neuromuskuläre erkrankungen im kindes- und jugendalter · physiotherapie orthesen zur...
TRANSCRIPT

Dr. Norbert Utzig
Neuromuskuläre Erkrankungen
im Kindes- und Jugendalter

Muskeldystrophie,
Dermatomyositis,
Myositis
Spinale Muskelatrophie
Poliomyelitis
Myasthenie
Neurale Muskelatrophie,
GBS
Formen neuromuskulärer Erkrankungen
degenerativ – immunvermittelt – infektiös – traumatisch – Tumor etc.
Friedreich-Ataxie
Multiple Sklerose
Myelitis

Neuromuskuläre Erkrankungen - Symptome
• Intrauterin: verminderte Kindsbewegungen
• Neonatal: Hypotonie, Ateminsuffizienz, schwaches Schreien,
Trinkschwäche
• Säugling: statomotorische Retardierung, Hypotonie
• Später: Gangstörung, Stolpern, verminderte grobe Kraft (Treppensteigen),
Ausdauer, verschmächtigtes Muskelrelief, Skoliose

Neuromuskuläre Erkrankungen - Diagnostik
• Anamnese
• Untersuchung
• CK, Transaminasen, Antikörper
• EMG / NLG
• Sonographie
• Liquordiagnostik
• Molekulargenetik
• (Nerv-) Muskelbiopsie
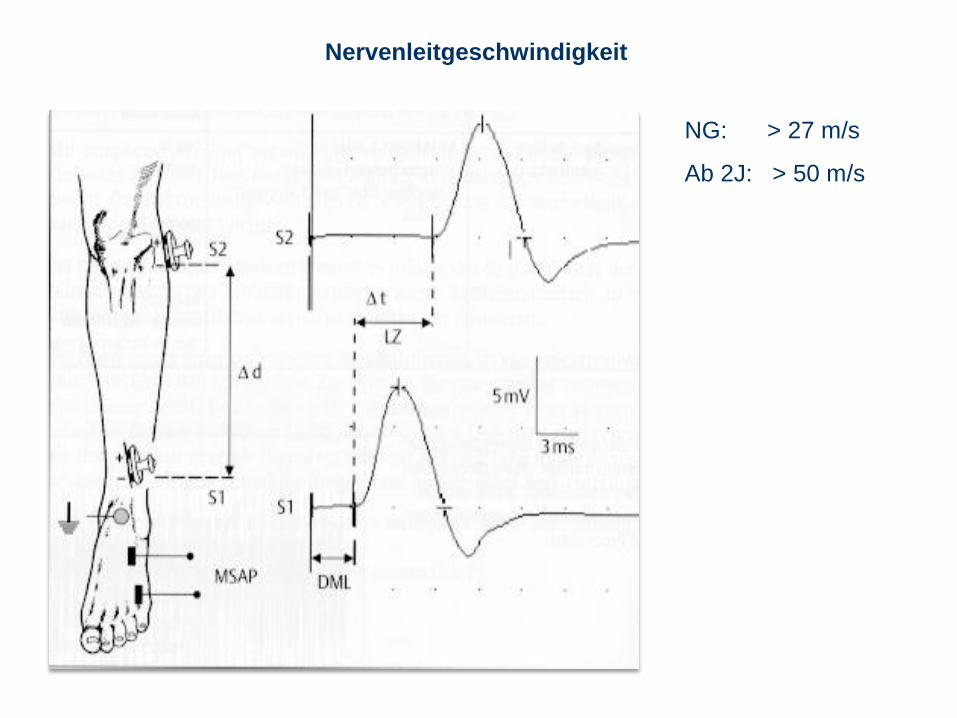
Nervenleitgeschwindigkeit
NG: > 27 m/s
Ab 2J: > 50 m/s
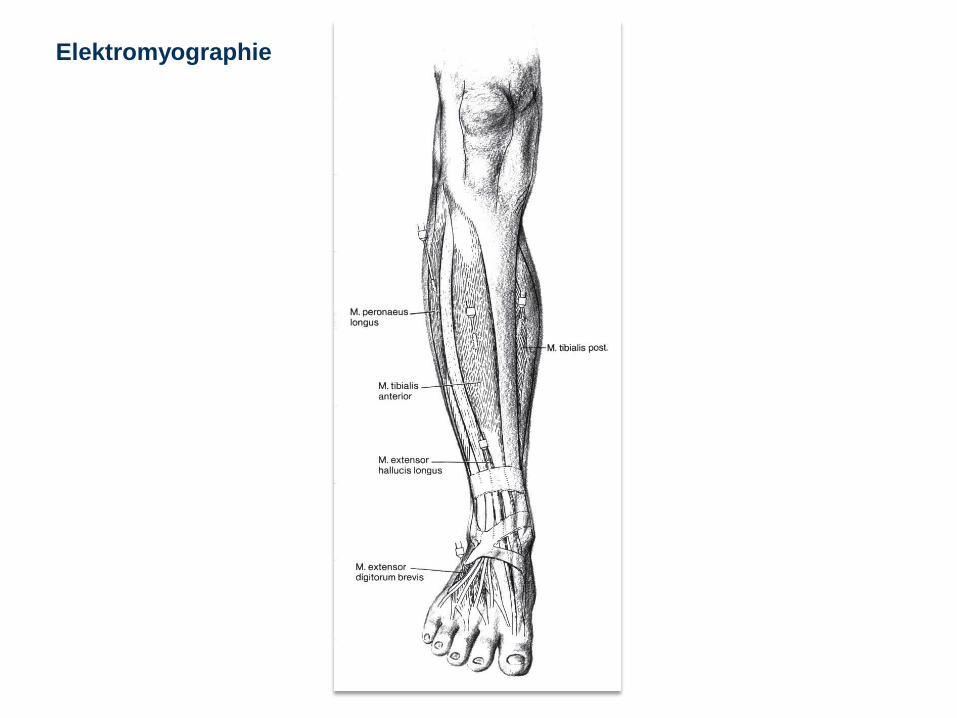
Elektromyographie

