ausbildung von Lungenmetastasen vermindert wird (Amirkhosravi et al., 1993). Daraus ergab sich die Fragestellung, durch welchen Mechanismus Thrombin die Aktivität von Typ-IV Kollagenasen bei Tumorzellen beeinflußt.
D.1 Thrombin erhöht die Aktivität von pro-MMP-9 Für die im folgenden beschriebenen Studien wurde die Osteosarkomzellinie U-2 OS
eingesetzt. Der Einfluß von Thrombin auf die Aktivität von MMP-2 und MMP-9 wurde in der Monolayer-Zellkultur mit dieser Osteosarkomzellinie untersucht. Die Osteosarkomzellinie wurde bis zu einer Konfluenz von 70% in DMEM-Medium kultiviert und danach für 24 h in serumfreiem Medium gehalten, da serumhaltiges Medium MMP’s enthält. Zur Vermeidung von autokrinen Effekten wurde das serumfreie Medium vor Beginn der Stimulation ausgetauscht und anschließend die Zellen konzentrationsabhängig mit Thrombin inkubiert. Nach weiteren 24 h wurde der Überstand mittels Zymographie untersucht. In Abb. 6 sieht man in den ersten beiden Spuren den Zellüberstand der Fibrosarkomzellinie HT 1080, unstimuliert und mit TPA stimuliert. Der Überstand dieser Zellen wurde als Positivkontrolle verwendet. HT 1080-Zellen haben eine basale Aktivität von pro-MMP-2 und pro-MMP-9, die durch TPA erhöht wird. Die Osteosarkomzellinie U-2 OS zeigt im Vergleich zu der basalen Aktivität von pro-MMP-2 und pro-MMP-9 nach Stimulation mit Thrombin eine konzentrationsabhängige Aktivierung von pro-MMP-9. Ein Anstieg der pro-MMP-9-Aktivität kann durch Zugabe von 0,25 bis 1,0 U/ml beobachtet werden. Bei Zugabe von 1,0 U/ml Thrombin ist das Maximum der Aktivierung erreicht. Daraus resultierend wurde in den nächsten Versuchen je 1,0 U/ml Thrombin eingesetzt.
37

D. Ergebnisse
Thrombin U/ml
0 0,25 0,5 1,0 2,0 3,0
MMP - 9
MMP - 2
TPA
0 +
Abbildung 6: Thrombin stimuliert die Aktivität von pro-MMP-9. Die Osteosarkomzellen wurden in 3 cm Petrischalen 24 h serumfrei kultiviert, anschließend für 24 h mit 0,25-3 U/ml Thrombin stimuliert. Die Zellkulturüberstände wurden abgenommen und auf die jeweilige Zellzahl normalisiert. Die Auftrennung der Proben erfolgte in einem gelatinhaltigem SDS-Gel (0,05 % Gelatin). Als Positivkontrolle wurden Überstände der Fibrosarkomzellie HT 1080, +/- TPA, verwendet.
Thrombin induziert die Expression einer Protease, die für die Invasion und
Metastasierung wichtig ist. In dem folgendem Experiment sollte deshalb festgestellt werde, ob die Steigerung von pro-MMP-9 rezeptorabhängig vermittelt wird.
D.2 Untersuchung des Signaltransduktionsweges nach Stimulation mit Thrombin
Nach der Stimulation mit Thrombin verhalten sich Tumorzellinien aggressiver (Henrikson et al., 1999). Die Steigerung der Aktivität von MMP’s nach Zugabe von Thrombin kann jedoch durch verschiedene Rezeptortyrosinkinasen vermittelt werden. Der Insulin-like growth factor-1 receptor (IGF-1R) wird durch Thrombin aktiviert (Rao et al.,1995) und hat Einfluß auf die Regulation von MMP-2 (Long et al.,1998). 1991 wurde der spezifische Thrombinrezeptor, ein Rezeptor mit sieben transmembranen Domänen, isoliert (Vu et al., 1991). Bisher ist bekannt, daß das Signal intrazellulär über α/β/γ-trimerische Rezeptoren und den MAP-Kinase-Weg weitergeleitet werden kann (Trejo et al., 1996).
Für die Untersuchung wurden die Osteosarkomzellen mit TRAP (thrombin receptor activating peptid) stimuliert. Dieses Peptid entspricht den ersten 6 Aminosäuren des extrazellulären Abschnittes des Rezeptors, der nach der proteolytischen Abspaltung durch Thrombin entsteht. Dieser Abschnitt bindet an den Rezeptor und bewirkt eine Konformationsänderung, die als positives Signal in das Zellinnere weitergeleitet wird. Es ist
38

D. Ergebnisse
eine hohe Selektivität der Peptidsequenz bei verschiedenen Gattungen zu beobachten (Gerszten et al., 1994). Falls sich nach Gabe von TRAP eine Aktivierung von MMP-9 in der U-2 OS-Zellinie wie durch Thrombin zeigt, unterstützt dies die Hypothese, daß Thrombin den Thrombinrezeptor spezifisch aktiviert.
Die Osteosarkomzellinie wurde nach 24 h in serumfreiem Medium parallel mit unterschiedlichen Thrombin- und TRAP-Konzentrationen stimuliert. Das Kulturmedium wurde nach 24 h in der Gelatinzymographie analysiert. In der vorliegenden Gelatinzymographie ist in der ersten Spur das Kulturmedium von unstimulierten U-2 OS-Zellen aufgetragen (Abb. 7). Die basale Aktivität von pro-MMP-9 ist bei den U-2 OS-Zellen wie schon in dem vorherigen Experiment niedrig. Als Positivkontrolle ist in den nächsten beiden Spuren der Überstand der Osteosarkomzelle nach Zugabe von 0,5 und 1,0 U/ml Thrombin aufgetrennt. Nach Zugabe von Thrombin stellt sich eine deutliche Erhöhung der Aktivität von pro-MMP-9 dar. In den folgenden Spuren wurden die Zellen mit aufsteigenden Konzentrationen von TRAP inkubiert. Mit aufsteigender Konzentration von TRAP ist ebenfalls eine zunehmende Aktivität von pro-MMP-9 zu erkennen. Durch Zugabe von TRAP wird wie durch die Zugabe von Thrombin eine Steigerung der pro-MMP-9-Aktivität beobachtet. Dies zeigt, daß Thrombin den Thrombinrezeptor spezifisch aktiviert und MMP-9 induziert. Die Aktivität von MMP-2 bleibt unverändert.
0 0 ,5 1 ,0 5 0 1 0 02 5
M M P -9
M M P -2
T h ro m b in U /m l T R A P µ M /m l
Abbildung 7: Stimulierung von U-2 OS durch TRAP. Die Zellinie wurde in 3 cm Petrischalen 24 h serumfrei gehalten, Stimulation mit 0,5-2,0 U Thrombin/ml und 5-100 µM TRAP. Die Zellkulturüberstände wurden nach weiteren 24 h abgenommen und auf die jeweilige Zellzahl normalisiert. Die Auftrennung der Proben erfolgte in einem gelatinhaltigen SDS-Gel. Als Positivkontrolle wurden Überstände der Fibrosarkomzellie HT 1080, mit TPA stimuliert, aufgetragen.
39

D. Ergebnisse
D.3 Thrombin stimuliert konzentrationsabhängig die RNA-Expression von MMP-9
Die in den vorausgegangenen Versuchen nachgewiesene Steigerung der Aktivität von pro-MMP-9 durch Thrombin kann transkriptionell und posttranskriptionell bewirkt werden. Durch Aktivierung des Promotors von MMP-9 wird die Transkription beeinflußt. Posttranskriptionell wird durch Zunahme der RNA- oder der Proteinstabilität eine Steigerung der MMP-Aktivität erreicht.
Der Einfluß von Thrombin auf die RNA-Expression wurde mit Hilfe der Northern-Blot-Analyse mit RNA-Proben, isoliert aus der U-2 OS-Zellinie, untersucht. Die Osteosarkomzellen wurden mit verschiedenen Konzentrationen von Thrombin inkubiert.
Nach 24 h wurde Gesamt-RNA aus U-2 OS-Zellen mit TRIZOL isoliert und in einem denaturierenden Agarosegel aufgetrennt. Mittels Kapillarblot erfolgte der Transfer auf eine Nylonmembran und nach Hybridisierung mit einer Gensonde für MMP-9 wurde eine Autoradiographie durchgeführt (Abb. 8). Dabei zeigte sich, daß im Vergleich zu der basalen Expression die Transkriptionsrate des MMP-9-Gens durch Thrombin gesteigert wurde. Der Anstieg der Expressionsrate ist konzentrationsabhängig von Thrombin.
Um zu zeigen, daß der Anstieg der MMP-9-Expression nicht durch unterschiedliche RNA-Mengen bedingt war, wurde die Nylonmembran anschließend gestrippt und mit einer Gensonde für GAPDH, einem house keeping Gen, hybridisiert. Dabei zeigte sich, daß je gleiche Mengen RNA aufgetragen wurden.
0 0 ,5 1 ,0T h ro m b in U /m l
2 ,0
M M P -9
G A P D H
Abbildung 8: Thrombin erhöht konzentrationsabhängig die Transkription von MMP-9. U-2 OS-Zellen wurden 24 h serumfrei kultiviert, mit 0,5-2,0 U Thrombin/ml stimuliert, und nach 24 h wurde die Gesamt-RNA isoliert. Die RNA wurde in einem 1 % formaldehydhaltigen Agarosegel aufgetrennt und mittels Kapillarblot auf eine Nitrozellulosemembran transferiert. Die Markierung erfolgte mit dem cDNA von MMP-9. Nachfolgend Stripping und Hybridisierung mit GAPDH.
40

D. Ergebnisse
D.4 Zeitabhängige Veränderung der Aktivität von pro-MMP-9 nach Stimulation mit Thrombin
Der Zeitraum zwischen der Zugabe eines Agens und der Reaktion der Zellen kann erheblich variieren. Nach Zugabe von Thrombin kann man eine Aktivierung von Blutplättchen innerhalb von Sekunden oder eine gesteigerte Proliferation von Endothelzellen nach Tagen beobachten. Es stellte sich die Frage, wann nach Zugabe von Thrombin auf Protein-Ebene ein Anstieg von pro-MMP-9 festzustellen ist, wann ein Maximum erreicht wird und wie lange die maximale Wirkung nachweisbar ist. Die Osteosarkomzellinie wurde bis zu einer Konfluenz von 70% in DMEM-Medium kultiviert und danach für 24 h in serumfreiem Medium gehalten. Die Osteosarkomzellinie U-2 OS wurde nach den bisherigen Ergebnissen einmalig mit je 1 U/ml Thrombin stimuliert, die Überstände anschließend in einem Zeitraum von 1-24 h abgenommen und im Zymogramm (Abb. 9) analysiert. Parallel zu den stimulierten Zellen wurden unstimulierte Zellen als Negativkontrolle mitgeführt. In der Gelatinzymographie zeigt sich nach 7 h eine geringe Zunahme der Aktivität von pro-MMP-9 und nach 10 h ist ein deutlicher Anstieg erkennbar. Erst nach 24 h ist die maximale Aktivität erreicht. In der Negativkontrolle ist über den gesamten Zeitraum keine nenneswerte Aktivität von pro-MMP-9 zu erkennen. Wie in den Voruntersuchungen ist bei pro-MMP-2 lediglich eine schwache Zunahme sichtbar.
- + - - ++
M M P -9
M M P -2
T h r o m b in U /m l
4 7 7 2 4 2 41 0
+
411
+-
Z e it in h
Abbildung 9: Zeitabhängige Stimulation der Aktivität von MMP-9 durch Thrombin. Die U-2 OS-Zellen wurden in 3 cm Petrischalen 24 h serumfrei kultiviert und mit 1 U Thrombin/ml stimuliert. Die Zellkulturüberstände wurden zwischen 1-24 h nach Stimulation abgenommen, nach Zellzahl normalisiert und in einem gelatinhaltigen SDS-Gel (0,05 % Gelatin) aufgetrennt. Als Positivkontrolle wurden Überstände der Fibrosarkomzellie HT 1080, mit TPA stimuliert, aufgetragen.
Die maximale Aktivität von pro-MMP-9 ist im Überstand nach 24 h meßbar. Es stellte sich im nächsten Schritt die Frage, mit welcher Kinetik die RNA-Expression mit der Aktivität korreliert.
41

D. Ergebnisse
D.5 Zeitabhängige Veränderung der Transkription von MMP-9 nach Stimulation mit Thrombin
Der konzentrationsabhängige Anstieg der pro-MMP-9-Aktivität nach Stimulation mit Thrombin wurde auf Protein- und RNA-Ebene untersucht. Neben der zeitabhängigen Zunahme des aktiven Enzyms wurde der Anstieg der Expression des MMP-9-Gens untersucht. Wann ist nach einmaliger Zugabe von Thrombin die RNA von MMP-9 zu erkennen und wie lange ist die Erhöhung auf RNA-Ebene nachweisbar? Bei 70 % Konfluenz wurde die U-2 OS-Zellinie für 24 h serumfrei kultiviert, das serumfreie Medium wurde vor Zugabe von Thrombin gewechselt und anschließend wurde 1 U Thrombin/ml
zugegeben. Nach 0.5, 2, 4 bis 28 h wurde die RNA mit dem TRIZOL Reagent isoliert. In dem aufgeführten Northern-Blot (Abb. 10) ist im Vergleich zu den Basalwerten nach 4 h ein deutliches RNA-Signal von MMP-9 detektierbar. In der Folgezeit ist eine Schwankung der MMP-9-RNA erkennbar. 8 h nach Stimulation ist eine maximale Expression der MMP-9-RNA detektierbar, nach 6 h und 12 h zeigt sich ein schwächeres Signal. In der Folgezeit steigt die Menge der MMP-9-RNA erneut und ist bis 28 h nach Stimulationsbeginn, annähernd der maximalen Transkriptionsrate, nachweisbar. Wie unter Kapitel C.5 beschrieben, wurde die Nylonmembran anschließend mit einer Gensonde für GAPDH hybridisiert, um die Gesamtmenge der RNA darzustellen. Eine einmalige Stimulation mit Thrombin reicht demzufolge aus, um die Transkriptionsrate von MMP-9 für mindestens 24 h auf ein im Vergleich zu den Basalwerten hohes Niveau zu steigern. Anhand der Zeitspanne zwischen dem Anstieg der MMP-9-RNA und dem Anstieg von pro-MMP-9 erkennt man, daß nach der Transkription bis zur Steigerung der Aktivität etwa 3 h vergehen.
M M P - 9
G A P D H
T h r o m b i n U / m l
0 0 , 5 2 4 6 2 8
T P A
2 4 8 1 2 2 42 0
Abbildung 10: Zeitabhängige Stimulation der Transkription von MMP-9 durch Thrombin. Die U-2 OS-Zellen wurden 24 h serumfrei kultiviert, mit 1 U Thrombin/ml stimuliert, und 0-28 h nach Stimulation wurde die Gesamt-RNA isoliert. Die RNA wurde in einem 1 % Agarosegel aufgetrennt und mit Kapillarblot auf eine Nitrozellulosemembran transferiert. Die Markierung erfolgt mit einer Gensonde für MMP-9. Als Kontrolle der verwendeten RNA-Menge dient eine Hybridisierung des Northern-Blots mit GAPDH. Als Positivkontrolle wurde Gesamt-RNA der Fibrosarkomzellinie HT 1080, mit TPA stimuliert, aufgetragen.
42

D. Ergebnisse
D.6 Thrombin aktiviert den MMP-9-Promoter auf Transkriptionsebene Da Thrombin auf RNA-Ebene eine Induktion von MMP-9 bewirkt, stellte sich die
Frage, ob der Anstieg durch eine vermehrte Transkription des MMP-9 -Gens erreicht wird. Die Regulation eines Gens wird durch die am 5’-Ende vorgeschaltete Promotorregion gesteuert. In diesem DNA-Abschnitt binden Transkriptionfaktoren und beeinflussen den Kopiervorgang von DNA auf RNA. Der Promotorregion wird ein Reportergen, wie CAT oder Luciferase, nachgeschaltet und in die Zellen transfiziert. Anschließend wird die Menge des entstandenen Reporterproteins nachgewiesen.
Um eine hohe Transfektionsrate der U-2 OS-Zellinie zu erhalten, wurde zunächst mit der X-Gal-Färbung die Effizienz verschiedener Transfektionsmethoden ausgewertet. Verglichen wurde u. a. die Kalziumphosphatmethode, das Lipofectin-Reagens von Gibco® und das Superfect-Reagens von Qiagen. In den Abbildungen kann man anhand der blaugefärbten Zellen die Transfektionseffizienz der einzelnen Ansätze erkennen. Mit dem SuperFect Transfektion Reagent von Qiagen wurde für diese Zellinie die höchste Transfektionsrate erreicht (Abb 11).
S u p erfec t L ip o fec tin K alz iu m -P h o s.
Abbildung11: X-Gal-Färbung der U-2 OS-Zellinie nach Transfektion mit verschiedenen Methoden. Die Zellen wurden nach dem von den Herstellern vorgegebenen Protokoll mit dem Expressionsvektor ß-Galaktosidase transfiziert und nach 24 h mit X-Gal angefärbt (siehe Methoden).
Zum Nachweis, daß die Transkription des kodierenden Genabschnitts für MMP-9
durch Thrombin erhöht ist, wurde in transienten Transfektionen die Aktivität des MMP-9-Promotors unstimuliert und nach Zugabe von Thrombin gemessen. Dazu wurden die U-2 OS-Zellen in Reportergenassays mit dem MMP-9-Promoter transient transfiziert. Als Positivkontrolle wurde das Onkogen H-Ras, ein Signaltransduktionsprotein, kotransfiziert. In früheren Untersuchungen konnte gezeigt werden, daß der MMP-9-Promoter durch H-Ras
43

