pathologies de hb - unifr.ch · - anticorps contre erythrocytes ... état héterozygote–...
TRANSCRIPT

1
Pathologies de Hb
Anémies Drépanocytose Thalassémies Polyglobulies
Anemies
anemies hémolytiques
v déficience d’Enzyme v anemie hemolytique autoimmune v anémie microangiopathique (TTP) v anemie Sickle-cell v Thalassemie v Malaria

2
Hämolytische Anämien Formen der Blutarmut aufgrund einer verkürzten Lebensdauer der Erythorcyten Ursachen: Blutkörperchen selber (Korpuskuläre Ursache) -Defekte der Erythrocyten membran -> Kugelzellenanämie, Elliptocytose (fehlen von Protein 4.1 auf Innenmembran (aldolase), Defekte in Ankyrin, Bande 3. Ca. 10 Tage Lebensdauer, dann entfernt in Milz -> Knochenmark Neubildung und Kompensation aber wenn zusätzlich Infektion vorhanden ist -> keine Kompensation mehr. -Enzymdefekte, Glucose-6-phosphat dehydrogenase Mangel, Pyrivatkinasemangel -Paroxysomale nächtliche Hämoglobinurie: Mutation in Synthese von GPI-Ankerprotein Unabhängig von Blutkörperchen (extrakorpuskuläre Ursachen) Immunhämolysen Mechanische Einwirkung Toxine (z.B. Schlangengift) Infektion (Mykoplasmenbefall)
v deficience d’Enzymes : Glucose 6-Phosphogluconate Pyruvate
G-6PD
Pyruvate kinase
Hemolyse: aigue – dommages oxydatifs
(deficience de G-6PD) ou chronique (deficience de PK)
3
• deficience en G6PD est généralement asymptomatique.
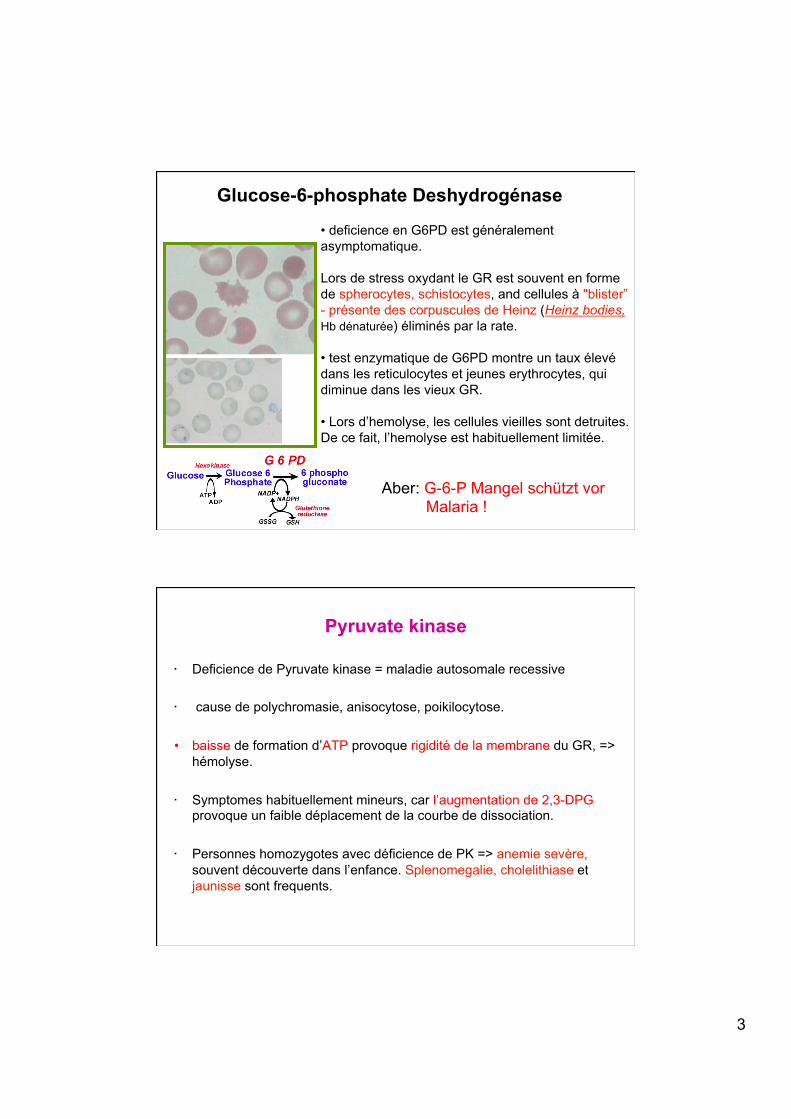
Lors de stress oxydant le GR est souvent en forme de spherocytes, schistocytes, and cellules à "blister” - présente des corpuscules de Heinz (Heinz bodies, Hb dénaturée) éliminés par la rate. • test enzymatique de G6PD montre un taux élevé dans les reticulocytes et jeunes erythrocytes, qui diminue dans les vieux GR.
• Lors d’hemolyse, les cellules vieilles sont detruites. De ce fait, l’hemolyse est habituellement limitée.
Glucose-6-phosphate Deshydrogénase
Aber: G-6-P Mangel schützt vor Malaria !
Pyruvate kinase
• Deficience de Pyruvate kinase = maladie autosomale recessive
• cause de polychromasie, anisocytose, poikilocytose.
• baisse de formation d’ATP provoque rigidité de la membrane du GR, => hémolyse.
• Symptomes habituellement mineurs, car l’augmentation de 2,3-DPG provoque un faible déplacement de la courbe de dissociation.
• Personnes homozygotes avec déficience de PK => anemie sevère, souvent découverte dans l’enfance. Splenomegalie, cholelithiase et jaunisse sont frequents.