Max B. 1J, Hypotonie, normales EMG

Eduard U. 8 Jahre, Muskeldystrophie Duchenne
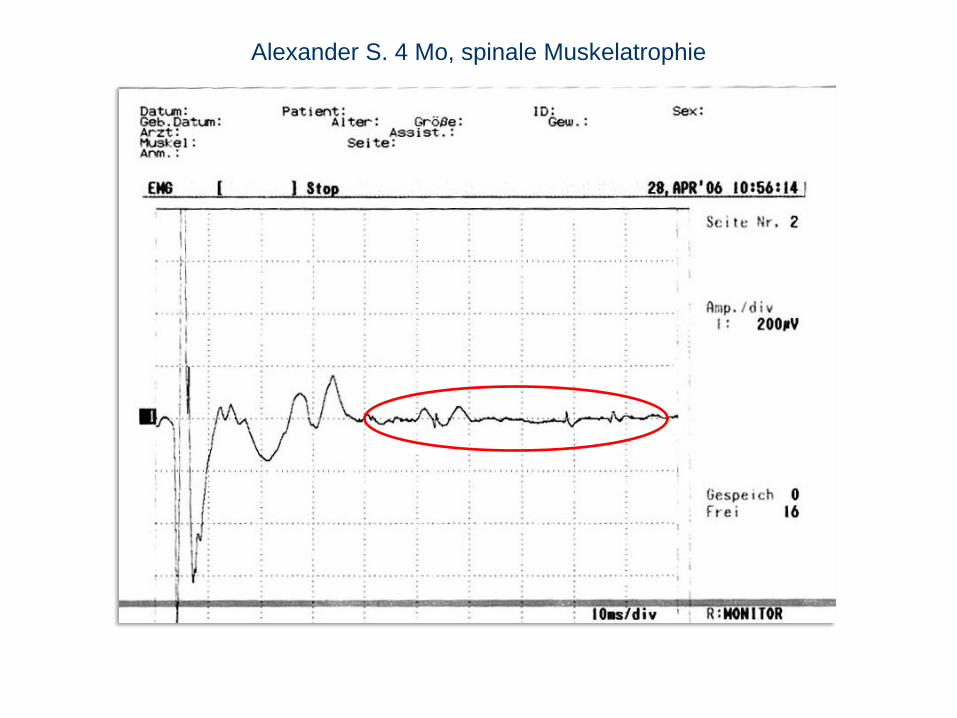
Alexander S. 4 Mo, spinale Muskelatrophie
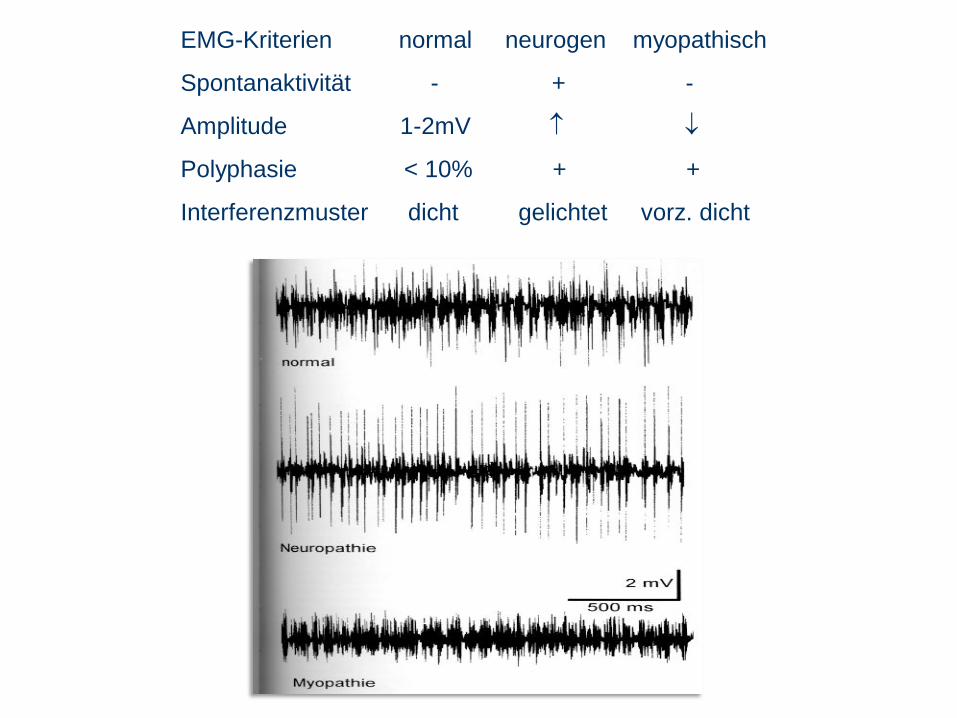
EMG-Kriterien normal neurogen myopathisch
Spontanaktivität - + -
Amplitude 1-2mV
Polyphasie < 10% + +
Interferenzmuster dicht gelichtet vorz. dicht

Spinale Muskelatrophie
Fehlen des neuronal apoptosis inhibiting protein (NAIP), rezessiv, Chr. 5q11, ->
Verlust der Vorderhornzellen (2. Motoneuron), Deletion im SMN1-Gen,
Copienzahl SMN 2 bestimmt Schwere.
SMA1: infantil (Werdnig-Hoffmann), Häufigkeit 1:10000 – 1:20000. Manifestation
0 - 2 J, extreme Hypotonie und Bewegungsarmut, distal etwas besser, dabei
Kontrast zu wachem Gesichtsausdruck (DD Postasphyxiesyndrom).
Keine Eigenreflexe, paradoxe Atmung, evtl. Fibrillieren der Zunge. Schwaches
Schreien, oft bereits intrauterin wenig Kindsbewegungen. Lebenserwartung
Jahre.
SMA 2: intermediär: freies Sitzen möglich
SMA3: juvenil (Kugelberg-Welander), Manifestation 1 - 30 Jahre, eher
schleichender Beginn und langsame Progredienz
SMA4: > 30 Jahre, wohl anderer Gendefekt

Spinale Muskelatrophie
EMG Spontanaktivität (Fibrillationen) und eher große Myopotentiale.
Nervenleitgeschwindigkeit normal. Fermente (CK, Leber, LDH, Aldolase) normal
bis leicht erhöht.
Diagnosesicherung in 94% durch Molekulargenetik (homologe Deletion im SMN1-
Gen). Selten Muskelbiopsie, dabei gruppierte Atrophie der Typ II Fasern.
Pränatale Diagnostik mit direkter Genanalyse.
Therapieversuche zur Erhöhung der Transkription von SMN2 (z.B. Valproat)
zunächst kein Erfolg. Jetzt Phase III Studie mit Olesoxime (Ansatz
Mitochondrienmembran).
Ganz aktuell: Nusinersen intrathekal (moduliert Splicing SMN2).
Motorisches Training im Mausmodell effektiv
(Überleben, Erhalt der Vorderhornzellen)