D. Ergebnisse
aktiviert wird (Gum et al., 1996). 24 h nach der Transfektion wurden die Osteosarkomzellen mit 1 U Thrombin/ml stimuliert. Die Zugabe von Thrombin 24 h nach der Transfektion bewirkt einen deutlichen Anstieg der Aktivität des MMP-9-Promotors (Abb. 12). Die Aktivierung erreicht annähernd die gleiche Höhe wie durch H-Ras. Um einen alleine durch den Vektor pSV-2-neo von H-Ras verursachten Effekt auszuschließen, wurde das Plasmid ohne eingesetzte Abschnitte parallel transfiziert und analysiert. In der Negativkontrolle konnte keine Aktivierung des Promotorabschnittes festgestellt werden. Während unstimulierte U-2 OS-Zellen eine 1,2-fache CAT-Konversion zeigten, wurde nach Stimulation mit Thrombin eine 2,4-fache Konversion gemessen (Abb 12).
MMP9-670 PromotorpSV2neoh-rasThrombin
+ + + ++ - - -- + - -- - - +
Abb. 12A CAT-Assay
44

D. Ergebnisse
0,00
0,50
1,00
1,50
2,00
2,50
3,00
3,50
Leervektor Positivkontrolle Negativkontrolle Thrombin
x-fa
che
Indu
ktio
n
Abb. B: Grafische Auswertung
Abbildung 12: Thrombin induziert den MMP-9-Promoter. Die U-2 OS-Zellinie wurde in 10 cm Petrischalen mit 5 µg eines CAT-Vektors, der durch den MMP-9-Promoter gesteuert wird, transient transfiziert und nach 24 h mit 1 U Thrombin/ml stimuliert. Nach 24 h wurden die Zellen geerntet, der CAT-Reporterassay durchgeführt und das Chloramphenicol durch Dünnschichtchromatographie aufgetrennt (Abb. A). Die Konversion von Chloramphenicol D-threo-[1,2-14C] in die verschiedenen acetylierten Derivate wurde mit dem Phosphor-Imager bestimmt. Die Induktion wird als X-fache zu der basalen MMP-9-Promoter-Aktivität angegeben (Abb. B). Als Positivkontrolle wurde die Zellinie mit 2,5 µg des Onkogens H-Ras transfiziert.
Die Inkubation der U-2 OS-Zellinie mit Thrombin aktiviert den MMP-9-Promotor.
Nach Aktivierung des Thrombinrezeptors kann das Signal über verschiedene Signaltransduktionswege in den Zellkern bis zu den Bindungsstellen des Promotors weitergeleitet werden. Im nächsten Schritt soll deshalb der verantwortliche Signaltransduktionsweg identifiziert werden.
45

D. Ergebnisse
D.7 Die MMP-9-Transkription wird über einen PI3-Kinase abhängigen Signaltransduktionsweg reguliert
Die Untersuchung des durch Thrombin induzierten Signaltransduktionsweges, der zur Aktivierung des MMP-9-Promoters führt, ist ein weiteres Ziel des Projektes. Wie bereits gezeigt (Kap. D.2), konnte die Aktivierung von MMP-9 durch das TRAP-Peptid nachgewiesen werden. Damit wurde gezeigt, daß Thrombin MMP-9 über den spezifischen Thrombinrezeptor aktiviert. Somit muß man davon ausgehen, daß Thrombin Signale über den Thrombinrezeptor in das Zellinnere überträgt. Nach Aktivierung des Thrombinrezeptors kann das Signal über verschiedene Transduktionswege bis in den Zellkern weitergeleitet werden. So wird durch die intrazellulären Untereinheiten des Thrombinrezeptors, Gαq und Gßγ, die MAP Kinase oder die PI 3-Kinase aktiviert.
Durch die Hemmung einzelner Schlüsselproteine, die an der Übertragung der Signale intrazellulär beteiligt sind, wurde versucht, den durch Thrombin ausgelösten Signaltransduktionsweg zu identifizieren. Die U-2 OS-Zellinie wird 1 h vor Stimulation mit verschiedenen synthetischen Hemmstoffen inkubiert und anschließend mit 1 U Thrombin/ml inkubiert. Getestet wurden 3 Inhibitoren, die bekannte Signaltransduktionskaskaden hemmen. Die verwendeten Hemmstoffe blockieren einzelne Schlüsselenzyme und schalten damit die gesamte Kaskade aus. Die eingesetzten Inhibitoren sind für den MAP-Kinase- und für den PI3-Kinase-Weg spezifisch. SB 203580 und PD 98059 blockieren Bestandteile der MAP-Kinasen. SB 203580 hemmt die p38-Kinase und PD 98059 hemmt die Aktivierung von MEK1. Durch den Einsatz von Ly 294002 wird selektiv die PI3-Kinase gehemmt. (Abb. 3, Einleitung Kapitel A.13).
24 h nach Stimulation wurde der Zellkulturüberstand mit der Gelatinzymographie untersucht. Bei den Experimenten hat sich gezeigt, daß der spezifische p38 MAPK–Inhibitor SB 203580 keinen Einfluß auf die Stimulation der MMP-9-Aktivität durch Thrombin hat. Ebensowenig PD 98059, ein spezifischer Inhibitor für MEK1. Dies zeigt, daß der MAPK-Signaltransduktionsweg vom Thrombinrezeptor in U-2 OS-Zellen nicht an der Aktivierung von MMP-9 beteiligt ist. Die Zugabe des spezifischen PI3-Kinase-Inhibitors Ly 294002 hingegen konnte die Aktivierung hemmen. Die Inhibierung ist konzentrationsabhängig (Abb. 13).
In der angeführten Gelatinzymographie ist in den ersten beiden Spuren der Zellüberstand von U-2 OS-Zellen nach 24 h ohne Stimulation aufgetrennt. Die basale Aktivität von pro-MMP-2 und –9 ist gering ausgeprägt. In den mittleren zwei Spuren wurde der Überstand von U-2 OS-Zellen 24 h nach Stimulation mit Thrombin aufgetrennt. Es ist eine deutliche Steigerung der Aktivität von pro-MMP-9 zu erkennen. Die Aktivität von pro-MMP-2 wird durch Thrombin wie auch in den vorausgegangenen Experimenten nicht beeinflußt. In den folgenden zwei Spuren ist der Zellüberstand von U-2 OS-Zellen, die vor der Thrombinstimulation mit unterschiedlichen Konzentrationen von Ly 294002 inkubiert wurden, aufgetrennt. Im Vergleich zur Thrombinstimulation ist bereits bei einer
46

D. Ergebnisse
Konzentration von 20 µM/ml Ly 294002 eine Reduktion der pro-MMP-9-Aktivität zu erkennen. Mit der erhöhten Konzentration von 40 µM/ml wird die stimulierende Wirkung von Thrombin nahezu vollständig aufgehoben.
M M P-9
M M P-2
Throm bin 1,0 U /m l - - + + + +
Ly 294002 µM /m l 20 40
Abbildung 13: Hemmung durch den PI3-Kinase-Inhibitor Ly 294002. Die U-2 OS-Zellen wurden 24 h serumfrei kultiviert und vor Stimulation mit den angegebenen Konzentrationen der Inhibitoren SB 203580, PD 98059 und LY 294002 inkubiert. 1 h nach Zusetzen der Hemmstoffe erfolgte die Stimulation mit 1 U Thrombin/ml. Die Zellkulturüberstände wurden 24h nach Stimulation abgenommen, bzgl. der Zellzahl normalisiert und in einem gelatinhaltigen SDS-Gel (0,1 % Gelatin) aufgetrennt. Das Experiment wurde dreimal wiederholt.
Mit dem Einsatz von Ly 294002 wird die Steigerung von pro-MMP-9 durch
Thrombin gehemmt. Demnach scheint der PI3-Kinaseweg für die Stimulation der MMP-9-Expression und die Aktivierung wichtig zu sein.
D.8 Identifizierung des MMP-9-Promotorabschnittes, der für die Stimulation durch Thrombin verantwortlich ist
Wie bereits unter Punkt X beschrieben, wird ein Gen in der Regel von der vorgeschalteten Promotorregion reguliert, in welcher verschiedene Transkriptionsfaktoren binden können. Nach Stimulation erfolgt eine Aktivierung spezifischer Signaltransduktionwege, und als Endprodukt bindet ein Transkriptionsfaktor oder ein Komplex von Faktoren an einer Konsensusbindungsstelle im Promotor und induziert die Transkription des Gens. Um die Bindungsstelle lokalisieren zu können, die nach Stimulation mit Thrombin das Ablesen des MMP-9-Gens beeinflußt, werden von der Promotorregion unterschiedlich lange Sequenzen verwendet und in das Reporterkonstrukt kloniert. Transfiziert man die Zellen mit den Deletionsmutanten und stimuliert mit Thrombin, so
47

D. Ergebnisse
kann eine Abnahme der Aktivität des Reportergens einen Hinweis auf die spezifische Bindungsstelle in der jeweiligen Sequenz geben.
In der Untersuchung wurden verschiedene Deletionen des MMP-9-Promotors von -670 bp Startpunkt der Transkription über -634, -599 und bis -73 verwendet. Als Vektor wurde das Reporterplasmid pGL3-Luciferase verwendet. Wichtige Bindungsstellen in diesem Bereich sind die beiden AP-1-Bindungsstellen auf Position -533 und- 79 bp (Sato et al., 1993a+b). Die Transfektion wurde mit dem SuperFect Transfektion Reagent von Qiagen, wie unter Kap. C.6 beschrieben, durchgeführt.
Nach Stimulation mit Thrombin konnte eine etwa 50%ige Reduktion der Luciferaseaktivität zwischen dem –670bp-Konstrukt und dem –634bp-Konstrukt festgestellt werden. Demnach sind in der Sequenz dieses Promotors Bindungsstellen für Transkriptionsfaktoren vorhanden, die für die Aktivierung mit Thrombin wichtig sind. Die Verwendung des kurzen –73bp Fragments des MMP-9-Promotors zeigt keine Aktivierung durch Thrombin.
Stimulatio n der MMP9-Pro motor Mutanten mit Thrombin
0
400
800
1200
1600
2000
2400
2800
MMP9-670 MMP9-634 MMP9-599 MMP9-73
RLU
Abbildung 14: Identifizierung von Bindungsstellen, die durch Thrombin aktiviert werden. Die U-2 OS-Zellinie wurde in 3 cm Petrischalen mit 5 µg eines Luciferase-Vektors, der durch den MMP-9-Promoter gesteuert wird, transient transfiziert und nach 24 h mit 1 U Thrombin/ml stimuliert. Nach 24 h wurden die Zellen geerntet und das Zellysat nach den gleichen Arbeitsschritten wie für den CAT-Assay hergestellt. Die Induktion wird als X-fache zu der basalen MMP-9-Promoter-Aktivität angegeben. Als Positivkontrolle wurde die Zellinie mit 2,5 µg H-Ras in dem pSV-2-neo-Vektor transfiziert.
48

D. Ergebnisse
D.9 Thrombin steigert die Invasivität von Tumorzellen Bei Tumorpatienten beobachtet man eine gesteigerte Koagulationsneigung und
damit ein erhöhtes Risiko, thromboembolische Komplikationen zu erleiden (Kakkar et al., 1995). Thrombin beeinflußt neben der Blutgerinnung wahrscheinlich weitere biologische Funktionen wie Proliferation und Migration von Zellen. Für die Metastasierung von Tumorzellen muß die Basalmembran überwunden werden. Durch Zugabe von Thrombin kann ein Anstieg der Aktivität von Metalloproteasen erreicht werden. Die verschiedenen Untergruppen der MMP-Familie sind für die Invasion und Metastasierung von Tumorzellen wichtig (Duffy et McCarthy, 1998). Es stellt sich deshalb die Frage, ob Thrombin die Invasion der Osteosarkomzellen aktiviert. In einem Invasionsassay mit Matrigel, das die Hauptbestandteile der Basalmembran enthält, wurde die U-2 OS-Zellinie mit Thrombin aktiviert. Für diesen Assay werden Membranen verwendet, die mit Matrigel beschichtet sind. Dieser Ansatz ist ein in vitro Assay, um die Migration von Zellen durch eine natürliche Barriere zu beobachten und zu quantifizieren. Damit kann das Metastasierungspotential nach Stimulierung mit Thrombin untersucht werden. Die Osteosarkomzellen U-2 OS wurden bei ca. 70 % Konfluenz abgelöst, gezählt und in serumfreien Medium auf der Oberseite der Matrigelmembran in Transwell-Kammern (Durchmesser 2,4 cm) ausgesät. In die untere Kammer wurde Kulturmedium von einer Fibroblastenzellinie, als Chemotaxis für die Osteosarkomzellen, gefüllt. Nach 1 h wurden die Zellen einmalig mit 1U Thrombin/ml stimuliert. Zusätzlich zu der Negativkontrolle mit unstimulierten Zellen wurde als Positivkontrolle eine Stimulation mit EGF mit 40 ng EGF /ml durchgeführt. Durch Zugabe von EGF wird der spezifische EGF-Rezeptor aktiviert. Eine Steigerung der Invasion ist dabei zu beobachten (Daub et al., 1997). Für eine exakte Bestimmung des Invasionsverhaltens der Tumorzellen wurden die Zellen ausgewertet, die die Matrigelschicht überwunden hatten. 72 h nach einmaliger Stimulation mit Thrombin wurde die obere Matrigel-Schicht entfernt. Die Zellen, die die Matrigelschicht überwunden hatten und an der Unterseite der Membran anlagen, wurden mit MTT gefärbt und quantitativ ausgewertet. Im Vergleich zu den unstimulierten Zellen oder mit EGF inkubierten Zellen konnte bei den mit Thrombin stimulierten Zellen auf der Unterseite der Matrigelmembran eine deutliche Steigerung an U-2 OS-Zellen festgestellt werden. Die Stimulation mit EGF hingegen bewirkte keine nennenswerte Veränderung des Migrationsverhaltens. Durch einmalige Zugabe von Thrombin wurde die Migration der Osteosarkomzellen durch Matrigel® deutlich gesteigert.
49

D. Ergebnisse
Invasion nach Stim ulation m it Throm bin
0
5
10
15
20
25
Negativkontrolle EGF Thrombin
Zelle
n/G
esic
htsf
eld
Abbildung 15: Steigerung der Invasion durch Thrombin. Die U-2 OS-Zellen wurden bei einer Konfluenz von ca. 70 % abgelöst und in mit Matrigel beschichteten 2,4 cm-Einsätzen ausgesät. Je Ansatz wurden 105 Zellen in serumfreiem Medium ausgesät. Als Chemotaxis wurde konditioniertes Medium einer NIH 3T3-Zellinie in der unteren Kammer eingesetzt. Die Zellen wurden mit 1 U Thrombin/ml oder 40 ng EGF/ml stimuliert, nach 72 h wurde die obere Matrigelschicht entfernt und die Zellen mit MTT gefärbt. Die Auswertung erfolgte photometrisch nach Lösung des Farbstoffes in DMSO.
D.10 Nachweis des Thrombinrezeptors in Osteosarkomgewebe Neben der Generierung von Fibrin bewirkt Thrombin eine Änderung in der
Zellmorphologie und dem Wachstum von Zellen. Thrombin, ein starker Promotor der Thrombozytenaggregation (Berndt et al.,1981), wirkt chemotaktisch auf Monozyten (Bar-Shavit et al., 1983) und hat eine mitogene Wirkung auf Endothelzellen (Chen et al.,1975). In den vorliegenden Ergebnissen zeigte sich zudem, daß Thrombin die proteolytische Aktivität von Tumorzellen steigert. Insofern sollte im nächsten Experiment der Thrombinrezeptor in Osteosarkomgewebe nachgewiesen und gezeigt werden, ob humanes Osteosarkomgewebe den Thrombinrezeptor exprimiert.
Aus Biopsiematerial von Osteosarkompatienten wurde mit dem Trizol® Reagent RNA präpariert. Nach reverser Transkription wurde eine PCR mit Primern aus der 5’- und 3’-Region der cDNA des Thrombinrezeptors durchgeführt. Die PCR-Produkte wurden anschließend in einem Agarosegel aufgetrennt. In dem dargestellten Agarosegel (Abb. 16) ist sowohl in der RNA der Osteosarkomzellinie als auch in den beiden Gewebeproben ein gleich grosses PCR-Produkt zu erkennen. Es korreliert von der Produktgröße mit dem zu erwartetenden cDNA-Abschnitt des Thrombinrezeptors. Neben dem Nachweis des
50

D. Ergebnisse
Thrombinrezeptors in der verwendeten U-2 OS-Zellinie konnte dieser Rezeptor somit auch in beiden Gewebeproben amplifiziert werden.
Osteo-SA
1
Marker
Osteo-SA
2
U2 O
S
Negativ-K
o.
1000 bp
500 bp
Abbildung 16: Nachweis des Thrombinrezeptors in Osteosarkomgewebe und in U-2 OS-Zellen. Gewebeproben von Osteosarkomgewebe wurden in einem gekühlten Mörser pulverisiert, in TRIZOL-Reagens aufgenommen und die RNA isoliert. Die RNA der U-2 OS-Zellinie wurde ebenfalls mit dem TRIZOL-Reagens isoliert. Nach Durchführung der RT-PCR wurden die Proben in einem 1 % Agarosegel analysiert.
51

E. Diskussion
E.1 Der Einfluß von Thrombin auf die Aktivität und die Expression von Typ-IV Kollagenasen
Metastasierung ist die Aussaat von malignen Zellen vom Primärtumor in ein anderes Organ und deren Anwachsen und weitere Ausbreitung (Sporn, 1996). Es ist der beängstingendste Aspekt von Krebserkrankungen, da trotz großer Fortschritte in der Früherkennung von Krebs, verbesserter chirurgischer Techniken, wirkungsvollerer Strahlentherapie und systemischer Chemotherapie die meisten Todesfälle durch Metastasen bedingt werden, die oft durch keine Therapie mehr beherrscht werden können. Zum Zeitpunkt der Diagnosestellung ist bei 20% der Patienten mit einem Osteosarkom schon eine Metastasierung nachzuweisen. Mikrometastasen, die mit den derzeitigen, bildgebenden Verfahren nicht erkennbar sind, müssen sogar bei 80 % aller Patienten angenommen werden (Saeter et al., 1995). Der Metastasierungsprozeß ist ein sehr komplexer Vorgang, der von der Tumorzelle sehr viele Anpassungsvorgänge verlangt und zum besseren Verständnis in verschiedene Stufen unterteilt wird (Hanahan et Weinberg, 2000). Nach lokaler Invasion in das umgebende Bindegewebe wandern Tumorzellen in die lokalen Blutgefäße ein und werden dort vom Blutstrom wegtransportiert (Quigley and Armstrong, 1998). Eine starke Vaskularisation erhöht für Tumorzellen die Wahrscheinlichkeit, Anschluß an das Blutgefäßsystem zu finden und zu metastasieren. Es kommt in dem nachfolgenden Kapillarstrombett (z. B. Lunge, Leber), in dem das Blut langsam fließt, zur Extravasation aus den Blutgefäßen und zur Tumorbildung in dem neuen Organ (Fidler et al., 1994). Es wiederholt sich dann der Zyklus aus Zellproliferation und lokaler Invasion der Tumorzellen, die mit Hilfe von tumorassoziierten Proteasen die extrazelluläre Matrix auflösen. Das Osteosarkom ist ein hochmaligner Tumor mit einem raschen infiltrativen Wachstum. Es werden frühzeitig vorwiegend pulmonale Metastasen gebildet.
Die Aktivierung der intrinsischen oder extrinsischen Blutgerinnungskaskade mündet in der Ausbildung des sogennanten Prothrombinase-Komplexes, welcher Prothrombin zu Thrombin umwandelt (Davie, 1991). Thrombin verwandelt zirkulierendes Fibrinogen in Fibrinmonomere, welche zu Fibrin polymerisieren und damit die Matrix für die Thrombusbildung darstellen. Daneben hat Thrombin auch eine direkte Wirkung auf Zellen. Es triggert die Mobilität von Entzündungszellen und Endothelzellen, und die für die Migration von Zellen wichtigen Adhäsionsmoleküle werden vermehrt gebildet (Chiang et Huang, 1996). Entscheidend für die Tumorbiologie ist der Einfluß von Thrombin auf Tumorzellen, da die Proliferation, die Invasion und die Metastasierung beeinflußt werden. So wird die Proliferation einer Tumorzellinie mit der Zugabe von Thrombin gesteigert und im Vergleich zu unstimulierten Zellen eine Matrigelmembran schneller überwunden (Chaikof et al., 1995; Henrikson et al., 1999). Durch die Inkubation mit Thrombin ist eine
52

E. Diskussion
vermehrte Metastasierung von Tumorzellen zu beobachten (Nierodzik et al., 1992; Wojtukiewicz et al., 1993, 2000).
Eine sehr wichtige Arbeit, die sich ebenfalls mit der Wirkung von Thrombin auf die Auflösung von extrazellulären Strukturen befaßt, ist von Fabunmi vorgestellt worden. Fabunmi untersuchte den Einfluß von Wachstumsfaktoren und Zytokinen auf die Typ-IV Kollagenasen (Fabunmi et al., 1996). Bei pathologischen Veränderungen der Gefäßwand wie bei der Atherosklerose wandern Gefäßmuskelzellen von der Tunica media in die Tunica intima und bilden eine Neointima. Während des Umbauvorganges muß die Membrana elastica interna als Basalmembran zwischen Media und Intima abgebaut werden. In der Arbeit wurden Gefäßmuskelzellen kultiviert und mit Thrombin in Konzentrationen von 2 U/ml stimuliert. Nach 4-12 h wurde der Überstand der Zellkultur in der Gelatin-Zymographie analysiert und die Aktivität von pro-MMP-9 bestimmt. Es zeigte sich eine Steigerung der Aktivität von pro-MMP-9 durch Thrombin, durch Zugabe von bFGF konnte dieser Effekt potenziert werden. Die Aktivität von pro-MMP-2 hingegen blieb unverändert. Vergleicht man das Ergebnis von Fabunmi et al. zu der vorliegenden Arbeit, ist in beiden Arbeiten eine Steigerung der Aktivität von pro-MMP-9 zu erkennen. Entsprechend unseren Ergebnissen wurde in der Arbeitsgruppe von Fabunmi in der Zymographie ebenfalls keine Steigerung von aktivem MMP-9 nachgewiesen.
Berücksichtigt man, daß verschiedene Zellinien verwendet wurden und die Experimente in unterschiedlichen Instituten durchgeführt wurden, ist die Aktivierung von pro-MMP-9 durch Thrombin als ein genereller Mechanismus in der Zellbiologie anzusehen. Zudem wird durch die Ergebnisse beider Arbeiten eine Reproduzierbarkeit der durchgeführten Experimente bewiesen.
Weder in seiner Arbeit noch in den hier vorgestellten Untersuchungen konnte ein Einfluß auf die Aktivierung von pro-MMP-2 zu MMP-2 gezeigt werden. Im Unterschied zu dem genannten Experiment wurde in der vorliegenden Arbeit die Osteosarkomzelllinie U-2 OS vor Inkubation 24 h mit serumfreiem Medium kultiviert. In unserer Arbeit wurde zusätzlich festgestellt, daß die Aktivität von pro-MMP-9 durch Thrombin konzentrationsabhängig von 0,25-1,0 U Thrombin/ml gesteigert wird.
Im Gegensatz dazu wurde in der Arbeit von Zucker nachgewiesen, daß aktives Thrombin bei Endothelzellen die Sekretion von pro-MMP-2 steigert (Zucker et al., 1998). Aktiviert man Endothelzellen von venösen Umbilikalgefäßen mit VEGF, wird vermehrt tissue factor gebildet. In Anwesenheit des Prothrombinase-Komplexes und Prothrombin wird dadurch aktives Thrombin freigesetzt. Die Stimulation mit aktivem Thrombin bewirkt eine erhöhte Aktivität von pro-MMP-2, wie in der Gelatin-Zymographie zu erkennen ist. Diese Steigerung konnte durch Gabe von Anti-Thrombin-III unterdrückt werden. Im Unterschied zu unserer Arbeit wurden die Zellen in serumhaltigem Medium kultiviert. Damit ist ein Einfluß von in diesem Kulturmedium vorhandenen Wachstumsfaktoren und Matrixmetalloproteasen nicht auszuschließen. In der Arbeit findet sich kein Hinweis auf
53

E. Diskussion
eine Analyse der Aktivität von MMP-9, da Endothelzellen neben MMP-2 hauptsächlich MMP-1 sezernieren. Die Steigerung der Aktivität von pro-MMP-2 durch Thrombin wurde in einer weiteren Arbeit an Mesenterialgefäßen von Ratten nachgewiesen (Fernandez-Patron et al., 2000). Die Erhöhung der Aktivität von pro-MMP-2 ist im Vergleich zu pro-MMP-9 jedoch gering ausgeprägt. In der Analyse mittels Northern-Blot konnte in der vorliegenden Arbeit nach Zugabe von Thrombin ebenfalls ein konzentrationsabhängiger Anstieg der Transkription des MMP-9-Gens von 0,5-2,0 U Thrombin/ml nachgewiesen werden.
Die Untersuchungen zur Zeitabhängigkeit nach Zugabe von Thrombin zeigten einen Anstieg der Aktivität von pro-MMP-9 nach 10 h, das Maximum wurde nach 24 h erreicht. In der bereits erwähnten Arbeit von Fabunmi wird ebenfalls ein Anstieg nach 8-10 h beobachtet, ein Maximum ist nach 24 h sichtbar (Fabunmi et al., 1996). In der Northern-Blot-Analyse ist ein Anstieg der MMP-9-RNA nach 4 h feststellbar, was auf eine transkriptionelle Regulation hinweist. Die erhöhten Transkriptionsprodukte der konzentrationsabhängigen Thrombinstimulation sind demzufolge nicht das Resultat einer verlängerten Halbwertszeit der MMP-9-RNA (Overall et al.,1991). Die Ergebnisse der transienten Transfektionen bestätigen, daß durch die Aktivierung mit Thrombin die Transkription des MMP-9-Gens gesteigert wird.
Als Vergleich zu dem zeitabhängigen Anstieg der mRNA von MMP-9 sind Arbeiten über den Einfluß von Thrombin auf die Transkription des uPA-Rezeptors und von Thrombomodulin zu erwähnen. In diesen Arbeiten wurde ebenfalls eine zeitabhängige Analyse durchgeführt. Nach Zugabe von Thrombin ist in der Northern-Blot-Analyse ein Anstieg der mRNA des uPA-Rezeptors nach 5 h und von Thrombomodulin nach 3 h zu erkennen (Reuning et al., 1994; Ma et al., 1997). Nach der Stimulation mit Thrombin läßt sich in der Zellkultur eine nahezu zeitgleiche Erhöhung der RNA von MMP-9, uPA und Thrombomodulin nachweisen. Es ist somit möglich, daß Thrombin eine größere Anzahl von Genen, insbesondere von Proteasen die für die Metastasierung wichtig sind, aktiviert.
Die in der vorliegenden Arbeit durchgeführten Zymographien zeigen eine Steigerung von pro-MMP-9, da die Aktivität der Typ-IV Kollagenasen durch Banden im Bereich von 92 und 72 kDa dargestellt wird. Diese entsprechen pro-MMP-9 mit einem Molekulargewicht von 92 kDa und pro-MMP-2 von 72 kDa. Nach der Aktivierung hat MMP-9 88 kDa und MMP-2 64 kDa (Sawicki et al., 1997; Coussens et al., 2000). Demnach wird von den Osteosarkomzellen nach Stimulation mit Thrombin die inaktive Vorstufe von MMP-9 vermehrt sezerniert. Damit stellt sich die Frage, durch welchen Mechanismus die Typ-IV Kollagenasen in vivo nach der Sekretion aktiviert werden oder welche Bedeutung die Vorstufen bei dem Vorgang der Invasion haben.
Einerseits kann es sein, daß MMP-9 auch in der pro-Form aktiv ist, dies jedoch nicht in der Zymographie detektiert werden kann, da Gelatin ein artifizielles Substrat ist. Eine Aktivierung von pro-MMP-9 nach der extrazellulären Präsentation durch umliegende Stromazellen wie Fibroblasten scheint nicht zu bestehen. In einer Arbeit von Lengyel et al.
54

E. Diskussion
wurde eine Karzinomzellinie mit einer Fibroblastenzellinie ko-kultiviert. In der Gelatin-Zymographie konnten lediglich die inaktiven Vorstufen nachgewiesen werden (Lengyel et al., 1995). Eine weitere Möglichkeit stellt die Aktivierung von pro-MMP-9 durch Entzündungszellen dar. In einem Mausmodell konnte gezeigt werden, daß Entzündungszellen bei der Präsentation von aktiven MMP-9 beteiligt sind (Coussens et al., 2000).
Neben der proteolytischen Aktivität könnte pro-MMP-9 auch durch Rekrutierung von Integrinen die Invasion von Tumorzellen verstärken. So wurde von Brooks eine Kolokalisation von MMP-2 und integrin alpha v beta 3 an der Zelloberfläche einer Melanomzellinie nachgewiesen (Brooks et al.,1996). Eine weitere Möglichkeit ist eine Funktion von pro-MMP-9, die durch den bisherigen Kenntnisstand noch nicht erklärbar ist.
E.2 Die Steigerung der Aktivität von MMP-9 durch Thrombin wird durch Aktivierung des Thrombinrezeptors vermittelt
Die Wirkung von Thrombin wird durch den spezifischen Thrombinrezeptor, einen siebenspännigen Rezeptor, übermittelt. Die Präsentation von TF an der Zelloberfläche und die damit verbundene Thrombingenerierung führt zu einer Aktivierung des Thrombinrezeptors (Fischer et al., 1995). In den letzten Jahren wurden vier verschiedene, spezifische Thrombinrezeptoren, nach der Aktivierung durch die Protease Thrombin auch protease activated receptor (PAR) genannt, nachgewiesen (Vu et al., 1991; Rasmussen et al., 1991; Nystedt et al., 1994; Ishihara et al., 1997; Kahn et al., 1998; Xu et al., 1998). Thrombin spaltet das carboxyterminale Ende des Rezeptors, das neu entstandene Rezeptorende lagert sich an der zweiten extrazellulären Domäne an und bewirkt eine Konformationsänderung des Rezeptors. Nach der Aktivierung wird intrazellulär über G-Proteine das Signal an Signaltransduktionswege weitergeleitet (Gerszten et al.,1994; Trejo et al., 1996).
Neben siebenspännigen Rezeptoren werden über Thrombin indirekt auch Rezeptortyrosinkinasen aktiviert. Mit dem Einsatz von Thrombin wird der EGF-Rezeptor aktiviert, die Aktivität der Typ-IV-Kollagenasen kann auch durch Oberflächenrezeptoren wie den IGF-1R verändert werden (Daub et al., 1997; Long et al., 1998). Mit der Verwendung des Agonisten TRAP (thrombin receptor activating peptide), welcher nach der Aktivierung das neue extrazelluläre Rezeptorende darstellt, wird die Weiterleitung des Signals über den Thrombinrezeptor und der Einfluß auf MMP-9 nachgewiesen (Vassallo et al., 1992; Gerszten et al.,1994). TRAP bewirkt somit eine spezifische Aktivierung des Thrombinrezeptors.
Durch das Zusetzen von TRAP mit der Aminosäuresequenz SFLLRN konnte in der Zymographie eine konzentrationsabhängige Steigerung der Aktivität von pro-MMP-9 erreicht werden. Damit wird die Spezifität der Aktivierung von MMP-9 durch Thrombin über den Thrombinrezeptor bewiesen. Die Steigerung entsprach der parallel durchgeführten
55