Spinale Muskelatrophie
Olesoxime bei SMA
Cholesterol-Oxim
Wirkung auf Mitochondrienmembran
10 mg/kg/d oral
Gute Verträglichkeit
Phase III – Studie: 165 Pat. SMA 2 und 3, Aufhalten der Progression gegenüber
Placebo
Nusinersen (Spinraza) bei SMA
Antisense-Oligonucleotid
moduliert Splicing SMN2
erhöht Gehalt an SMN1-Protein
intrathekale Anwendung
HWZ 160 Tage
Orphan Drug Status seit 12/2016 in EU / USA
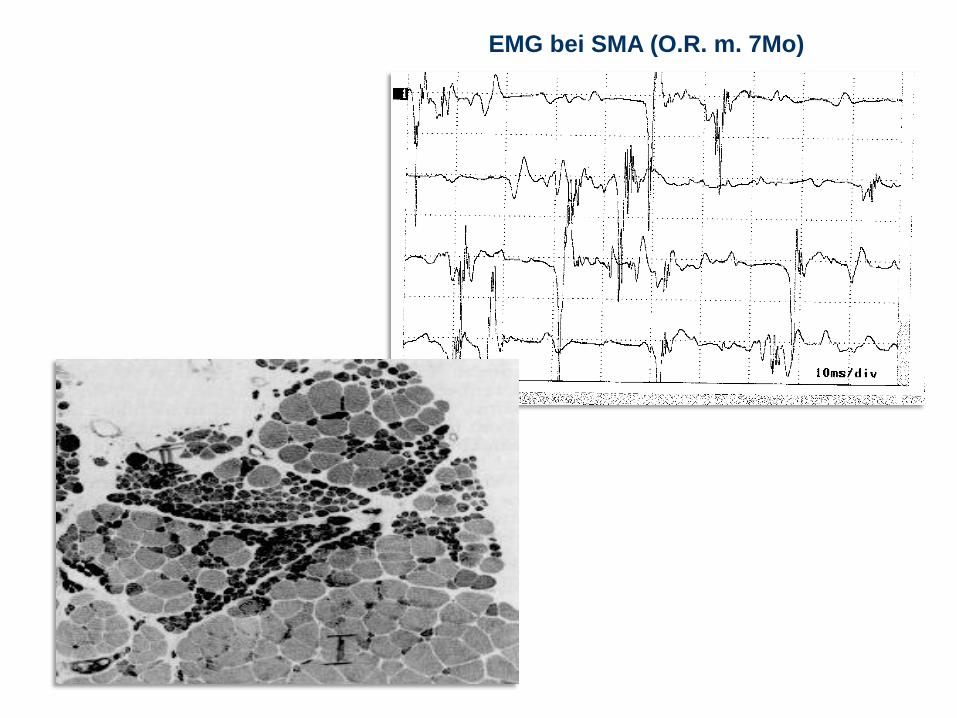
EMG bei SMA (O.R. m. 7Mo)

Neurale Muskelatrophie (Hereditär sensorisch-motorische Neuropathie)
Häufigster Typ HSMN I (Charcot-Marie-Tooth). Autosomal dominant (Trisomie
PMP22-Gen Chr.17), u.U. SMRet im Sgl.Alter, Hypotonie, später oft Fußprobleme
durch Hohlfuß, Fehlbelastung, “Storchenbeine”, auch ataktische Komponente.
Diagnostik: Klinisches Bild, FA, NLG vermindert (10-20m/s), NLG der Eltern,
Molekulargenetik, Suralisbiopsie mit De/Remyelinisierung (Zwiebelschalen). DD
Friedreichsche Ataxie.
Therapie: KG, Orthesen, op. Korrektur der Fußfehlstellung

Muskelerkrankungen

MYOPATHIEN
Angeborene Myopathien
Erworbene Myopathien

MYOPATHIEN
Angeborene Myopathien
- Metabolische Myopathien
- Progressive Muskeldystrophien
- Strukturmyopathien

MYOPATHIEN
Angeborene Myopathien
- Metabolische Myopathien
- Progressive Muskeldystrophien
- Strukturmyopathien
Myopathien i.e.S. im Verlauf langsamer als Muskeldystrophien,
teils keine Progredienz
Diagnose durch Stoffwechseluntersuchung und Biopsie
Beispiele:
Myotubuläre Myopathie
Glycogenose VI
Muskel-Carnitin-Defizit
Endokrin
Mitochondrial
Fasertypendisproportion.

MYOPATHIEN
Erworbene Myopathien
- Entwicklungsstörungen
- traumatische, toxische Myopathien
- Endokrine Myopathien
- Entzündliche Myopathien

Muskeldystrophien

- Muskeldystrophie Typ Duchenne
- Muskeldystrophie Typ Becker
- Muskeldystrophie Emery-Dreifuss
- Fazio-skapulo-humerale Muskeldystrophie
Erb-Landouzy-Déjerine
- Myotone Muskeldystrophie Steinert-Curshmann-Batten-Gibb
- MD Batten-Turner
- MD Cornelia de Lange
- MD Fukayama
- MD Santavuori
- MD Leyden-Möbius
- MD Erb
- Okuläre MD
- Distale MD
Muskeldystrophien

Muskeldystrophie
Degenerativer Prozess der Muskelzelle, z.B. Wandbestandteil fehlt oder
verändert (z.B. Dystrophin).
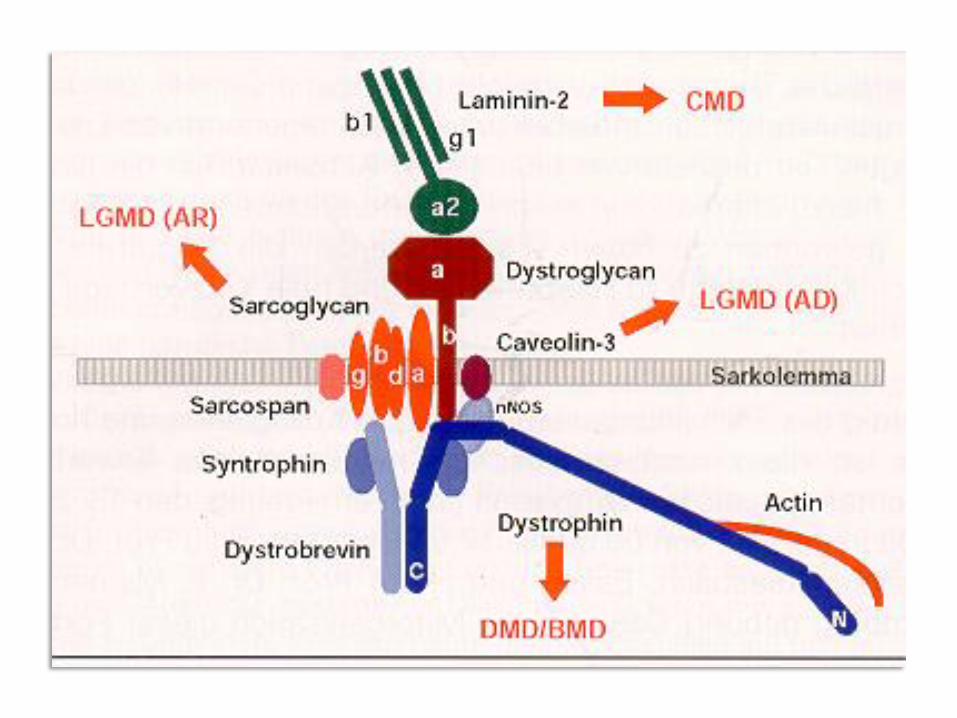

Muskeldystrophie
M. Duchenne: "Muskelschwund", Xp21 rezessiv, Leseraster der DNA ist
verschoben -> Dystrophin fehlt (Deletion, Nonsense oder Stopp-Codons).
Häufigkeit 1:3500 mNG, ca. 1/3 Neumutationen
Beginn meist Ende Kleinkindzeit bis Schulalter, manchmal bereits verzögerte
statomot. Entwicklung als Säugling. Schwäche der gesamten Muskulatur,
besonders Haltemuskulatur Rumpf und Oberschenkel, Hyperlordose mit
Spitzfußhaltung, Gowers-Zeichen (Emporklettern an sich selbst) positiv.
Pseudohypertrophie der Waden.
Gefährdung bez. Skoliose und Kontrakturen, besonders Achillessehnen,
Kniebeuger, Hüftbeuger. Rollstuhl ab 10.-14.LJ. Frühzeitige OP (Rideaukonzept)
verlängert Gehfähigkeit und verzögert damit Skolioseentwicklung. Etwa 1/3 der
Kinder auch mental beeinträchtigt.
Auch der Herzmuskel ist betroffen: Kardiomyopathie mit Herzinsuffizienz und
Rhythmusstörungen

Muskeldystrophie
Massive Erhöhung der Muskelfermente (CK), bereits im Säuglingsalter, auch
Leberfermente und LDH hoch.
Im EMG kleine aufgesplitterte hochpolyphasische Potentiale.
Diagnosesicherung durch Molekulargenetik (ca.2/3 Deletion 1/3 Punktmutationen
im Dystrophin-Gen), wenn nicht diagnostisch Biopsie und Histologie
(Muskelfasernekrosen mit fettig-bindegewebigem Umbau, Dystrophin fehlt).
M. Becker – Kiener: DNA-Leseraster erhalten, Dystrophin teilweise vorhanden,
benignere Form mit Beginn im Schul/Jugendlichenalter, sehr variable
Progredienz. Gefahr: Herzrhythmusstörungen.

EMG bei MDD (A.M.m. 10Mo)
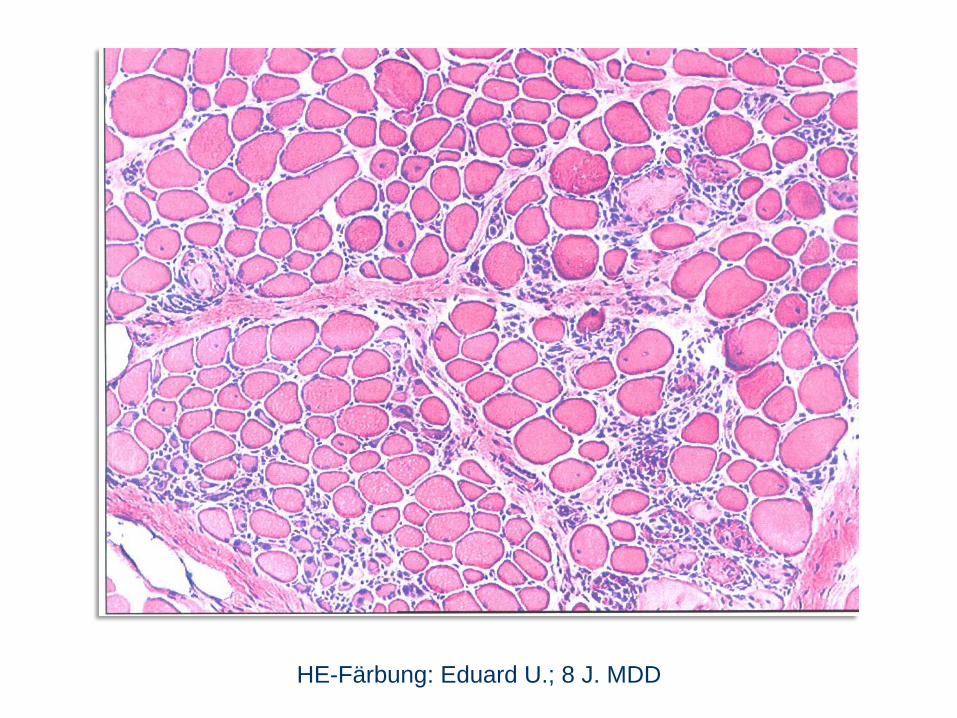
HE-Färbung: Eduard U.; 8 J. MDD
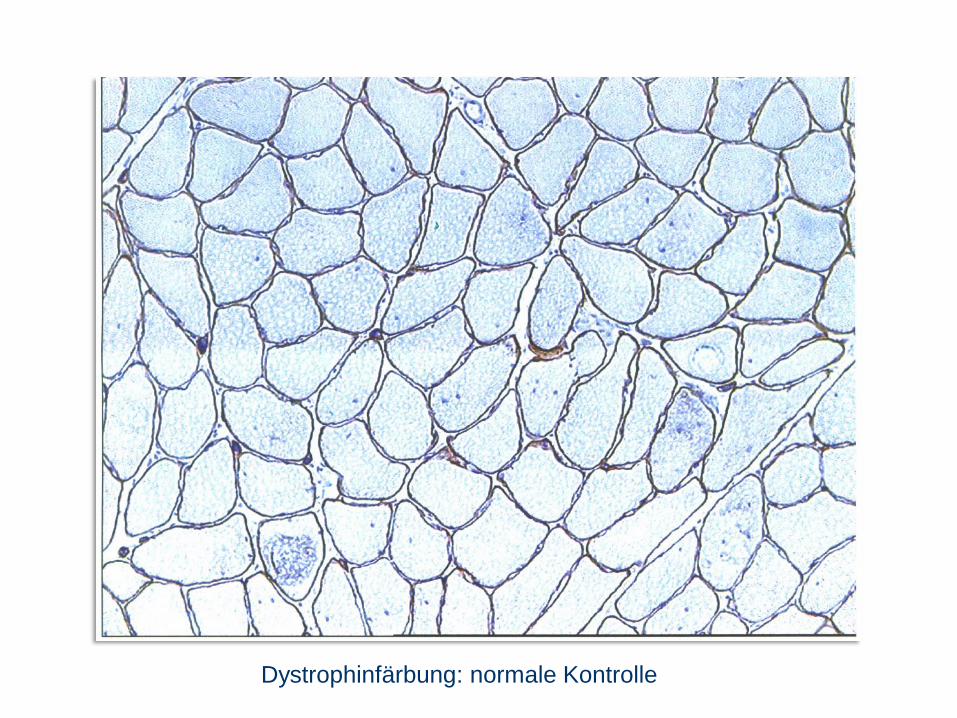
Dystrophinfärbung: normale Kontrolle