E. Diskussion
Stimulation mit Thrombin. Von den vier verschiedenen Thrombinrezeptoren wird nur PAR-1 durch das verwendete Peptid aktiviert. PAR-2 wird nur durch Trypsin, Tryptase oder die Blutgerinnungsfaktoren VIIa und Xa aktiviert (Molino et al., 1997). PAR-3 ist bei der Aktivierung von PAR-4 als Kofaktor beteiligt (Nakanishi-Matsui et al., 2000). PAR-4 wird durch das Peptid AYPGKF spezifisch aktiviert und kann durch das in meinen Versuchen verwendete Peptid SFLLRN nicht aktiviert werden (Faruqi et al., 2000). Die eingesetzte Konzentration von TRAP entspricht den Werten, die in der Literatur angegeben werden (Reuning et al., 1994; Kanthou et al., 1995, Andersen et al., 1999).
E.3 Die Bedeutung des PI3-Kinase-Weges bei der Signalübermittlung Die Aktivierung von PAR-1 beeinflußt in der Zellkultur die Proliferation und die
proteolytische Aktivität. Durch Inkubation mit TRAP ist eine beschleunigte Zellteilung bei Gefäßzellen und Fibroblasten zu beobachten (Kanthou et al., 1995; Trejo et al., 1996). Nach Aktivierung des Thrombinrezeptors wird das Signal über G-Proteine an verschiedene Signaltransduktionswege weitergeleitet (Hung et al., 1992; Offermanns et al., 1994; Barr et al., 1997). Durch selektive Blockierung von Schlüsselenzymen der wichtigsten Signaltransduktionswege konnte in der vorliegenden Arbeit nachgewiesen werden, daß nach Aktivierung des Thrombinrezeptors das Signal über die PI-3 Kinase weitergeleitet wird. Mit der Zugabe von Ly 294002, einem Inhibitor der PI-3 Kinase, wurde die Erhöhung der pro-MMP-9-Aktivität durch Thrombin verhindert. Bisherige Arbeiten zeigten, daß die Gβγ-Untereinheit von PAR-1 die PI-3 Kinase aktivieren kann (Clapham et Neer, 1997). PAR-1 kann neben der PI-3 Kinase aber auch den MAPK-Signaltransduktionsweg aktivieren (Kramer et al., 1995; Trejo et al., 1996). ERK, JNK und p38 sind die Schlüsselenzyme der MAPK, und die Aktivität von MMP-9 kann durch diese Proteinkinasen reguliert werden (Lakka et al., 2000). Durch Zugabe der Inhibitoren PD 98059 und SB 203580 werden mit MEK1 und der p38-Kinase wichtige Kinasen des MAPK gehemmt. In der Gelatinzymographie zeigte sich, daß beide Inhibitoren die thrombinspezifische Aktivierung von pro-MMP-9 nicht beeinflussen konnten. Somit wird nach Aktivierung des Thrombinrezeptors durch Thrombin das Signal lediglich über die PI3-Kinase weitergeleitet. Ellerbroek untersuchte die Signaltransduktion und die Veränderung der Aktivität von MMP-9 nach Aktivierung des EGF-Rezeptors bei einer Ovarialkarzinom-Zellinie. Durch Inhibierung der PI3-Kinase konnte ein Rückgang des membrangebundenen pro-MMP-9 beobachtet werden, Inhibitoren der MAPK hatten keinen Einfluß. Die PI3-Kinase reguliert demzufolge die Aktivität von membrangebundene pro-MMP-9 und damit die zielgerichtete Auflösung der extrazellulären Matrix (Ellerbroek et al. 2001). Eine mögliche Bestätigung zu diesen in vitro-Versuchen stellt der Nachweis einer erhöhten Aktivität der PI3-Kinase in Gewebeproben von kolorektalen Tumoren und Ovarialkarzinomen dar (Philipps et al., 1997, Shayesteh et al., 1999).
56

E. Diskussion
Der MAPK-Signaltransduktionsweg spielt aber eine wichtige Rolle für die Regulation der MMP-9-Expression und der Invasion. In einer Arbeit von Simon wurde eine Reduktion der MMP-9-Expression durch die Zugabe von PD 98059 oder SB 203580 erreicht, wobei in diesen Experimenten ebenfalls nicht mit Thrombin stimuliert wurde (Simon et al., 1996; Gum et al., 1997). Simon zeigte in einer Folgearbeit, daß bei einer Karzinomzellinie durch Zugabe von PMA die Aktivität von pro-MMP-9 und von Urokinase gesteigert wird. Mit dem Einsatz von SB 203580 wurde gezeigt, daß die Signaltransduktion dabei über die p38-Kinase erfolgt. Des Weiteren wurde in einem Invasionsassay aufgezeigt, daß mit der Steigerung von MMP-9 durch PMA das Invasionsverhalten der Karzinomzellen erhöht wird (Simon et al., 1998).
In der vorliegenden Arbeit wurde erstmals gezeigt, daß nach Aktivierung des Thrombinrezeptors die Aktivität von pro-MMP-9 über PI3-Kinase reguliert wird.
E.4 Identifizierung des regulierenden Abschnittes des MMP-9-Promotors nach Stimulation durch Thrombin
Die Expression der meisten Matrixmetalloproteinasen ist in normalem Gewebe niedrig und wird für einen Umbau der extrazellulären Matrix induziert. Die Expression von Matrixmetalloproteinasen erfolgt auf Transkriptionsebene und wird über den Promotor gesteuert. Im Unterschied zu den übrigen Metalloproteasen weist der Promotor des MMP-9-Gens eine zweite AP-1-Bindungsstelle auf (Sato and Seiki, 1993). Bei der Stimulation von Fibroblastenzellinien mit Wachstumsfaktoren und Zytokinen konnte gezeigt werden, daß der Transkriptionsfaktor NF-κB für die Aktivierung der Transkription des MMP-9-Gens wichtig ist (Bond et al., 1998).
In der vorliegenden Arbeit wurde mit Hilfe von CAT-Assays eine Analyse des MMP-9-Promotors nach Stimulation mit Thrombin durchgeführt. Es zeigte sich unter Verwendung von verschieden langen Promotorabschnitten des MMP-9-Promotors ein deutlicher Aktivitätsverlust zwischen der Position –670bp und –634bp. Demzufolge muß in diesem Bereich die Bindungsstelle für die Regulation nach Aktivierung der PI3-Kinase liegen. Eine Beteiligung von AP-1 und NF-κB kann daher ausgeschlossen werden, da die Bindungsstellen für AP-1 auf Position –533 und –79 und für NF-κB bei –600 liegen (Benbow et Brinckerhoff, 1997; Sato et Seiki, 1993). Eine Computeranalyse des Promotorabschnittes von –670 bis –634 zeigte, daß u. a. eine Bindungsstelle für den Transkriptionsfaktor PEA3 (polyoma virus enhancer A-binding protein-3) in diesem Bereich liegt (Munaut, 1999). Die Bedeutung dieser Bindungsstelle auf die Promotoraktivität von MMP-9 wurde von Gum nachgewiesen (Gum et al., 1996).
57

E. Diskussion
E.5 Thrombin steigert die Invasion von Tumorzellen Bösartige Tumore besitzen die Fähigkeit, durch invasives Wachstum
Gewebegrenzen zu überwinden und in benachbarte Kompartimente einzudringen. In der MRT von Osteosarkomen sieht man bereits im Frühstadium eine Infiltration der umgebenden Strukturen wie Muskelgruppen und Gefäß-Nervenbündel. Dabei müssen das Periost und die Muskelfaszien, die durch die Basalmembran gebildet werden, aufgelöst werden. Die Basalmembran muß auch bei der Überwindung der Gefäßwand und der Ausbildung von hämatogenen Metastasen aufgelöst werden (Kim et al., 1998). Die Hauptbestandteile der Basalmembran werden durch die Präsentation von Typ-IV-Kollagenasen abgebaut (Nakajima et al., 1991). In der vorliegenden Arbeit wurde auch untersucht, inwieweit Thrombin die Invasion von Tumorzellen durch eine künstliche, extrazelluläre Matrix beeinflußt. In dem durchgeführten Invasionsassay wurde eine Matrigelmembran als Modell der Basalmembran verwendet. Die Stimulation von Osteosarkomzellen mit Thrombin führt zu einer deutlichen Zunahme der Invasion. In einer Arbeit von Bar-Shavit wurde nachgewiesen, daß Gefäßmuskelzellen durch Thrombin, welches in konjugierter Form vorliegt, aktiviert werden. Damit kann Thrombin, gebunden an die extrazelluläre Matrix, die Invasion von Tumorzellen beeinflussen (Bar-Shavit, 1990).
Diese Versuche weisen darauf hin, daß Thrombin auch in vivo die Invasion von Tumorzellen regulieren kann. Die Analyse der Blutgerinnung bei Tumorpatienten zeigt eine Aktivierung der extrinsischen Gerinnungskaskade durch tissue factor, wodurch aktives Thrombin freigesetzt wird (Kakkar A. K. et al., 1995). Durch die Aktivierung des Thrombinrezeptors wird die Aktivität von pro-MMP-9 erhöht, eine vermehrte Auflösung der extrazellulären Matrix ist die Folge. Die Aggressivität und frühzeitige Metastasierung von Osteosarkomen kann durch diesen Aktivierungsmechanismus erklärt werden.
In der Arbeit von Simon konnte in vivo nachgewiesen werden, daß mit einer Erhöhung der Aktivität von MMP-9 die Invasion einer Karzinomzellinie gesteigert wird (Simon et al., 1999). Das invasive Verhalten der Mammakarzinomzellinie kann durch eine Reduktion der Thrombinrezeptoren gesenkt werden. Durch Transfektion mit Antisense-Sequenzen wird die Synthese der Rezeptoren gehemmt, die Karzinomzellen weisen im Vergleich zu den unbehandelten Zellen eine geringere Invasivität auf (Even-Ram et al., 1998). Henrikson zeigte unter Verwendung der Matrigelmembran, daß nach Zugabe von Thrombin oder TRAP das Invasionsverhalten einer aggressiven Mammakarzinom-Zellinie gesteigert wird. Bei weniger aggressiven Zellinien war jedoch keine Zunahme zu erkennen (Henrikson et al., 1999).
Die Aktivierung des Thrombinrezeptors durch Thrombin beeinflußt neben MMP-9 auch andere Proteasenklassen. So konnte durch Zugabe von Thrombin oder TRAP (SFLLRN) ein Anstieg der mRNA des Urokinase-Rezeptors detektiert werden. Die zeitabhängige Analyse der mRNA des uPA-Rezeptors im Northern-Blot zeigte nach Stimulation einen Anstieg nach 4-5 h (Reuning et al., 1994; Yoshida et al., 1994). In der
58

E. Diskussion
vorliegenden Arbeit wurde ebenfalls ein Anstieg der mRNA von MMP-9 nach 4 h nachgewiesen. Es besteht damit die Möglichkeit, daß Tumorzellen durch die Aktivierung mit Thrombin zeitgleich die Aktivität von verschiedenen Proteasensystemen hochregulieren. Eine gesteigerte Auflösung der Komponenten der extrazellulären Matrix und damit verbunden eine gesteigerte Invasivität wäre die Folge (DeClerck et Laug, 1996).
Heparin blockiert aktives Thrombin und unterdrückt die Aktivierung des Thrombinrezeptors. Durch eine geringere Steigerung der Aktivität von MMP-9 sollte ein Rückgang der Invasivität von Tumorzellen zu beobachten sein. Amirkhosravi konnte in einem Tierexperiment diese These bestätigen (Amirkhosravi et al., 1993). Wird eine Fibrosarkomzellinie Mäusen intravenös injiziert, ist eine Steigerung der Blutgerinnung zu beobachten. Mit der Gabe von Heparin wurde im Vergleich zu unbehandelten Mäusen eine Reduktion der Blutgerinnung festgestellt und in der Folgezeit bildeten sich weniger Lungentumore aus.
In einer klinischen Studie analysierte die Arbeitsgruppe von Tempelhoff die Auswirkung von niedermolekularem Heparin bei Patientinnen mit einem gynäkologischen Malignom. Die Überlebenszeit der mit niedermolekularem Heparin behandelten Patientinnen war signifikant verlängert (Tempelhoff et al., 1999). Diese klinische Beobachtung bestätigt die Ergebnisse der vorliegende Arbeit, wonach Thrombin die Invasivität von Tumorzellen erhöht.
E.6 Ausblick Die Bedeutung von Thrombin und von Matrixmetalloproteinasen bei der Ausbildung
maligner Tumore wurde in verschiedenen in vitro- und in vivo-Versuchen nachgewiesen. Pacheco konnte eine Korrelation zwischen der Expression von Typ-IV-Kollagenasen in Gewebeproben von Mammakarzinomen und der Überlebenszeit der Patientinnen nachweisen (Pacheco et al., 1998). Experimentell konnte nachgewiesen werden, daß die Aktivität von MMP-9 der Metastasierungsrate von Tumorzellen entspricht (Bernhard et al., 1990). Die Aktivierung des Thrombinrezeptors durch Thrombin bewirkt bei Tumorzellen ebenfalls eine Steigerung der Metastasierung. Durch Zugabe von Thrombin wurde eine vermehrte Ausbildung von pulmonalen Metastasen beobachtet (Nierodzik et al., 1998). In der vorliegenden Arbeit wurde nachgewiesen, daß die Osteosarkomzellinie U-2 OS durch Zugabe von Thrombin vermehrt MMP-9 synthetisiert. In dem Invasionsassay weisen mit Thrombin stimulierte Zellen, wahrscheinlich durch eine Erhöhung der Aktivität von MMP-9, eine gesteigerte Invasivität auf. Thrombin beeinflußt jedoch neben den Typ-IV Kollagenasen weitere Proteasensysteme und Adhäsionsmoleküle, die für die Metastasierung von Tumorzellen wichtig sind. Durch eine cDNA-Micro-Chip-Analyse könnte eine Vielzahl von Genen, die für Proteolyse und Adhäsion wichtig sind, analysiert werden.
Mit dem Nachweis der Wirkungsweise von Thrombin auf die Expression von MMP-9 können Therapiekonzepte entwickelt werden, die Induktion von MMP-9 durch Thrombin
59

E. Diskussion
gezielt zu hemmen. Aktives Thrombin kann durch den Einsatz von Heparin oder Hirudin gebunden werden. So konnte in einer klinischen Studie gezeigt werden, daß mit dem Einsatz von niedermolekularen Heparin die Langzeitüberlebensrate bei gynäkologischen Malignompatientinnen verbessert wird (Tempelhoff et al., 1999). Die perioperative Gabe mit niedermolekularem Heparin erscheint nach diesen Ergebnissen eine sinnvolle Ergänzung zu den bisherigen Therapiekonzepten von Tumorerkrankungen zu sein (Zacharski et al., 1990).
Außer durch den Einsatz von Heparin kann nach Aktivierung des spezifischen Thrombinrezeptors die Aktivität von pro-MMP-9 durch eine Blockade des Signaltransduktionsweges oder durch Inhibitoren der Typ-IV Kollagenasen gehemmt werden. Wie in der vorliegenden Arbeit nachgewiesen, wird mit der spezifischen Hemmung der PI3-Kinase durch Ly 294002 eine Reduktion von pro-MMP-9 beobachtet. Im nächsten Schritt sollte der Einfluß des PI3-Kinase-Inhibitors Ly 294002 auf die Metastasierung im Tiermodell untersucht werden.
Mit der Herstellung von synthetischen MMP-Inhibitoren wurde eine weitere Therapiemöglichkeit entwickelt, die tumorassoziierte Auflösung der extrazellulären Matrix zu hemmen (Talbot et Brown, 1996). Mit Hilfe von spezifischen Inhibitoren kann die proteolytische Aktivität von Untergruppen der MMP-Familie gehemmt werden. Maekawa zeigte eine selektive Hemmung von MMP-2, -9 und –14 mit dem Inhibitor BPHA (N-biphenylsulfonyl-phenylalanine hydroxiamic acid). Durch eine tägliche orale Verabreichng von 200 mg/kg konnte im Tiermodell das Tumorwachstum von zwei verschiedenen Tumorzellinien zwischen 45-48 % reduziert werden (Maekawa et al., 1999). Die in Phase III befindlichen Untersuchungen der Inhibitoren BB-2516 (British Biotech), AG3340 (Agouron) und BAY 12-9566 (Bayer) werden zeigen, inwieweit die Hemmung von MMP-9 einen klinischen Vorteil bei der Behandlung von malignen Tumoren hat.
Die Bedeutung von Typ-IV Kollagenasen und Thrombin bei der Invasion und Metastasierung von Tumorzellen wurde in zahlreichen Arbeiten nachgewiesen. Die Ergebnisse dieser Arbeit eröffnen Möglichkeiten, die Aktivierung von MMP-9 durch Thrombin zu hemmen und damit den tumorassoziierten Abbau der extrazellulären Matrix durch Tumorzellen zu reduzieren.
60