Eduard U.; 8 J. MDD

Panel-Diagnostik bei Muskelerkrankungen
(MGZ München)

Therapieansätze bei Muskeldystrophie Duchenne
Heilung derzeit nicht möglich. Steroide verlangsamen die Progredienz
(Gehfähigkeit + 2 - 5 Jahre) aber mit erheblichen Nebenwirkungen
Physiotherapie
Orthesen zur Stabilisierung und Kontrakturprophylaxe, Hilfsmittel für Mobilität und
Kommunikation (E-Rollstuhl, Schreibcomputer)
Operative Korrektur von Fehlstellungen (Skoliose) und Kontrakturen (Rideau)
Therapie der Kardiomyopathie
(Prophylaxe mit ACE-Hemmern frühzeitig, ab 5 – 6 Jahre)
Nicht-invasive Beatmung (bei nächtlicher Hypoventilation)
Gentherapie

Steroidtherapie bei Muskeldystrophie Duchenne
Beginn mit 4 - 6 Jahren (Plateauphase)
Prednisolon initial 0,75mg/kg/d
Deflazacort initial 0,9mg/kg/d
Gehfähigkeit + 2 - 5 Jahre
Weniger WS-Chirurgie
Verbesserte Herz/Lungenfunktion
Spätere nichtinvasive Beatmung
Höhere Lebensqualität
Höhere Lebenserwartung

Nebenwirkungen der Steroidtherapie
Gewichtszunahme (häufigster Abbruchgrund)
Frakturen (Prednison), teils subklinisch
Linsentrübung (Deflazacort), teils subklinisch
Kleinwuchs
Bluthochdruck
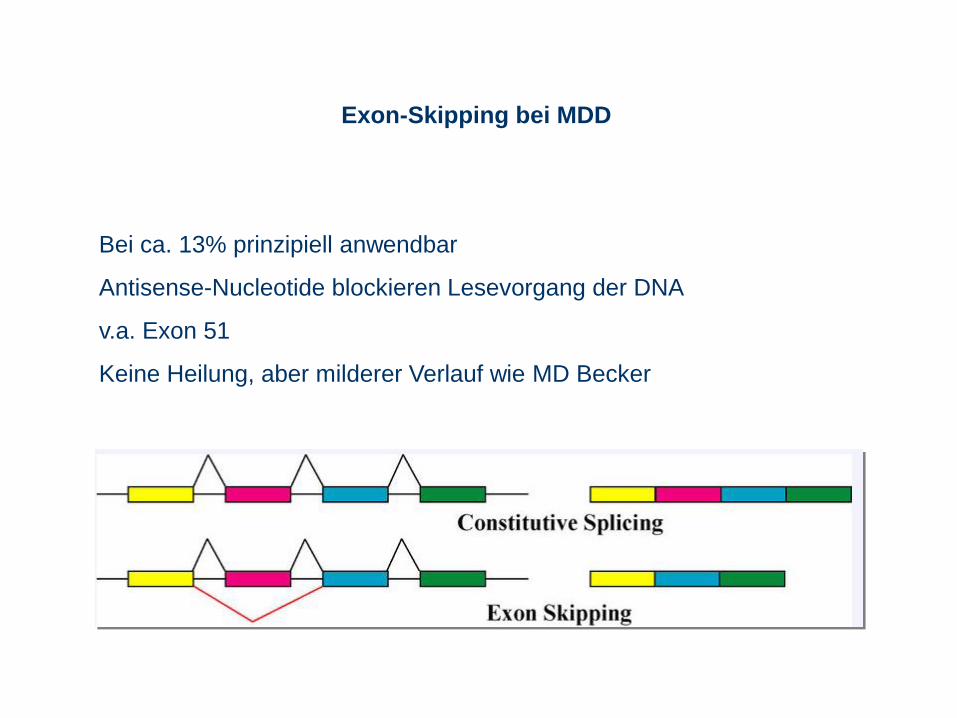
Exon-Skipping bei MDD
Bei ca. 13% prinzipiell anwendbar
Antisense-Nucleotide blockieren Lesevorgang der DNA
v.a. Exon 51
Keine Heilung, aber milderer Verlauf wie MD Becker
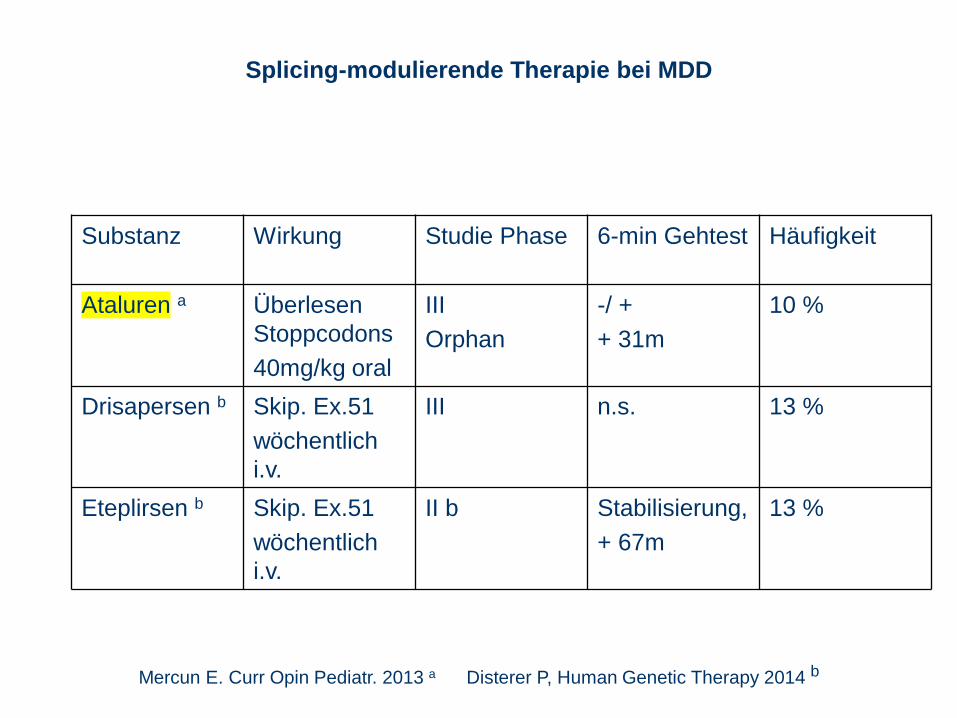
Substanz Wirkung Studie Phase 6-min Gehtest Häufigkeit
Ataluren a Überlesen
Stoppcodons
40mg/kg oral
III
Orphan
-/ +
+ 31m
10 %
Drisapersen b Skip. Ex.51
wöchentlich
i.v.
III n.s. 13 %
Eteplirsen b Skip. Ex.51
wöchentlich
i.v.
II b Stabilisierung,
+ 67m
13 %
Splicing-modulierende Therapie bei MDD
Mercun E. Curr Opin Pediatr. 2013 a Disterer P, Human Genetic Therapy 2014 b

Operative Maßnahmen bei Muskelerkrankungen
Prophylaxe/Therapie von Kontrakturen
(Rideau-Konzept): Achillessehen, mediane Kniebeuger, M. rectus femoris,
M. tensor fasciae latae , evtl. Tractus iliotibialis (Gehfähigkeit + 3J)
Skoliose-Korrektur (Cobb 20-40°): Luque-Rahmen ab 10J, Teleskopstab n.
Naumann.

Interdisziplinäre Betreuung bei progredienten
neuromuskulären Erkrankungen
Krankengymnastik
Ergotherapie
Sozialpädagogik
Psychologie
Orthopädie
Orthopädietechnik
Humangenetik
Neuropädiatrie




Genetik

Genetik
G G
Rezessive Vererbung -> Wiederholungsrisiko 1:4
G K K G K K
G KG K

Genetische Beratung
• Informationspflicht des Arztes über das Wiederholungsrisiko bei
hereditären Erkrankungen
• Information über den voraussichtlichen Verlauf und
Therapiemöglichkeiten
• Information über Möglichkeiten einer pränatalen Diagnostik
• keinerlei Wertung
• Non-direktiv, Entscheidung über Erhalt oder Abbruch einer
Schwangerschaft liegt allein bei den betroffenen Eltern
• Präimplantationsdiagnostik (in Deutschland nach Ethikvotum
ab 1.2.2014)

Methoden der pränatalen Diagnostik
Chorionzottenbiopsie: 10. Woche p.m.
Amniozentese: 15. Woche p.m.

Stefanie N., 8 Jahre
Anfang 12/03: afebrile Infektion der oberen Luftwege
Mitte 12/03: zunehmende Schwäche der Beine, mehrfach Sturz beim
Treppensteigen, Inappetenz und Gewichtsabnahme 5 kg
Extern LP: 9 Leuko, Alb 548, Verlegung wegen Zittrigkeit (Myoklonien?) und
zunehmender Paresen
Ende 12/03: (Kinderneurologie HGW): Hypotonie, kein freies Stehen, Ataxie,
PSR, ASR, TSR neg., Blasenentleerungsstörung
V.a. Polyradikuloneuritis (Guillain-Barré-Syndrom)
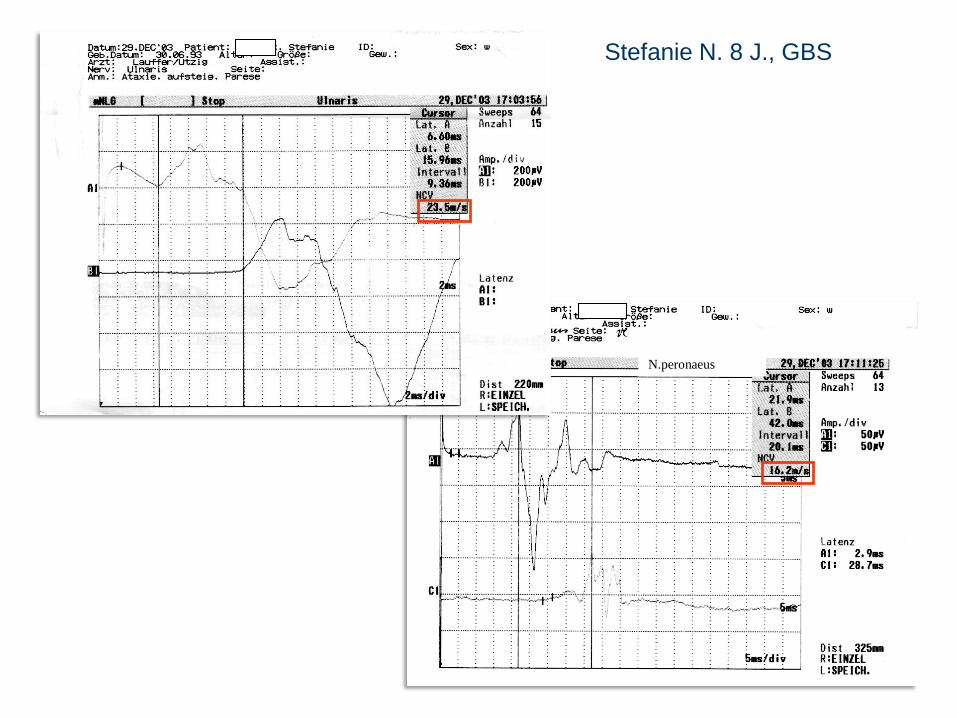
N.peronaeus
Stefanie N. 8 J., GBS

Stefanie N., 8J
NLG: vermindert mit erhöhter distaler Latenz
Liquor: 7 Leucos, Protein mit 3,5 g/l extrem erhöht
Therapie: i.v. Immunglobuline 0,4g/kg/d für 5 Tage
Ende 1/04: steht frei, kurze Gehstrecken möglich, Ataxie rückläufig
2/04: Verlegung in Reha-Klinik

Akute Polyradikuloneuritis I
(Guillain-Barre-Syndrom)
Oft vorausgehende Infektion (CMV, Mycoplasmen), zelluläre und humorale
immunologische Kreuzreaktion mit Myelin des peripheren Nervensystems, nach
1-4 Wo aufsteigende Lähmungen, aber auch Atemlähmung innerhalb 24h
möglich!
Begleitende Rücken/ Beinschmerzen und Sensibilitätsstörungen möglich.
Bei Kindern oft zunächst vermehrtes Stolpern. MER erloschen.
Diagnostik: NLG mit Desynchronisierung, Reizschwelle , auch
Leitungsblock. Liquor-EW .