F. Zusammenfassung Die Verbindung zwischen einer malignen Erkrankung und gesteigerter
Blutgerinnung ist seit langem nachgewiesen. Thromboembolische Komplikationen treten bei Patienten mit einer malignen Erkrankung gehäuft auf. Auf Tumorzellen wurde die Überexpression des spezifischen Rezeptors für die Serinprotease Thrombin nachgewiesen, aber die Rolle von Thrombin bei malignen Erkrankungen erst ansatzweise geklärt. Eine Ursache für die gesteigerte Invasion von Tumorzellen ist die Stimulation von Proteasen. Aufgrund der Spezifität für extrazelluläre Matrixproteine haben die Matrixmetalloproteasen eine zentrale Bedeutung bei der Invasion von Tumorzellen. Die Auflösung der Basalmembran ist dabei ein wichtiger Schritt zur Ausbildung von Metastasen. Typ-IV Kollagenasen (MMP-2 und –9) degradieren spezifisch Kollagen Typ-IV und damit die kollagenen Hauptbestandteile der Basalmembran.
Wir untersuchten den Einfluß von Thrombin auf die Expression der Typ-IV Kollagenase (MMP-9, 92 kDa Gelatinase) in einer Osteosarkomzellinie U-2 OS. Die Osteosarkom-Zellen produzieren kein MMP-9, wie in der Gelatinzymographie und der Northern-Blot-Analyse gezeigt werden konnte. Zugabe von humanem Thrombin induziert einen konzentrationsabhängigen Anstieg der MMP-9-Sekretion. Weniger als 0,5 U Thrombin/ml bewirkt einen Anstieg der Aktivität, und durch Inkubation mit 1 U Thrombin/ml wird ein etwa fünffacher Anstieg der pro-MMP-9-Aktivität beobachtet. Parallel zu der Sekretion des MMP-9-Proteins steigt die Transkription des MMP-9-Gens nach Gabe von Thrombin, wie in der Northern-Blot-Analyse und in den transienten Transfektionen gezeigt wurde. Die Induktion der Transkription von MMP-9 beginnt nach 4 h und persistiert für mindestens 24 h. Die pro-MMP-9-Aktivität steigt nach 7 h und erreicht das Maximum nach 24 h.
Um die spezifische Beteiligung des Thrombinrezeptors in der Stimulation von MMP-9 nachzuweisen, wurde das Thrombinrezeptor-aktivierende Peptid (TRAP) verwendet. 24 h nach Inkubation mit verschiedenen Konzentrationen von TRAP wurde im Überstand der U-2 OS-Zellen ein konzentrationsabhängiger Anstieg der MMP-9-Aktivität beobachtet.
Transiente Transfektionen mit verschiedenen 5’-Deletionen des MMP-9-Promotors zeigten, daß eine Sequenz zwischen –670 und –634 für die Induktion von MMP-9 auf der Transkriptionsebene verantwortlich ist.
Die Stimulation von MMP-9 durch Thrombin wird mit Ly 294002, einem Inhibitor der PI-3-Kinase, verhindert. Die Zugabe von verschiedenen Inhibitoren des MAPK-Kinaseweges (SB 203580, PD 98059) beeinflußte die Induktion durch Thrombin nicht.
61

F. Zusammenfassung
In einem Metastasierungsmodell der extrazellulären Matrix mit einer Matrigelmembran wurde gezeigt, daß die Invasion der Osteosarkomzellen durch Stimulation mit Thrombin gesteigert wird.
In der vorliegenden Arbeit wird gezeigt, daß Thrombin eine Protease (MMP-9) induziert, die für die Auflösung der Basalmembran wichtig ist. Diese Erkenntnisse tragen dazu bei, den Mechanismus der thrombininduzierten Invasion von Tumorzellen besser zu verstehen.
62

G. Literaturverzeichnis
J. Alam and J. L. Cook. Reporter genes: Application to the study of mammalian gene transcription. Anal.Biochem. 188:245-254, 1990.
A. Albini, Y. Iwamoto, H. K. Kleinman, G. R. Martin, S. A. Aaronson, J. M. Kozlowski, and R. N. McEwan. A rapid in vitro assay for quantitating the invasive potential of tumor cells. Cancer Res. 47 (June):3239-3245, 1987.
G. Algire, H. W. Chalkely, and F. Y. Legallais. Vascular reactions of normal and malignant tumors in vivo. Vascular reactions of mice to wounds and to normal and neoplastic transplants. J.Nat.Cancer Inst. 6:73-73, 1945.
L. A. Allan and M. Fried. p53-dependent apoptosis or growth arrest induced by different forms of radiation in U2OS cells: p21WAF1/CIP1 repression in UV induced apoptosis. Oncogene 18 (September):5403-5412, 1999.
J. C. Alwine, D. J. Kemp, and G. R. Stark. Method for detection of specific RNAs in agarose gels by transfer to diazobenzyloxymethyl-paper and hybridization with DNA probes. Proc.Natl.Acad.Sci. 74 (December):5350-5354, 1977.
M. Amirkhosravi and J. L. Francis. Procoagulant effects of the MC28 fibrosarcoma cell line in vitro and in vivo. Br.J.Haematol. 85:736-744, 1993.
H. Andersen, D. L. Greenberg, K. Fujikawa, W. Xu, D. W. Chung, and E. W. Davie. Protease-activated receptor 1 is the primary mediator of thrombin-stimulated platelet procoagulant activity. Proc.Natl.Acad.Sci. 96 (September):11189-11193, 1999.
N. Araki, A. Uchida, and T. Kimura. Involvement of the retinoblastoma gene in primary osteosarcomas and other bone and soft-tissue tumors. Clin Orthop 270:271-277, 1991.
J. L. Aschner, J. M. Lennon, J. W. Fenton, M. Aschner, and A. B. Malik. Enzymatic activity is necessary for thrombin-mediated increase in endothelial permeability. Am.J.Physiol. 259 (October):270-275, 1990.
A. Ashkenazi and V. M. Dixit. Apoptosis control by death and decoy receptors. Curr.Opin.Cell Biol. 11:255-260, 1999.
R. Bar-Shavit, A. Kahn, G. D. Wilner, and J. W. Fenton. Monocyte chemotaxis: stimulation by specific exosite region in thrombin. Science 220 (728):731, 1983.
R. Bar-Shavit, M. Benezra, A. Eldor, E. Hy-Am, J. W. Fenton, G. D. Wilner, and I. Vlodavsky. Thrombin immobilized to extracellular matrix is a potent mitogen for vascular smooth muscle cells: nonenzymatic mode of action. Cell Regul. 1 (May):453-463, 1990.
A. J. Barr, L. F. Brass, and D. R. Manning. Reconstitution of receptors and GTP-binding regulatory proteins (G proteins) in Sf9 cells. A direct evaluation of selectivity in receptor-G protein coupling. J.Biol.Chem. 272:2223-2229, 1997.
S. H. Barsky, S. Togo, S Garbisa, and L. Liotta. Type IV collagenase immunoreactivity in invasive breast carcinoma. Lancet 1:296-297, 1983.
C. B. Basbaum and Z. Werb. Focalized proteolysis: Spatial and temporal regulation of extracellular matrix degradation at the cell surface. Curr.Opin.Cell Biol. 8:731-738, 1996.
P. Basset, J. P. Bellocq, C. Wolf, I. Stoll, P. Hutin, J. M. Limacher, O. L. Podhajcer, M. P. Chenard, M. C. Rio, and P. Chambon. A novel metalloproteinase gene specifically expressed in stromal cells of breast carcinomas. Nature 348:699-704, 1990.
63

G. Literaturverzeichnis
J. S. Bedford and J. B. Mitchell. The effect of hypoxia on the growth and radiation response of mammalian cells in culture. Br.J.Radiol. 47:687-696, 1974.
B. Belisle and A. Abo. N-Formyl peptide receptor ligation induces rac-dependent actin reorganization through Gβγ subunits and class Ia phosphoinositide 3-kinase. J.Biol.Chem. 275 (August):26225-26232, 2000.
C Belloc, H. S. Lu, C. Soria, R. Fridman, Y. Legrand, and S. Menashi. The effect of platelets on invasivness and protease production of human mammary tumor cells. Int.J.Cancer 60:413-417, 1995.
U. Benbow and C. E. Brinckerhoff. The AP-1 site and MMP gene regulation: what is all the fuss about. Matrix Biol. 15:519-526, 1997.
G. Bergers, K. Javaherian, K. M. Lo, J. Folkman, and D. Hanahan. Effects of angiogenesis inhibitors on multistage carcinogenesis in mice. Science 284 (April):808-812, 1999.
M. C. Berndt and D. R. Phillips. Platelet membrane proteins: composition and receptor function. In: Platelets in Biology and Pathology, edited by J. L. Gordon, Amsterdam:Elsevier/North Holland Biomedical Press, 1981, p. 43-74.
E. J. Bernhard, R. J. Muschel, and E. N. Hughes. Mr 92,000 gelatinase release correlates with the metastatic phenotype in transformed rat embryo cells. Cancer Research 50:3872-3877, 1990.
E. J. Bernhard, S. B. Gruber, and R. J. Muschel. Direct evidence linking expression of matrix metalloproteinase 9 (92-kDa gelatinase/collagenase) to the metastatic phenotype in transformed rat embryo cells. Proc.Natl.Acad.Sci. 91:4293-4297, 1994.
H. Birkedal-Hansen, W. G. I. Moore, M. K. Bodden, L. J. Windsor, B. Birkedal-Hansen, A. DeCarlo, and J. A. Engler. Matrix metalloproteinases: A review. Critical Reviews in Oral Biology and Medicine 4:197-250, 1993.
A. G. Bodnar, M. Quellete, M. Frolkis, S. E. Holt, C. Chiu, G. B. Morin, C. B. Harley, J. W. Shay, S. Lichtsteiner, and W. E. Wright. Extension of life-span by introduction of telomerase into normal cells. Science 279:349-352, 1998.
M. Bond, R. P. Fabunmi, A. H. Baker, and A. C. Newby. Synergistic upregulation of metalloproteinase-9 by growth factors and inflammatory cytokines: an absolute requirement for transkription factor NF-kB. FEBS 435:29-34, 1998.
N. Bouck, V. Stellmach, and S. C. Hsu. How tumors become angiogenic. Adv.Cancer Res. 69:135-174, 1996.
S. Brem, H. Brem, J. Folkman, D. Finkelstein, and A. Patz. Prolonged tumor dormancy by prevention of neovascularization in the vitreous. Cancer Res. 36 (August):2807-2812, 1976.
P. C. Brooks, S. Stromblad, L. C. Sanders, T. L. von Schalscha, R. T. Aimes, W. G Stetler-Stevenson, J. P. Quigley, and D. A. Cheresh. Localization of matrix metalloproteinase MMP-2 to the surface of invasive cells by interaction with integrin alpha v beta 3. Cell 85 (May):683-693, 1996.
P. D. Brown, A. T. Levy, I. M. Margulies, L. A. Liotta, and W. G Stetler-Stevenson. Independent expression and cellular processing of Mr 72.000 type IV collagenase and interstitial collagenase in human tumorigenic cell lines. Cancer Res. 50:6184-6191, 1990.
T. M. Bryan, A. Englezou, L. Dalla-Pozza, M. A. Dunham, and R. R. Reddel. Evidence for an alternative mechanism for maintaining telomere length in human tumors and tumor-derived cell lines. Nature Med. 3:1271-1274, 1997.
T. M. Bryan and T. R. Cech. Telomerase and the maintenance of chromosome ends. Curr.Opin.Cell Biol. 11:318-324, 1999.
64

G. Literaturverzeichnis
A. J. Butt, S. M. Firth, and R. C. Baxter. The IGF axis and programmed cell death. Immunol.Cell Biol. 77:256-262, 1999.
E. Camerer, W. Huang, and S. R. Coughlin. Tissue factor- and factor X-dependent activation of protease-activated receptor 2 by factor VIIa. Proc.Natl.Acad.Sci. 97 (May):5255-5260, 2000.
M. Campanacci. Bone and soft tissue tumors. edited by M. Campanacci, Wien New York:Springer, 1990,
R. Canete-Solar, L. Litzky, I. Lubensky, and R. J. Muschel. Localization of the 92 kD gelatinase mRNA in squamous cell and adenocarcinomas of the lung using in situ hybridization. Am J Pathol 144:518-527, 1994.
E. L. Chaikof, R. Caban, C.-N. Yan, G. N. Rao, and M. S. Runge. Growth-related responses in arterial smooth muscle cells are arrested by thrombin receptor antisense sequences. J Biol Chem 270 (March):7431-7436, 1995.
J. H. Chen, M. Ishii, L. Wang, K. Ishii, and S. R. Coughlin. Thrombin receptor activation: confirmation of the intramolecular tethered liganding hypothesis and discovery of an alternative intermolecular liganding mode. J Biol Chem 269:16041-16045, 1994.
L. B. Chen and J. M. Buchanan. Mitogenic activity of blood components. I. Thrombin and prothrombin. Proc.Natl.Acad.Sci. 72 (USA):131-135, 1975.
H. S. Chiang and T. F. Huang. Thrombin enhances the adhesion and migration of human colon adenocarcinoma cells via increased beta-3-integrin expression on the tumor cell surface and their inhibition by the snake venom peptide, rhodostomin. Br.J.Cancer 73 (April):902-908, 1996.
G. Christofori and H. Semb. The role of the cell-adhesion molecule E-cadherin as a tumour-suppressor gene. TIBS 24 (February):73-76, 1999.
D. E. Clapham and E. J. Neer. G protein beta gamma subunits. Ann.Rev.Pharmacol.Toxicol. 37:167-203, 1997.
M. H. Cobb, S. Xu, J. E. Hepler, M. Hutchinson, J. Frost, and D. J. Robbins. Regulation of the MAP kinase cascade. Cell.Mol.Biol.Res. 40:253-256, 1994.
R. W. Colman, V. J. Maeder, E. W. Salzman, and J. Hirsch. Overview of hemostasis. In: Hemostasis and Thrombosis, edited by Lippincott, Philadelphia:Colman,R.W.;Hirsh,J.;Marder,V.J.;Salzman,E.W., 1987, p. 3-17.
A. Compagni, P. Wilgenbus, M. A. Impagnatiello, M. Cotten, and G. Christofori. Fibroblast growth factors are required for efficent tumor angiogenesis. Cancer Res. 60 (December):7163-7169, 2000.
B. R. Conklin and H. R. Bourne. Structural elements of Ga subunits that interact with Gßg,receptors and effectors. Cell 73:631-641, 1993.
A. J. Connolly, H. Ishihara, M. L. Kahn, R. V. Farese, and S. R. Coughlin. Role of the thrombin receptor in development and evidence for a second receptor. Nature 381 (June):516-519, 1996.
T. M. Connolly, C. Condra, D. M. Feng, J. J. Cook, M. T. Stranieri, C. F. Reilly, R. F. Nutt, and R. J. Gould. Species variability in platelet and other cellular responsiveness to thrombin receptor-derived peptides. Thrombosis and Haemostasis 72:627-633, 1994.
C. Cordon-Cardo and C. Prives. At the crossroads of inflammation and tumorigenesis. J.Exp.Med. 190:1367-1370, 1999.
J. Cornuz, S. D. Pearson, M. A. Creager, E. F. Cook, and L. Goldmann. Importance of findings on the initial evaluation for cancer in patients with symptomatic idiopathic deep venous thrombosis. Ann.Intern.Med. 125:785-793, 1996.
65

G. Literaturverzeichnis
S. R. Coughlin. Protease-activated receptors and platelet function. Thrombosis and Haemostasis 82 (2):353-356, 1999.
C. M. Counter, A. A. Avilion, C. E. LeFeuvre, N. G. Stewart, C. W. Greider, C. B. Harley, and S. Bacchetti. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO 11:1921-1929, 1992.
L. M. Coussens and Z. Werb. Matrix metalloproteinase expression and neoplasia. Chemistry Biolog. 3 (895):904, 1996.
L. M. Coussens, W. W. Raymond, G. Bergers, M. Laig-Websters, O. Behrendsten, Z. Werb, G. H. Caughey, and D. Hanahan. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 13:1382-1397, 1999.
L. M. Coussens, C. L. Tinkle, D. Hanahan, and Z. Werb. MMP-9 supplied by bone-marrow-derived cells contributes to skin carcinogenesis. Cell 103 (Oktober):481-490, 2000.
H Daub, C. Wallasch, A. Lankenau, A. Herrlich, and A. Ullrich. Signal characteristics of G protein-transactivated EGF-receptor. EMBO 16 (23):7032-7044, 1997.
M. G. Davey and E. F. Luscher. Actions of thrombin and other coagulant and proteolytic enzymes on blood platelets. Nature 216 (December):857-858, 1967.
E. W. Davie and O. D. Ratnoff. Waterfall sequence for intrinsic blood clotting. Science 145:1310-1312, 1964.
E. W. Davie, K. Fujikawa, and W. Kisiel. The coagulation cascade: initiation, maintenance and regulation. Biochemistry 30:10363-10370, 1991.
Y. A. DeClerck, T. D. Yean, B. J. Ratzkin, H. S. Lu, and K. E. Langley. Purification and characterization of two related but distinct metalloproteinase inhibitors secreted by bovine aortic endothelial cells. J Biol Chem 264 (17445):17453, 1989.
Y. A. DeClerck and W. E. Laug. Cooperation between matrix metalloproteinases and the plasminogen activator-plasmin system in tumor progression. Enzyme Prot. 49:72-84, 1996.
D. T. Denhardt. Signal-transducing protein phosphorylation cascades mediated by Ras/Rho proteins in the mammalian cell: the potential for multiplex signalling. Biochem J 318:729-747, 1996.
J. R. deWet, K. V. Wood, M. DeLuca, D. R. Helinski, and S. Subramani. Firefly luciferase gene: structure and expression in mammalian cells. Mol.Cell.Biol. 7 (February):725-737, 1987.
J. Domin and M. D. Waterfield. Using structure to define the function of phosphoinositide 3-kinase family members. FEBS 410 (June):91-95, 1997.
M. B. Donati. Cancer and thrombosis. Haemostasis 24:128-131, 1994.
M. J. Duffy and K. McCarthy. Matrix metalloproteinases in cancer: Prognostic markers and targets for therapy (Review). International Journal of Oncology 12:1343-1348, 1998.
H. F. Dvorak, G. R. Dickersin, A. M. Dvorak, E. J. Manseau, and K. Pyne. Human breast carcinoma: fibrin deposits and desmoplasia inflammatory cell type and distribution. Microvasculature and infarction. J Nat Cancer Inst 67:335-345, 1981.
S. E. Egan, B. W. Giddings, M. W. Brooks, L. Buday, A. M. Sizeland, and R. A. Weinberg. Association of Sos Ras exchange protein with Grb2 is implicated in tyrosine kinase signal transduction and transformation. Nature 363:45-51, 1993.
S. M. Ellerbroek, J. M. Halbleib, M. Benavidez, J. K. Warmka, E. V. Wattenberg, M. S. Stack, and L. G. Hudson. Phosphatidylinositol 3-kinase activity in epidermal growth factor-stimulated matrix
66