NLG bei GBS (K.S., w., 12 J.)
46 m/s
53 m/s
Befundnormalisierung

Myasthenie I

Myasthenie I
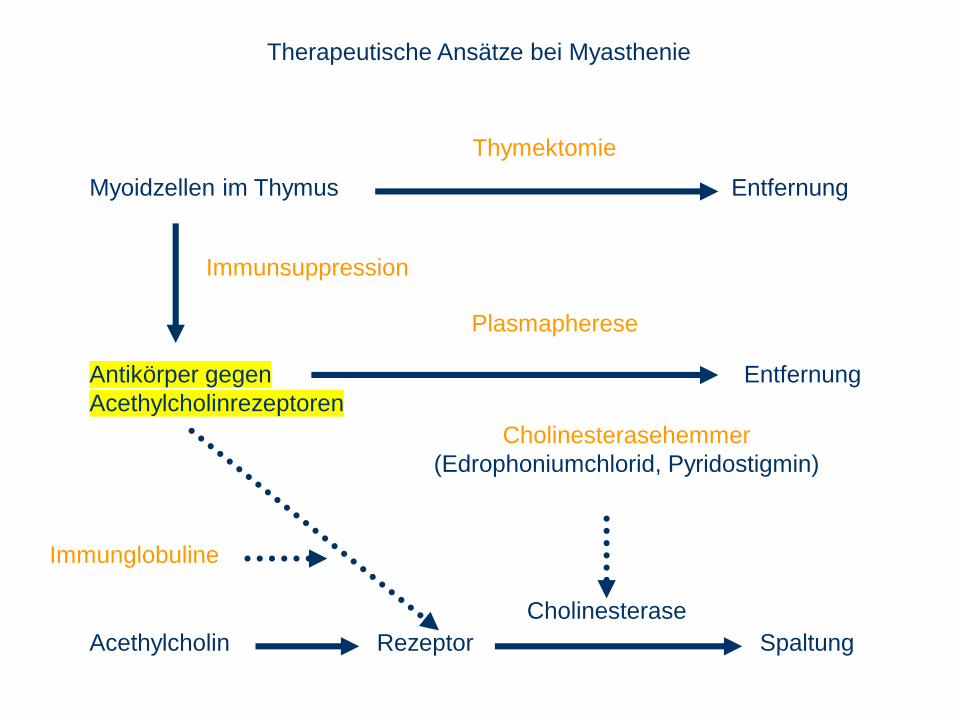
Störung der neuromuskulären Übertragung durch Antikörper gegen
Acetylcholinrezeptoren.
Muskelähnliche Zellen (Myoidzellen) im Thymus später auch in Lymphknoten
rufen Antikörperbildung hervor, Kreuzreaktion mit AChRezeptoren.
Beginn eher rasch, mitunter nach einem unspezifischen Infekt, verschiedene
Formen mit Schwäche der Haltemuskulatur (generalisierte M.),
Augenmuskelparesen (okuläre M.), Schluck- und Sprechstörung (Dysarthrie),
kann bis zur Ateminsuffizienz gehen als myasthenische Krise.
Typisch sind Belastungsabhängigkeit und tageszeitliche Bindung. Abends
und nach Belastung schlechter.
Passive Übertragung der AK von Mutter auf Kind führt zu passagerer
neonataler (“Leih”) Myasthenie.
Acethylcholin Rezeptor Spaltung
Myoidzellen im Thymus Entfernung
Antikörper gegen Entfernung
Acethylcholinrezeptoren
Therapeutische Ansätze bei Myasthenie
Cholinesterasehemmer
(Edrophoniumchlorid, Pyridostigmin)
Immunsuppression
Plasmapherese
Cholinesterase
Thymektomie
Immunglobuline

Myasthenie II
Diagnose:
Nachweis von AK gegen Ach-Rezeptoren und Skelettmuskel
Bei Neurographie evtl. Dekrement
Beweis durch Tensilon-Test, Hemmer der ACholEsterase, Wirkung nur wenige
Minuten. Monitoring und Beatmungsbereitschaft, Atropin bereithalten!
Therapie:
Cholinesterasehemmer (Mestinon)
Kortikoide
Thymektomie (muß frühzeitig durchgeführt werden sonst wirkungslos)
Biopsie nicht erforderlich
DD: Kongenitale Myasthenie = Rezeptordefekt für Acetylcholin
Antikörper negativ
> keine Thymektomie

EMG bei Myasthenia gravis (J.K., w., 12 J.)

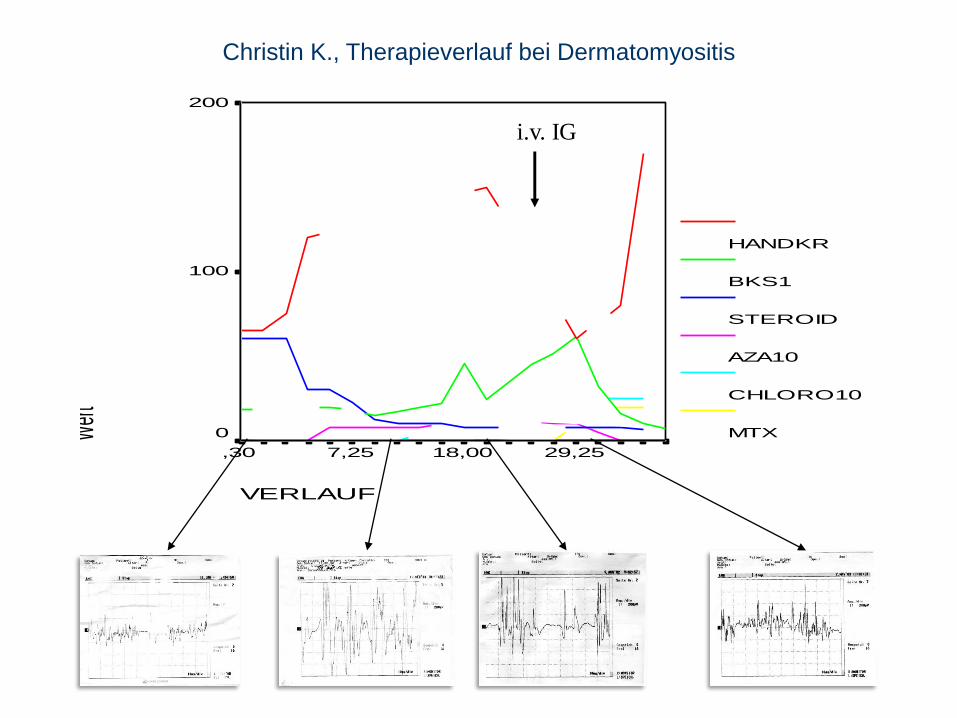
Dermatomyositis I
5 – 15 J., Mädchen doppelte Häufigkeit
primär humoral vermittelte Immunreaktion (molekulares Mimikri) führt zum
Verlust von Kapillaren und zu Atrophie von Muskelfaszikeln
-> proximal betonte Muskelschwäche und Schmerzen.
Beginn mit Fieber, Müdigkeit, livide Verfärbung und Ödem der Oberlider,
Schmetterlingsförmiges Gesichtsexanthem, V-förmiges Erythem am Stamm,
Rötung über Gelenken, Teleangiektasien parungual.
Schluck- und Atemstörung möglich.
Verkalkungen subkutan und intramuskulär bei längerem Verlauf in 30-70%.
VERLAUF
29,2518,007,25,30
Wer
t200
100
0
HANDKR
BKS1
STEROID
AZA10
CHLORO10
MTX
Christin K., Therapieverlauf bei Dermatomyositis
i.v. IG

Dermatomyositis II
Diagnose: BKS , CK , EMG (myopathisch und neuropathisch), Biopsie.
Therapie: Im akuten Stadium Ruhigstellung, frühzeitige und langanhaltende
Gabe von Kortikoiden (über 2 Jahre), weitere Optionen MTX, AZA, CSA, CPA,
IvIG, Plasmapherese. Nach Abklingen der Akutsymptomatik KG zur
Kontrakturprophylaxe.
Komplikationen: artikulär, gastrointestinal, pulmonal, kardial, retinal.

Polymyositis
Zellgebundene Immunreaktion
seltener als DM
bei Kindern meist kein paraneoplastisches Phänomen
ab 14 Jahren
symmetrische proximale Schwäche
langsame Progression.
Diagnose und Therapie wie DM

Poliomyelitis
Typ I-III, Enterovirus, RNA, Picorna
Endemiegebiete in Afrika und Asien, keine Lebendimpfung mehr empfohlen
(Impfpolio 1:106), Prophylaxe mit Salk-Impfung
Asymptomatisch in 90-95%
Zunächst grippales Krankheitsbild, nach 14T 2. Fieberphase und Paresen
durch Schädigung der Vorderhornzellen. Disposition nach Abwehrlage. 2/3
spinal, 1/3 bulbär. Überwiegend untere Extrem, asymmetrisch
Besserung in ersten 4-6 Wo, anschließend nur noch in 1-2% möglich
Bleibende Paresen in 0,001 – 0,1%.
Diagnose: Liquordiagnostik (seröse Meningitis), PCR, Virusisolierung,
Serologie. EMG, NLG.
DD: Myelitis durch andere neurotrope Enteroviren, Borrelien, GBS

Läsion des Plexus brachialis
Neonatal: 1:1000, bei Schädellage z.B. durch Lateralflexion des Kopfes, bei BEL
durch Zug am Arm.
Oberer Plexus C5- C7 fast immer betroffen, Arm adduziert, innenrotiert,
Ellenbogen gestreckt, UA proniert, Hand geschlossen, keine Dorsalextension. Bei
oberer + unterer Parese (+ C8-Th1) Arm schlaff, auch keine Fingerbeweglichkeit.
Oft begleitende Klavikulafraktur.
Späteres Trauma: Verkehrsunfälle, Lagerung OP. Erholung wenn Kontinuität
erhalten, sonst OP erforderlich. Regeneration 1mm/d, (Plexus lumbalis selten
betroffen).
Diagnose: Untersuchung, EMG, MRT, Myelo-CT (leere Tasche bei Wurzelausriss).
Therapie: KG zur Vermeidung von Kontrakturen, teils Elektrostimulation,
Vermeidung von Zug auf Plexus, Spontanremission bis 70%, falls in 3 Monaten
keine Besserung bei kompletten Paresen Mikrochirurgische OP.

Multiple Sklerose
Encephalomyelitis disseminata
Multilokuläre, demyelinisierende Gehirn- und Rückenmarkserkrankung
ungeklärter Ätiologie
Autoimmunreaktion gegen Myelinprotein (aMBP) nach Virusinfektion (?)
(HHV6, Polyomaviren, Chlamydien)
Genetische Disposition ? (HLA-System, T-Zell-Rezeptoren,
Bcl-2-Expression in Oligodendrozyten)
Umweltfaktoren ? (Einfluss des geographischen Aufenthaltes)
Hohe Prävalenz in Europa/USA, Nord-Süd-Gefälle polwärts
Prävalenz in Deutschland 50 / 100 000, F : M = 2 : 1
Manifestation v.a. 2. und 3. Lebensjahrzehnt
Manifestation vor dem 10. Lj. in 0,2 – 2 % der Fälle

Verlaufsformen:
schubförmig rezidivierend-remittierend (ca. 70%)
schubförmig progredient (ca. 25%)
primär chronisch-progredient (ca. 1 – 5%)
Prognose:
30% benigner Verlauf
10% maligner Verlauf

Leitsymptome:
zentrale Paresen (45 / 85 %)
Sensibilitätsstörungen (42 / 86 %)
Optikusneuritis (33 / 62 %)
zerebelläre Symptome (24 / 79 %)
Augenmotilitätsstörungen (14 / 36 %)
Andere Hirnstammsymptome (10 / 29 %)
Blasen/Mastdarmstörungen (9 / 61 %)
Psychische Störungen (4 / 39 %)

Differentialdiagnosen bei multilokulärem ZNS-Befall:
Neuroborreliose
Neurosyphilis
Sarkoidose
Kollagenosen und Vaskulitiden (Antiphospholipidsyndrom)
Rezidivierende Ischämien
AIDS
Gefäßmalformationen

Diagnostik:
Labor: Serologie, Auto-AK, ACE
Liquor: lymphozytäre Pleozytose, oligoklonale IgG-Banden
MRT: Entmarkungsherde
Evozierte Potentiale: VEP, MEP, SSEP, AEP
Urinstatus, Uroflowmetrie, Urographie, Blasendruck

MS – Therapie:
im Schub Glukokortikoide
Langzeitmedikation bei schubförmigen Verlauf
Beta-Interferon (hIFN-ß)
Azathioprin
Mitoxantrone
Monoklonale Antikörper
bei chronisch-progredientem Verlauf Cyclophosphamid, Methotrexat
Versuch: Linomide (synthet. Immunmodulator), Immunglobuline
Physiotherapie, Hilfsmittel, Blasentraining, HWI-Prophylaxe
Regelmäßige Lebensführung, keine Hitze, kein Alkohol
psychotherapeutische, soziale Unterstützung