G. Literaturverzeichnis
metalloproteinase-9 production and cell surface association. Cancer Res. 61 (March):1855-1861, 2001.
V. Ellermann and O. Bang. Experimentelle Leukämie bei Hühnern. Zentralbl.Bakteriol.Abt. 46:595-595, 1908.
W. F. Enneking, S. S. Spanier, and M. A. Goodman. A system for the surgical staging of musculoskeletal sarcoma. Clin Orthop 153:106-120, 1980.
N. Esumi, D. Fan, and I. J. Fidler. Inhibition of murine melanoma experimental metastasis by recombinant desulfatohirudin, a highly specific thrombin inhibitor. Cancer Res. 51:4549-4556, 1991.
S. Even-Ram, B. Uziely, P. Cohen, S. Grisaru-Granovsky, M. Maoz, Y. Ginzburg, R. Reich, I. Vlodavsky, and R. Bar-Shavit. Thrombin receptor overexpression in malignant and physiological invasion process. Nature Med. 4 (August):909-914, 1998.
R. P. Fabunmi, A. H. Baker, E. J. Murray, R. F. G. Booth, and A. C. Newby. Divergent regulation by growth factors and cytokines of 95 kDa and 72 kDa gelatinases and tissue inhibitors of metalloproteinases-1, -2 and -3 in rabbit aortic smooth muscle cells. Biochem J 315:335-342, 1996.
T. R. Faruqi, E. J. Weiss, M. J. Shapiro, W. Huang, and S. R. Coughlin. Structure-function analysis of protease-activated receptor 4 tethered ligand peptides. Determinants of specificity and utility in assays of receptor function. J.Biol.Chem. 275 (June):19728-19734, 2000.
M. Faure, T. A. Voyno-Yasenetskaya, and H. R. Bourne. cAMP and ßγ subunits of heterotrimeric G proteins stimulate the mitogen-activated protein kinase pathway of COS-7 cells. J Biol Chem 269:7851-7854, 1994.
P. Fedi, S. R. Tronick, and S. A. Aaronson. Growth factors. In: Cancer medicine, edited by J. F. Holland, R. C. Bast, D. L. Morton, E. Frei, D. W. Kufe, and R. R. Weichselbaum, Baltimore:Williams and Wilkins, 1997, p. 41-64.
P. L. Felgner, T. R. Gadek, M. Holm, R. Roman, H. W. Chan, M. Wenz, J. P. Northtrop, G. M. Ringold, and M. Danielsen. Lipofection: a highly efficient, lipid-mediated DNA-transfection procedure. Proc.Natl.Acad.Sci. 84 (November):7413-7417, 1987.
C. Fernandez-Patron, M. W. Radomski, and S. T. Davidge. Role of matrix metalloproteinase-2 in thrombin-induced vasorelaxation of rat mesenteric arteries. Am.J.Physiol.Heart Circ.Physiol. 278:1473-1479, 2000.
R. Ferracini, M. F. DiRenzo, K. Scotlandi, N. Baldini, M. Olivero, P. L. Lollini, O. Cremona, M. Campanacci, and P. M. Comoglio. The Met/HGF receptor is over-expressed in human osteosarcomas and is activated by either a paracrine or an autocrine circuit. Oncogene 10:739-749, 1995.
I. J. Fidler and L. M. Ellis. The implications of angiogenesis for the biology and therapy of cancer metastasis. Cell 79:185-188, 1994.
C. Fisch, A. W. Jones, and W. D. Gambill. Acute thrombophlebitis associated with carcinoma of the stomach. Gastroenterology 18:290-290, 1951.
E. G. Fischer, W. Ruf, and B. M. Mueller. Tissue factor-initiated thrombin generation activates the signaling thrombin receptor on malignant melanoma cells. Cancer Research 55:1629-1632, 1995.
K. P. Foley and R. N. Eisenmann. Two MAD tails: what the recent knockouts of Mad1 and Mx1 tell us about the MYC/MAX/MAD network. Biochim.Biophys.Acta 1423:37-47, 1999.
S. M. Frisch and H. Francis. Disruption of epithelial cell-matrix interactions induces apoptosis. J.Cell.Biol. 124 (February):619-626, 1994.
67

G. Literaturverzeichnis
M. Gebhardt, A. Goorin, J. Traina, A. Perez-Atayde, J. Anderson, J. G. Watts, and N. Jaffe. Long-term results of limb-salvage and amputation in extremity osteosarcoma. In: New developments for limb salvage in muskuloskeletal tumors., edited by Springer, New York: 1989, p. 99-109.
R. E. Gerszten, J. H. Chen, M. Ishii, K. Ishii, L. Wang, T. Nanevicz, C. W. Turck, T.-K. H. Vu, and S. R. Coughlin. Specifity of the thrombin receptor for agonist peptide is defined by its extracellular surface. Nature 368 (April):648-651, 1994.
F. G. Giancotti and E. Ruoslathi. Integrin signaling. Science 285 (August):1028-1032, 1999.
F. Gonzalez, A. Seth, D. Raden, D. Bowman, F. Fay, and R. Davis. Serum-induced translocation of mitogen-activated protein kinase to the cell surface ruffling membrane and the nucleus. J.Cell.Biol. 122:1089-1101, 1993.
A. Goorin and J. W. Andersen. Experience with multiagent chemotherapy for osteosarcoma. Clin Orthop 270:22-28, 1991.
S. G. Gordon. Cancer cell procoagulants and their role in malignant disease. Seminars in Thrombosis and Hemostasis 18:424-433, 1992.
R. Gorlick, A. G. Huvos, G. Heller, A. Aledo, G. P. Beardsley, J. H. Healey, and P. A. Meyers. Expression of HER2/erb-2 correlates with survival in osteosarcoma. J.Clin.Oncol. 17 (September):2781-2788, 1999.
R. J. A. Grand, A. S. Turnell, and P. W. Grabham. Cellular consequences of thrombin-receptor activation. Biochem J 313:353-368, 1996.
D. R. Green and J. C. Reed. Mitochondria and apoptosis. Science 281:1309-1312, 1998.
R. J. Grimer and R. S. Sneath. Diagnosing malignant bone tumours. J Bone Joint Surg Br 72 (Br):22-28, 1990.
J. Gross and C. M. Lapiere. Collagenolytic activity in amphibian tissue: a tissue culture assay. Proc.Natl.Acad.Sci. 48:1014-1022, 1962.
R. Gum, E. Lengyel, J. Juarez, J. H. Chen, H. Sato, M. Seiki, and D. Boyd. Stimulation of 92-kDA gelatinase B promoter activity by ras is mitogen-activated protein kinase kinase 1-independent and requires multiple transcription factor binding sites including closely spaced PEA3/ets and AP-1 sequences. Journal of Biological Chemistry 271 (18):10672-10680, 1996.
R. Gum, H. Wang, E. Lengyel, J. Juarez, and D. Boyd. Regulation of the 92 kDa type 4 collagenase expression by the jun aminoterminal kinase- and the extracellular signal-regulated kinase-dependent signaling cascades. Oncogene 14:1481-1493, 1997.
R. Halaban. Melanoma cell autonomous growth: the Rb/E2F pathway. Cancer Metastasis Rev 18:333-343, 1999.
T. L. Halvorsen, G. Leibowitz, and F. Levine. Telomerase activity is sufficient to allow transformed cells to escape from crisis. Mol.Cell.Biol. 19:1864-1870, 1999.
D. Hanahan and R. A. Weinberg. The hallmarks of cancer. Cell 100 (January):57-70, 2000.
M. F. Hansen. Molecular genetic considerations in osteosarcoma. Clin Orthop 270:237-246, 1991.
J. H. Hartwig, G. M. Bokoch, C. L. Carpenter, P. A. Janmey, L. A. Taylor, A. Toker, and T. P. Stossel. Thrombin receptor ligation and activated rac uncap actin filament barbed ends through phosphoinositide synthesis in permeabilized humen platelets. Cell 82 (August):643-653, 1995.
F. Hefti and G. Jundt. Is the age of osteosarcoma patients increasing? J Bone Joint Surg Br 77 (Suppl II) (Br):207-208, 1995.
68

G. Literaturverzeichnis
K. P. Henrikson, S. L. Salazar, J. W. Fenton, and B. T. Pentecost. Role of thrombin receptor in breast cancer invasivness. Br.J.Cancer 79 (3/4):401-406, 1999.
J. M. Herbert, I. Lamarche, and F. Dol. Induction of vascular smooth muscle cell growth by selective activation of the thrombin receptor. Effect of heparin. FEBS 301:155-158, 1992.
I. D. Hiles, M. Otsu, S. Volinia, M. J. Fry, I. Gout, R. Dhand, G. Panayotou, F. Ruiz-Larrera, A. Thompson, and N. F. Totty. Phosphatidylinositol 3-kinase: structure and expression of the 110 kd catalytic subunit. Cell 70 (August):19-29, 1992.
K. V. Honn, B. F. Sloane, and P. G. Cavanaugh. Role of the coagulation system in tumor-cell-induced platelet aggregation and metastasis. Haemostasis 18:37-46, 1988.
E. W. Howard, E. C. Bullen, and M. J. Banda. Preferential inhibition of 72 and 92 kDa gelatinases by tissue inhibitor of metalloproteinase-2. J Biol Chem 266 (13070):13075, 1991.
P. Huhtala, A. Tuuttila, L. T. Chow, J. Lohi, J. Keski-Oja, and K Tryggvason. Complete structure of the human gene for 92-kDa type IV collagenase. Divergent regulation of expression for the 92- and 72-kilodalton enzyme genes in HT-1080 cells. J.Biol.Chem. 266 (September):16485-16490, 1991.
P. Huhtala, M. J. Humphries, J. B. McCarthy, P. M. Tremble, Z. Werb, and C. H. Damsky. Cooperative signaling by alpha 5 beta 1 and alpha 4 beta 1 integrins regulates metalloproteinase gene expression in fibroblasts adhering to fibronectin. J.Cell.Biol. 129 (May):867-879, 1995.
D. T. Hung, T.-K. H. Vu, N. A. Nelken, and S. R. Coughlin. Thrombin-induced events in non-platelet cells are mediated by the unique proteolytic mechanism established for the cloned platelet thrombin receptor. J Cell Biol 116 (3):827-832, 1992.
D. T. Hung, Y. H. Wong, T.-K. H. Vu, and S. R. Coughlin. The cloned platelet thrombin receptor couples to at least two distinct effectors to stimulate both phosphoinositide hydrolysis and inhibit adenylyl cyclase. J.Biol.Chem. 353:20831-20834, 1992.
A. G. Ide, N. Baker, and S. L. Warren. Vascularization of the Brown-Pearce rabbit epithelioma transplant as seen in the transparent ear chamber. Am.J.Roentgenol. 42:891-891, 1939.
Y. T. Ip and R. Davis. Signal transduction by the c-Jun N-terminal kinase (JNK)-from inflammation to development. Curr.Opin.Cell Biol. 10:205-219, 1998.
H. Ishihara, A. J. Connolly, D. Zeng, M. L. Kahn, Y. W. Zheng, C. Timmons, T. Tram, and S. R. Coughlin. Protease-activated receptor 3 is a second thrombin receptor in humans. Nature 386:502-506, 1997.
M. L. Kahn, Y. W. Zheng, W. Huang, V. Bigornia, D. Zeng, S. Moff, R. V. Farese, C. Tam, and S. R. Coughlin. A dual thrombin receptor system for platelet activation. Nature 394:690-694, 1998.
A. K. Kakkar, N. DeRuvo, V. Chinswangwatanakul, S. Tebbutt, and R. C. N. Williamson. Extrinsic-pathway activation in cancer with high factor VIIa and tissue factor. Lancet 346:1004-1005, 1995.
V. V. Kakkar, C. T. Howe, and A. N. Nicholaides. Deep vein thrombosis of the leg. Is there a high risk group? Am.J.Surg. 120:527-531, 1970.
C. Kanthou, O Benzakour, G. Patel, J. Deadman, V. V. Kakkar, and F. Lupu. Thrombin receptor activating peptide (TRAP) stimulates mitogenesis, c-fos and PDGF-A gene expression in human vascular smooth muscle cells. Thrombosis and Haemostasis 74(5):1340-1347, 1995.
J. Kim, W. Yu, K. Kovalski, and L. Ossowski. Requirement for specific proteases in cancer cell intravasation as revealed by a novel semiquantitative PCR-based assay. Cell 94 (August 7):353-362, 1998.
69

G. Literaturverzeichnis
K. J. Kim, B. Li, J. Winer, M. Armanini, N. Gillett, H. S. Phillips, and N. Ferrara. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 362 (April):841-844, 1993.
A. Kimichi, X. F. Wang, R. A. Weinberg, S. Cheifetz, and J. Massague. Absence of TGF-beta receptors and growth inhibitory responses in retinoblastoma cells. Science 240 (April):196-199, 1988.
K. W. Kinzler and B. Vogelstein. Lessons from hereditary colorectal cancer. Cell 87:159-170, 1996.
H. K. Kleinman, M. L. McGarvey, J. R. Hassell, V. L. Star, F. B. Cannon, G. W. Laurie, and G. R. Martin. Basement membrane complexes with biological activity. Biochemistry 25 (January):312-318, 1986.
A. Klippel, M.-A. Escobedo, M. S. Wachowicz, G. Apell, T. W. Brown, M. A. Giedlin, W. M. Kavanaugh, and L. T. Williams. Activation of phosphatidylinositol 3-kinase is sufficient for cell cycle entry and promotes cellular changes characteristic of oncogenic transformation. Mol.Cell.Biol. 18:5699-5711, 1998.
G. M. Kraehn, J. Utikal, M. Udart, K. M. Greulich, P. Kaskel, U. Leiter, and R. U. Peter. Extra c-myc oncogene copies in high risk cutaneous malignant melanoma and melanoma metastases. Br.J.Cancer 84 (January):72-79, 2001.
R. M. Kramer, E. F. Roberts, B. A. Strifler, and E. M. Johnstone. Thrombin induces activation of p38 MAP kinase in human platelets. J Biol Chem 270 (November):27395-27398, 1995.
S. Krugmann, P. T. Hawkins, N. Pryer, and S. Braselmann. Characterizing the interactions between the two subunits of the p101/110γ phosphoinositide 3-kinase and their role in the activation of this enzyme by Gßγ subunits. J.Biol.Chem. 274 (June):17152-17158, 1999.
J. M. Kyriakis, T. L. Force, U. R. Rapp, J. V. Bonventre, and J. Avruch. Mitogen regulation of c-Raf-1 protein kinase activity toward mitogen-activated protein kinase kinase. J Biol Chem 268 (16009):16019, 1993.
S. S. Lakka, S. L. Jasti, A. P. Kyritsis, W. K. Yung, F. Ali-Osman, G. L. Nicolson, and J. S. Rao. Regulation of MMP-9 (type IV collagenase) production and invasiveness in gliomas by extracellular signal-regulated kinase and jun amino-terminal kinase signaling cascades. Clin.Exp.Metas. 18:245-252, 2000.
J. Lawler. The functions of thrombospondin-1 and -2. Curr.Opin.Cell Biol. 12 (October):634-640, 2000.
E. Lengyel, R. Gum, J. Juarez, G. Clayman, M. Seiki, H. Sato, and D. Boyd. Induction of Mr 92,000 type IV collagenase expression in a squamous cell carcinoma cell line by fibroblasts. Cancer Res. 55:963-967, 1995.
T. S. Lewis, P. S. Shapiro, and N. G. Ahn. Signal transduction through MAP kinase cascade. Adv.Cancer Res. 74:49-139, 1998.
L. A. Liotta, S. Abe, and P. Robey. Preferential digestion of basement membrane collagen by an enzyme derived from a metastatic murine tumor. Proc.Natl.Acad.Sci. 76 (USA):2268, 1979.
L. A. Liotta, K Tryggvason, S Garbisa, I. Hart, C. M. Foltz, and S. Shafie. Metastatic potential correlates with enzymatic degradation of basement membrane collagen. Nature 284:67-68, 1980.
L. A. Liotta, K Tryggvason, S Garbisa, P. Robey, and S. Abe. Partial purification and characterization of a neutral protease which cleaves type IV collagen. Biochemistry 20:100-104, 1981.
L. A. Liotta and W. G Stetler-Stevenson. Metalloproteinases and cancer invasion. Cancer Biology 1:99-106, 1990.
70

G. Literaturverzeichnis
L. Long, O. Jonasson, S. Roberts, R. McGrath, E. McGrew, and W. E. Cole. Cancer cells in blood. Results of a simplified isolation technique. Arch.Surg. 80:910-919, 1960.
L. Long, R. Navab, and P. Brodt. Regulation of the Mr72,000 Type IV collagenase by the type I insulin-like growth factor receptor. Cancer Res. 58 (August):3243-3247, 1998.
R. Lorenzet, G. Peri, D. Locati, P. Allavena, M. Colucci, and N. Semeraro. Generation of procoagulant activity by mononuclear phagocytes: a possible mechanism contributing to blood clotting activation within maligant tissues. Blood 62:271-273, 1983.
R. M. Lyons and H. L. Moses. Transforming growth factors and the regulation of cell proliferation. Eur.J.Biochem. 187:467, 1990.
S.-F. Ma, J. G. N. Garcia, U. Reuning, S. P. Little, N. U. Bang, and E. P. Dixon. Thrombin induces thrombomodulin mRNA expression via the proteolytically activated thrombin receptor in cultured bovine smooth muscle cells. J.Lab.Clin.Med. 129 (June):611-618, 1997.
R. G. MacFarlane. An enzyme cascade in the blood clotting mechanism,and its function as a biochemical amplifier. Nature 202:498-499, 1964.
R. Maekawa, H. Maki, H. Yoshida, K. Hojo, H. Tanaka, T. Wada, N. Uchida, Y. Takeda, H. Kasai, H. Okamoto, H. Tsuzuki, Y. Kambayashi, F. Watanabe, K. Kawada, K. Toda, M. Ohtani, K. Sugita, and T. Yoshioka. Correlation of antiangiogenic and antitumor efficacy of N-biphenyl sulfonyl-phenylalanine hydroxiamic acid (BPHA), an orally-active, selective matrix metalloproteinase inhibitor. Cancer Res. 59 (March):1231-1235, 1999.
K. Malarkey, C. M. Belham, A. Paul, A. Graham, A. McLees, P. H. Scott, and R. Plevin. The regulation of tyrosin kinase signalling pathways by growth factor and G-protein-coupled receptors. Biochem J 309:361-375, 1995.
K. G. Mann, M. E. Nesheim, W. R. Church, P. Haley, and S. Krishnaswamy. Surface-dependent reactions of the vitamin K-dependent enzyme complexes. Blood 76:1-16, 1990.
F. W. Marsden, F. O. Stephens, S. W. McCarthy, and A. M. Ferrari. IIB osteosarcoma. Current management, local controll, and survival statistics-The Australian experience. Clin Orthop 270:113-120, 1991.
L. J. McCawley, S. Li, E. V. Wattenberg, and L. G. Hudson. Sustained activation of the mitogen-activated protein kinase pathway. A mechanism underlying receptor tyrosine kinase specificity for matrix metalloproteinase-9 induction and cell migration. J Biol Chem 274 (February):4347-4353, 1999.
C. A. McNamara, I. J. Sarembok, L. W. Gimple, J. W. Fenton, S. R. Coughlin, and G. K. Owens. Thrombin stimulates proliferation of cultured rat aortic smooth muscle cells by a proteolytically activated receptor. J.Clin.Invest. 91 (January):94-98, 1993.
P. Mignatti and D. B. Rifkin. Biology and biochemistry of proteinases in tumor invasion. Physiol Rev 73:161-195, 1993.
J. Mirra, R. Gold, and P. Picci. Osseous tumors of intramedullary origin. In: Bone tumors-clinical, radiologic and pathologic correlations, edited by J. Mirra, Philadelphia:Lea & Febiger 303, 1989,
M. Molino. Interactions of mast cell tryptase with thrombin receptors and PAR-2. J.Biol.Chem. 272:4043-4049, 1997.
E. R. Morgan and M. Haugen. Late effect of cancer therapy. Cancer Treat.Res. 92:343-375, 1997.
E. Morgunova, A. Tuuttila, U. Bergmann, M. Isupov, Y. Lindqvist, G. Schneider, and K Tryggvason. Structure of human pro-matrix metalloproteinase-2: activation mechanism revealed. Science 284:1667-1670, 1999.
71

G. Literaturverzeichnis
M. Morrison. An analyis of the blood picture in 100 cases of malignancy. J.Lab.Clin.Med. 17:1071-1071, 1932.
T. Mosmann. Rapid colorimetic assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J.Immunol.Methods 65 (December):55-63, 1983.
A. M. Moursi, R. K. Globus, and C. H. Damsky. Interactions between integrin receptors and fibronectin are required for calvarial osteoblast differentiation in vitro. J.Cell Sci. 110:2187-2196, 1997.
B. M. Mueller, R. A. Reisfeld, T. S. Edgington, and W. Ruf. Expression of tissue factor by melanoma cells promotes efficient hematogenous metastasis. Proc.Natl.Acad.Sci. 89 (USA):11832-11836, 1992.
C. Munaut, T. Salonurmi, S. Kontusaari, P. Reponen, T. Morita, J.-M. Foidart, and K Tryggvason. Murine matrix metalloproteinase 9 gene. 5'-upstream region contains cis-acting elements for expression in osteoclasts and migrating keratinocytes in transgenic mice. J.Biol.Chem. 274 (February):5588-5596, 1999.
J. C. Murray. Coagulation and cancer. Br.J.Cancer 64:422-424, 1991.
M. Nakajima, D. Welch, and P. N. Belloni. Degradation of basement membran type IV collagen and lung subendothelial matrix by rat mammary adenocarcinoma cell clones of differing metastatic potentials. Cancer Research 47:4869-4869, 1987.
M. Nakajima and A. M. Chop. Tumor invasion and extracellular matrix degradative enzymes: regulation of activity by organ factors. Semin.Cancer Biol. 2 (April):115-127, 1991.
H. Nakamura, H. Ueno, K. Yamashita, T. Shimada, E. Yamamoto, M. Noguchi, N. Fujimoto, H. Sato, M. Seiki, and Y. Okada. Enhanced production and activation of progelatinase A mediated by membrane-type 1 matrix metalloproteinase in human papillary thyroid carcinomas. Cancer Research 59:467-473, 1999.
M. Nakanishi-Matsui, Y. W. Zheng, D. J. Sulciner, E. J. Weiss, M. J. Ludeman, and S. R. Coughlin. PAR3 is a cofactor for PAR4 activation by thrombin. Nature 404 (April):609-613, 2000.
E. J. Neer. Heterotrimeric G proteins: organizer of transmembrane signals. Cell 80:249-257, 1995.
J. P. Neglia and M. E. Nesbit. Care and treatment of long-term survivors of childhood. Cancer 71(10 Suppl) (May):3386-3391, 1993.
E. V. Negrescu, K. L. DeQuintana, and W. Siess. Platelet shape change induced by thrombin receptor activation. J Biol Chem 270 (January):1057-1061, 1995.
M. L. Nierodzik, F. Kajumo, and S. Karpatkin. Effect of thrombin treatment of tumor cells on adhesion of tumor cells to platelets in vitro and tumor metastasis in vivo. Cancer Res. 52 (June):3267-3272, 1992.
M. L. Nierodzik, A. Klepfish, and S. Karpatkin. Role of platelets, thrombin, integrin IIb-IIIa, fibronectin and von Willebrand factor on tumor adhesion in vitro and metastasis in vivo. Thrombosis and Haemostasis 74 (1):282-290, 1995.
M. L. Nierodzik, R. M. Bain, L.-X. Liu, M. Shivji, K. Takeshita, and S. Karpatkin. Presence of the seven transmembrane thrombin receptor on human tumour cells: effect of activation on tumour adhesion to platelets and tumour tyrosine phosphorylation. Br.J.Haematol. 92 (September):452-457, 1996.
M. L. Nierodzik, K. Chen, and K. Takeshita. Protease-activated receptor-1 (PAR-1) is required and rate limiting for thrombin-enhanced experimental pulmonary metastasis. Blood 92:3694-3700, 1998.
72

G. Literaturverzeichnis
J. Nishida, M. Abe, T. Shiraishi Shimamura, G. Tumura, T. Satoh, and S. Ehara. Familial occurence of teleangiectatic osteosarcoma: Cousin cases. J Pediatr Orthop 14:119-122, 1994.
S. K. Nordeen. Luciferase reporter gene vectors for analysis of promotors and enhancers. Biotechniques 6 (May):454-458, 1988.
S. Nystedt, K. Emilsson, C. Wahlestedt, and J. Sundelin. Molecular cloning of a potential proteinase activated receptor. Proc.Natl.Acad.Sci. 91:9208-9212, 1994.
S. Offermanns, K.-L. Laugwitz, K. Spicher, and G. Schultz. G proteins of the G12 family are activated via thromboxane A2 and thrombin receptors in human platelets. Proc.Natl.Acad.Sci. 91 (USA):504-508, 1994.
A. F. Olumi, G. D. Grossfeld, S. W. Hayward, P. R. Carroll, T. D. Tlsty, and G. R. Cunha. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 59:5002-5011, 1999.
C. M. Overall, J. L. Wrana, and J. Sodek. Transcriptional and post-transcriptional regulation of 72-kDa gelatinase/type IV collagenase by transforming growth factor-1 in human fibroblasts. J Biol Chem 266:14064-14071, 1991.
M. M. Pacheco, M. Mourao, E. B. Mantonavi, I. N. Nishimoto, and M. M. Brentani. Expression of gelatinases A and B, stromelysin-3 and matrilysin genes in breast carcinomas: clinico-pathological correlations. Clin.Exp.Metas. 16 (577):585, 1998.
C. Papin, A. Eychene, A. Brunet, G. Pages, J. Pouyssegur, G. Calothy, and J. V. Barnier. B-Raf protein isoforms interact with and phosphorylate MEK-1 on serine residues 218 and 222. Oncogene 10:1647-1651, 1995.
E. Pearlstein, C. Ambrogio, C. Gasic, and S. Karpatkin. Inhibition of platelet aggregating activity of two human adenocarcinomas of the colon and an anaplastic murine tumor with a specific thrombin inhibitor, dansylarginine-N-(-3-ethyl-1,5-pentanediyl)amide. Cancer Research 41:4535-4539, 1981.
K. Perrem, T. M. Bryan, A. Englezou, T. Hackl, E. L. Moy, and R. R. Reddel. Repression of an alternative mechanism for lengthening of telomeres in somatic cell hybrids. Oncogene 18 (June):3383-3390, 1999.
W. A. Philipps, F. St. Clair, A. D. Munday, R. J. S. Thomas, and C. A. Mitchell. Increased levels of phosphatidylinositol 3-kinase activity in colorectal tumors. Cancer 83 (July):41-47, 1998.
A. Piccioli, P. Prandoni, B. M. Ewenstein, and S. Z. Goldhaber. Cancer and venous thromboembolism. Am.Heart J. 132:850-855, 1996.
T. Plath, K. Detjen, M. Welzel, Z. von Marschall, D. Murphy, M. Schirner, B. Wiedenmann, and S. Rosewicz. A novel function for the tumor suppressor p16(INK4a): induction of anoikis via upregulation of the alpha(5)beta(1) fibronectin receptor. J.Cell.Biol. 150 (September):1467-1478, 2000.
S. Pons, T. Asano, E. Glasheen, M. Miralpeix, Y. Zhang, T. L. Fisher, M. B. Myers, X. J. Sun, and M. F. White. The structure and function of p55PIK reveal a new regulatory subunit for phosphtidylinositol 3-kinase. Mol.Cell.Biol. 15:4453-4465, 1995.
J. Ponten and E. Saksela. Two established in vitro cell lines from human mesenchymal tumours. Int.J.Cancer 2:434-447, 1967.
C. Pyke, E. Ralkiaer, P. Huhtala, T. Hurskainen, K. Dano, and K. Tryggvason. Localization of messenger RNA for Mr 72,000 and 92,000 type IV collagenase in human skin cancers by in situ hybridization. Cancer Res. 52:1336-1341, 1992.
C. Pyke, E. Ralkiaer, K Tryggvason, and K. Dano. Messenger RNA for two type IV collagenase is located in stroma cells in human colon cancer. Am J Pathol 142 (359):365, 1993.
73

G. Literaturverzeichnis
J. P. Quigley and P. B. Armstrong. Tumor cell intravasation alu-cidated: the chick embryo opens the window. Cell 94 (August):281-284, 1998.
G. N. Rao, P. Delafontaine, and M. S. Runge. Thrombin stimulates phosphorylation of insulin-like growth factor-I-receptor, insulin receptor substrate-I and phospholipase CγI in rat aortic smooth muscle cells. J.Biol.Chem. 270:27871-27875, 1995.
L. V. M. Rao. Tissue factor as a tumor procoagulant. Cancer Metastasis Rev 11:249-266, 1992.
V. H. Rao, R. K. Singh, D. C. Delimont, R. H. Finnell, J. A. Bridge, J. R. Neff, B. P. Garvin, D. L. Pickering, W. G. Sanger, B. A. Buehler, and G. B. Schaefer. Transcriptional regulation of MMP-9 expression in stromal cells of human giant cell tumor of bone by tumor necrosis factor-alpha. International Journal of Oncology 14 (2):291-300, 1999.
U. B. Rasmussen, V. Vouret-Craviari, S. Jalla, Y. Schlesinger, G. Pages, A. Pavirani, J. P. Lecocq, and E. Van Obberghen-Schilling. cDNA cloning and expression of a hamster alpha-thrombin receptor coupled to Ca2+ mobilization. FEBS 288 (August):123-128, 1991.
M. W. Renshaw, X.-D. Ren, and M. A. Schwartz. Growth factor activation of MAP kinase requires cell adhesion. EMBO 16:5592-5599, 1997.
U. Reuning, S. P. Little, E. P. Dixon, and N. U. Bang. Effect of thrombin, the thrombin receptor activation peptide, and other mitogens on vascular smooth muscle cell urokinase receptor mRNA levels. Blood 84 (December):3700-3708, 1994.
F. R. Rickles and R. L. Edwards. Activation of blood coagulation in cancer: Trousseau's syndrome revisited. Blood 62:14-31, 1983.
F. R. Rickles, M. Levine, and R. L. Edwards. Hemostatic alterations in cancer patients. Cancer Metastasis Rev 11 (November):237-248, 1992.
H. Rochefort and E. Liaudet-Coopman. Cathepsin D in cancer metastasis: a protease and a ligand. APMIS 107:86-95, 1999.
G. Rodriguez-Tarduchy, M. K. Collins, I. Garcia, and A. Lopez-Rivas. Insulin-like growth factor-1 inhibits apoptosis in IL-3-dependent hemopoietic cells. J.Immunol. 149:535, 1992.
P. Rous. A sarcoma of the fowl transmissible by an agent separable from the tumor cells. J.Exp.Med. 13:397-397, 1911.
E. Ruoslathi and B. Öbrink. Common principles in cell adhesion. Exp.Cell.Res. 227:1-11, 1996.
A. Saaristo, T. Karpanen, and K. Alitalo. Mechanisms of angiogenesis and their use in the inhibition of tumor growth and metastasis. Oncogene 19 (December):6122-6129, 2000.
G. Saeter, J. Hoie, A. E. Stenwig, A. K. Johansson, E. Hannisdal, and O. P. Solheim. Systemic relapse of patients with osteogenic sarcoma. Prognostic factors for long term survival. Cancer 75:1084-1093, 1995.
T. Sakai, T. Lund-Hansen, L. Paborsky, A. H. Pedersen, and W. Kisiel. Binding of human factors VII and VIIa to a human bladder carcinoma cell line (J 82). J.Biol.Chem. 264:9980-9988, 1989.
H. Sato, M. Kita, and M. Seiki. v-src activates the expression of 92-kDa type IV collagenase through the AP-1 site and the GT Box homologous to retinoblastoma control elements. Journal of Biological Chemistry 268:23460-23468, 1993.
H. Sato and M. Seiki. Regulatory mechanism of 92 kDa type IV collagenase gene expression which is associated with invasiveness of tumor cells. Oncogene 8:395-405, 1993.
G. Sawicki, E. Salas, J. Murat, H. Miszta-Lane, and M. W. Radomski. Release of gelatinase A during platelet activation mediates aggregation. Nature 386 (April):616-619, 1997.
74

G. Literaturverzeichnis
H. J. Schaeffer and M. J. Weber. Mitogen-activated protein kinases: specific messages from ubiquitous messengers. Mol.Cell.Biol. 19 (April):2435-2444, 1999.
K. Scotlandi, N. Baldini, M. Olivero, M. F. DiRenzo, M. Martano, M. Serra, M. C. Manara, P. M. Comoglio, and R. Ferracini. Expression of Met/hepatocyte growth factor receptor gene and malignant behavior of musculoskeletal tumors. Am J Pathol 149 (Oktober):1209-1219, 1996.
N. Semeraro and M. Colucci. Tissue factor in health and disease. Thrombosis and Haemostasis 78:759-764, 1997.
M. J. Shapiro, E. J. Weiss, T. R. Faruqi, and S. R. Coughlin. Protease-activated receptors 1 and 4 are shut off with distinct kinetics after activation by thrombin. J.Biol.Chem. 275 (August):25216-25221, 2000.
S. S. Shapiro and S. McCord. Prothrombin. In: Progress in hemostasis and thrombosis,Anonymous New York:Grune&Stratton, 1978, p. 177-177.
L. Shayesteh, Y. Lu, W. L. Kuo, R. Baldocchi, T. Godfrey, C. Collins, D. Pinkel, B. Powell, G. B. Mills, and J. W. Gray. PIK3CA is implicated as an oncogene in ovarian cancer. Nat.Genetics 21 (January):99-102, 1999.
C. Simon, J. Juarez, G. L. Nicolson, and D. Boyd. Effect of PD 98059, a specific inhibitor of mitogen-activated protein kinase kinase, on urokinase expression and in vitro invasion. Cancer Res. 56:5369-5374, 1996.
C. Simon, H. Goepfert, and D. Boyd. Inhibition of the p38 mitogen-activated protein kinase by SB 203580 blocks PMA-induced Mr 92,000 type IV collagenase secretion and in vitro invasion. Cancer Res. 15 (March):1135-1139, 1998.
C. Simon, M. J. Hicks, A. J. Nemechek, R. Mehta, B. W. O'Malley, H. Goepfert, C. M. Flaitz, and D. Boyd. PD 098059,an inhibitor of ERK1 activation,attenuates the in vivo invasivness of head and neck squamous cell carcinoma. Br.J.Cancer 80 (July):1412-1419, 1999.
M. Skobe and N. E. Fusenig. Tumorigenic conversion of immortal human keratinocytes through stromal cell activation. Proc.Natl.Acad.Sci. 95:1050-1055, 1998.
D. J. Slamon, G. M. Clark, S. G. Wong, W. J. Levin, A. Ullrich, and W. L. McGuire. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235:177-182, 1987.
E. M. Southern. Detection of specific sequences among DNA fragments separated by gel electrophoresis. J.Mol.Biol. 98 (503):517, 1975.
M. B. Sporn and A. B. Roberts. Transforming growth factor-ß: Multiple actions and potential clinical applications. JAMA 262:938, 1989.
M. B. Sporn. The war on cancer. Lancet 347:1377-1381, 1996.
A. Stark, A. Kreicbergs, U. Nilsonne, and C. Silfverswoerd. The age of osteosarcoma patients is increasing. J Bone Joint Surg Br 72 (Br):89-93, 1990.
L. R. Stephens, K. T. Hughes, and R. F. Irvine. Pathway of phosphatidylinositol(3,4,5)-triphosphate synthesis in activated neutrophils. Nature 351 (May):33-39, 1991.
A. Strasser, A. W. Harris, M. L. Bath, and S. Cory. Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature 348:331-333, 1990.
T. M. Sweeney, M. C. Kibbey, M. Zain, M. Friedman, and H. K. Kleinman. Basement membrane and the SIKVAV laminin-derived peptide promote tumour growth and metastases. Cancer Metastasis Rev 10:245-254, 1991.
75

G. Literaturverzeichnis
D. C. Talbot and P. D. Brown. Experimental and clinical studies on the use of matrix metalloproteinase inhibitors for the treatment of cancer. European J.Cancer 32:2528-2533, 1996.
I. F. Tannock. The relation between cell proliferation and the vascular system in a transplanted mouse mammary tumour. Br.J.Cancer 22:258-273, 1968.
G.-F. Tempelhoff, G. Hommel, F. Niemann, D. Schneider, and L. Heilmann. Langzeitmortalität bei 324 gynäkologischen Malignompatientinnen in Abhängigkeit von der perioperativen Thromboseprophylaxe mit entweder niedermolekularen Heparin oder unfraktioniertem Heparin-Eine prospektive doppelblinde randomisierte Erhebung. Gynäkol.Geburtshilfl.Rundsch. 39:7-51, 1999.
C. Thomsson, M. Schmitt, L. Goretzki, P. Oppelt, L. Pache, P. Dettmar, and H. Graeff. Prognostic value of the cystein proteases cathepsin B and cathepsin L in human breast cancer. Clin.Cancer Res. 1:741-746, 1995.
N. A. Thornberry and Y. Lazebnik. Caspases: enemies within. Science 281:1312-1316, 1998.
J. A. Trejo, A. J. Connolly, and S. R. Coughlin. The cloned thrombin receptor is necessary and sufficient for activation of mitogen-activated protein kinase and mitogensis in mouse lung fibroblasts. J.Biol.Chem. 271 No.35 (No.35):21536-21541, 1996.
A. Trousseau. Phlegmasia alba dolens. In: Clinique Medicale de l'Hotel-Dieu de Paris., edited by Balliere, Paris: 1864, p. 654-712.
K Tryggvason, M. Hoyhtya, and C. Pyke. Type IV collagenase in invasive tumors. Breast Cancer Res Treat 24:209-218, 1993.
Y. Tsujimoto, J. Cossman, E. Jaffe, and C. M. Croce. Involvement of the bcl-2 gene in follicular lymphoma. Science 228:1440-1440, 1985.
R. R. Vassallo, T. Kieber-Emmons, K. Chichowski, and L. F. Brass. Structure-function relationships in the activation of platelet thrombin receptors by receptor-derived peptides. J Biol Chem 27 (6081):6085, 1992.
D. L. Vaux, S. Cory, and T. M. Adams. Bcl-2 promotes the survival of hematopoietic cells and cooperates with c-myc to immortalize pre-B cells. Nature 335:440-442, 1988.
T.-K. H. Vu, D. T. Hung, V. I. Wheaton, and S. R. Coughlin. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell 64 (1057):1068, 1991.
T.-K. H. Vu, V. I. Wheaton, D. T. Hung, and S. R. Coughlin. Domains specifying thrombin-receptor interaction. Nature 353:674-677, 1991.
Y. Wang, M. S. Simonson, J. Pouyssegur, and M. J. Dunn. Endothelin rapidly stimulates mitogen-activated proetin kinase activity in rat mesangial cells. Biochem J 287:589-594, 1994.
C. P. Webb, L. Van Aelst, M. H. Wigler, and F. Vande Woude. Signaling pathways in Ras-mediated tumorigenicity and metastasis. Proc.Natl.Acad.Sci. 95 (July):8773-8778, 1998.
A. Wehmeirer, D. Tschope, and J. Esser. Circulating activated platelets in MPD. Thromb.Res. 61:271-278, 2000.
R. A. Weinberg. The retinoblastoma protein and cell cycle control. Cell 81:323-330, 1995.
A. Weiss and J. Schlessinger. Switching signals on or off by receptor dimerization. Cell 94 (August):277-280, 1998.
H. J. Weiss, V. T. Turitto, H. R. Baumgartner, Y. Nemerson, and T. Hoffmann. Evidence for the presence of tissue factor activity on subendothelium. Blood 73:968-975, 1989.
76

G. Literaturverzeichnis
M. Whitman, D. R. Kaplan, B. Schaffhausen, L. Cantley, and T. M. Roberts. Association of phosphatidylinositol kinase activity with polyoma middl-T competent for transformation. Nature 315 (May):239-242, 1985.
S. M. Wilhelm, I. E. Collier, B. L. Marmer, A. Z. Eisen, G. A. Grant, and G. I. Goldberg. SV 40-transformed human lung fibroblasts secrete a 92 kDa type IV collagenase which is identical to that secreted by normal human macrophages. J Biol Chem 264 (17213):17221, 1989.
K. Winkler, S Bielack, G. Delling, H. Jürgens, R. Kotz, and M. Salzer-Kuntschik. Treatment of osteosarcoma: experience of the Cooperative Osteosarcoma Study Group (COSS). Cancer Treat.Res. 62:269-277, 1993.
D. S. Witherow, Q. Wang, K. Levay, J. L. Cabrera, J. Chen, G. B. Willars, and V. Z. Slepak. Complexes of the G protein subunit gbeta 5 with the regulators of G protein signaling RGS7 and RGS9. Characterization in native tissue and in transfected cells. J.Biol.Chem. 275 (August):24872-24880, 2000.
J. F. Woessner. Quantification of matrix metalloproteinases in tissue samples. Methods Enzymol. 248:510-528, 1995.
M. Z. Wojtukiewicz, L. R. Zacharski, T. E. Moritz, K. Hur, R. L. Edwards, and F. R. Rickles. Prognostic significance of blood coagulation tests in carcinoma of the lung and colon. Blood Coag.Fibrinolysis 3:429-437, 1992.
M. Z. Wojtukiewicz, D. G. Tang, J. J. Ciarelli, K. K. Nelson, D. A. Walz, C. A. Diglio, E. F. Mammen, and K. V. Honn. Thrombin increases the metastatic potential of tumor cells. Int.J.Cancer 54:793-806, 1993.
M. Z. Wojtukiewicz, D. G. Tang, K. K. Nelson, D. A. Walz, C. A. Diglio, and K. V. Honn. Thrombin enhances tumor cell adhesive and metastatic properties via increased alpha IIb beta 3 expression on the cell surface. Thromb.Res. 68 (November):233-245, 2000.
W. F. Xu, H. Andersen, T. E. Whitmore, S. R. Presnell, D. P. Yee, A. Ching, T. Gilbert, E. W. Davie, and D. C. Foster. Cloning and characterization of human protease-activated receptor 4. Proc.Natl.Acad.Sci. 95 (June):6642-6646, 1998.
Y. Yarden and A. Ullrich. EGF and erbB2 receptor overexpression in human tumors. Growth factor receptor tyrosine kinases. Ann Rev Biochem 57:443-478, 1988.
Y. Yodo and T. Abe. Fibrinopeptide A (FPA) level and cancer fibrinogen kinetics in patients with malignant disease. Thrombosis and Haemostasis 46:706-706, 1981.
E. Yonish-Rouach, D. Grunwald, S. Wilder, A. Kimichi, E. May, J. J. Lawrence, P. May, and M. Oren. p53-mediated cell death: relationship to cell cycle control. Mol.Cell.Biol. 13 (March):1415-1423, 1993.
E. Yoshida, E. N. Verrusio, H. Mihara, D. Oh, and H. C. Kwaan. Enhancement of the expression of urokinase-type plasminogen activator from PC-3 human prostate cancer cells by thrombin. Cancer Res. 54 (June):3300-3304, 1994.
K. Yoshioka, S. Nakamori, and K. Itoh. Overexpression of small GTP-binding protein RhoA promotes invasion of tumor cells. Cancer Res. 59 (April):2004-2010, 1999.
T. Yoshizaki, H. Sato, M. Furukawa, and J. S. Pagano. The expression of matrix metalloproteinase 9 is enhanced by Epstein-Barr virus latent membrane protein 1. Proc.Natl.Acad.Sci. 95 (March):3621-3626, 1998.
P. D. Yurchenco, Y. S. Cheng, and H. Colognato. Laminin forms an independent network in basement membranes. J Cell Biol 117:1119-1133, 1992.
77

G. Literaturverzeichnis
L. R. Zacharski, A. R. Schned, and G. D. Sorenson. Occurrence of fibrin and tissue factor antigen in human small cell carcinoma of the lung. Cancer Res. 43:3963-3968, 1983.
L. R. Zacharski, V. Constatini, M. Z. Wojtukiewicz, V. A. Memoli, and J. Kudryk. Anticoagulants as cancer therapy. Semin.Oncol. 17:217-227, 1990.
X.-F. Zhang, J. Settleman, J. M. Kyriakis, E. Takeuchi-Suzuki, S. J. Elledge, M. Marshall, J. T. Bruder, U. R. Rapp, and J. Avruch. Normal and oncogenic p21 proteins bind to the amino-terminal regulatory domain of c-Raf-1. Nature 364:308-313, 1993.
C. Zhong, D. J. Hayzer, M. A. Corson, and M. S. Runge. Molecular cloning of the rat vascular smooth muscle thrombin receptor. Evidence for in vitro regulation by basic fibroblast growth factor. J Biol Chem 267 (August):16975-16979, 1992.
S. Zucker, H. Mirza, C. E. Conner, A. F. Lorenz, M. H. Drews, W. F. Bahou, and J. Jesty. Vascular endothelial growth factors induces tissue factor and matrix metalloproteinase production in endothelial cells: conversion of prothrombin to thrombin results in progelatinase A activation and cell proliferation. Int.J.Cancer 75:780-786, 1998.
78

H. Abkürzungsverzeichnis Abb. Abbildung ADP Adenosin-5’-diphosphat AP-1 Aktivator Protein-1 APS Ammoniumpersulfat ATP Adenosin-5’-triphosphat bcl-2 B-cell leukemia/lymphoma 2 bp base pair BSA Bovines Serumalbumin Ca2+ Kalzium CAT Chloramphenicol-Acetyl-Transferase cDNA komplementäre Desoxyribonukleinsäure Ci Curie cpm Counts per minute CTP Cytidin-5’-triphosphat ddH2O Doppelt deionisiertes Wasser DEPC Diethylpyrocarbonat DMSO Dimethylsulfoxid DTT Dithiothreitol EDTA Ethylendiaminotetraessigsäure ELISA Enzyme-linked immunosorbent assay ERK Extracellular signal-regulated kinases EtBr Ethidiumbromid FCS Fötales Kälberserum g Erdbeschleunigung GAPDH Glycerinaldehyd-3-phosphatdehydrogenase GPCR G-protein coupled receptor GDP Guanosin-5’-diphosphat GTP Guanosin-5’-triphosphat h Stunden HER-2 Human epidermal growth factor receptor HEPES 4-(2-Hydroxyethyl)-1-Piperazinethansulfonsäure IGF Insulin-like growth factor IL-1ß Interleukin-1ß KAc Kaliumacetat kB Kilobasen kD Kilodalton MAPK Mitogen-activated protein kinase min Minuten MMP Matrixmetalloproteinasen MOPS 3-(N-Morpholino)propansulfonsäure Mr Molekulargewicht mRNA Messenger Ribonukleinsäure MTT (3-[4,5-Dimethylthiazol-2-yl]-2,5-diphenyltetrazolium-Bromid NaAc Natriumacetat NaCl Natriumchlorid NaN3 Natriumazid NTP Nukleotidtriphosphat nm Nanometer
79

H. Abkürzungsverzeichnis
OD optische Dichte ONPG Ortho-Nitrophenylgalactopyranosid PAR protease activated receptor PBS phosphatgepufferte Kochsalzlösung PCR Polymerase Chain Reaction PDGF Platelet derived growth factor PEG Polyethylenglykol PI Phosphatidylinositol PI3K Phosphatidylinositol 3-Kinase PMA Phorbolmyristatacetat RT Raumtemperatur RT-PCR Reverse Transkriptase-Polymerase Chain Reaction S Sekunden SDS Natriumlaurylsulfat SSC Natriumchlorid-Natriumcitrat-Puffer TAE Tris-Acetat-EDTA-Puffer TBE Tris-Borsäure-EDTA-Puffer TE Tris-EDTA-Puffer TEMED N, N, N´, N´-Tetramethylenethylendiamin TF Tissue factor TGFα Tumor growth factor α TNFα Tumor Nekrose Faktor α TNF-R1 Tumor Nekrose Faktor-Rezeptor 1 TRAP Thrombin Rezeptor aktivierendes Peptid Tris Tris-Hydroxymethyl-Aminomethan TTP Thymidin-5’-triphosphat U Units Upm Umdrehungen pro Minute V Volt VEGF Vascular Endothelial growth factor W Watt
80

I. Danksagung
An dieser Stelle möchte ich mich herzlich bedanken bei:
Herrn Univ. Prof. Dr. R. Gradinger, Herrn Univ. Prof. Dr. M.Schmitt und Herrn Univ. Prof.
Dr. H. Graeff für die Arbeitsmöglichkeit in der Klinischen Forschergruppe der Frauenklinik
der TUM und die Unterstützung dieser wissenschaftlichen Dissertation.
Herrn PD Dr. H. Rechl und Herrn PD Dr. Ernst Lengyel für die umfassende Betreuung
dieser wissenschaftlichen Doktorarbeit. Mit viel Geduld haben sie mir
molekularbiologisches Denken und Arbeiten vermittelt und verstanden, mein Interesse an
zellulären Zusammenhängen zu fördern.
Frau Dr. A. Fenn-Rottler für die tatkräftige Mitarbeit, Teamfähigkeit und Zuverlässigkeit.
Frau Kerstin Dehne, Frau Dr. Sandra Hapke, Frau Katrin Härthing, Frau Claudia Jäger,
Herrn Dr. Bernd Mühlenweg und Frau Dr. Sabine Ried für die Zusammenarbeit innerhalb
der Arbeitsgruppe und Hilfsbereitschaft im Labor.
Allen Kolleginnen und Kollegen in der Klinischen Forschergruppe der Frauenklinik der
TUM.
Allen, die mir während der Arbeit fachlichen und menschlichen Beistand geleistet haben.
81