4
C/ Anémies mégaloblastiques par carence vitaminique Folates et/ou B12
• Carence en vit B12:
-troubles de l’absorption (gastrectomie, anomalie du récepteur iléal du Facteur Intrinsèque, lésions inflammatoires intestinales ou pancréatiques)
Maladie de Biermer: MAI avec Auto Ac anti FI
-rarement une carence par apport insuffisant
• Carence en folates:
-très souvent insuffisance d’apport lors accroissement des besoins (grossesse, hémolyse, hémorragie, infections), ou malnutrition
-troubles de l’absorption (maladies inflammatoires intestinales, résection,…)
-défaut d’utilisation des folates: alcoolisme, médicaments antifoliques
Signes cliniques habituels de l'anémie, mais d'installation très progressive (plusieurs mois), associés à glossite, diarrhées, stérilité, et un syndrome neurologique si carence en vit. B12.
Absorption de Vitamin B12 : Absorption diminuée: - pas de facteur intrinsèque (anemie pernicieuse) - pas d’absorption sur ileum terminal - croissance bacterienne importante
Vitamin B12 Intrinsic factor
Absorption
Stomach
Terminal ileum
Perniziöse Anämie Morbus Biermer, Vitamin B12 Mangel B12 -> braucht es um N5-Tetrahydrofolat zu machen
5
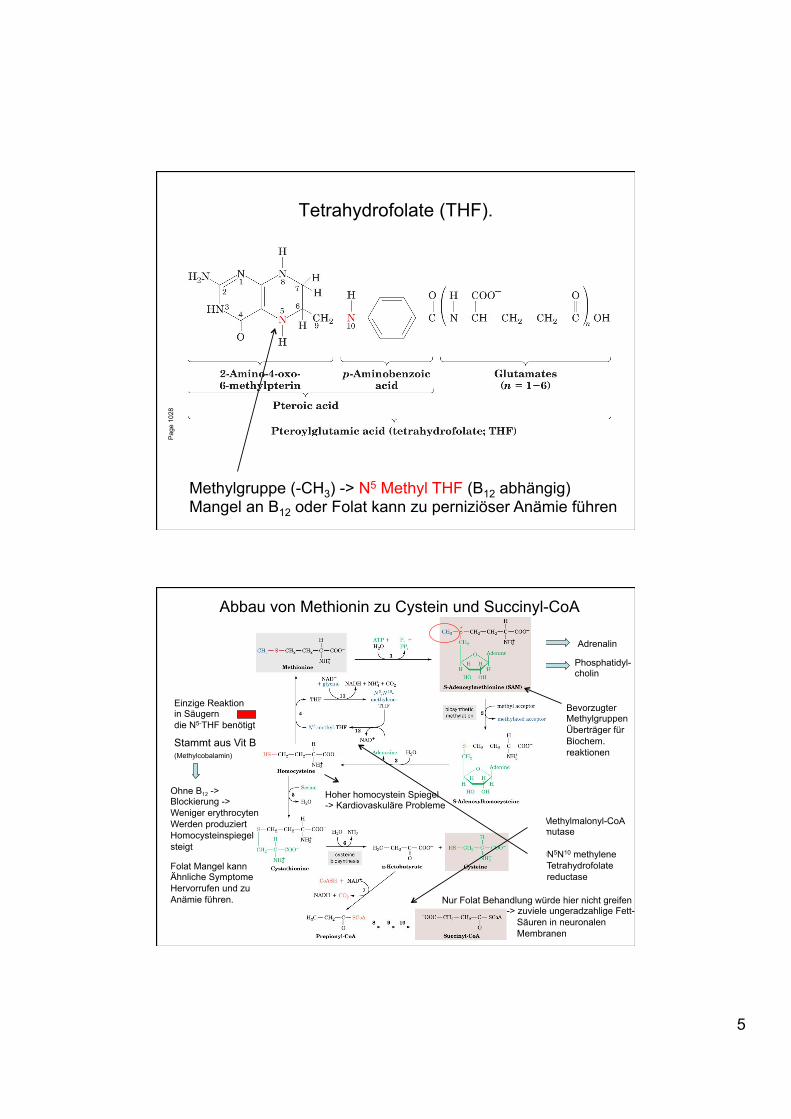
Tetrahydrofolate (THF). P
age
1028
Methylgruppe (-CH3) -> N5 Methyl THF (B12 abhängig) Mangel an B12 oder Folat kann zu perniziöser Anämie führen
Abbau von Methionin zu Cystein und Succinyl-CoA
Stammt aus Vit B12 (Methylcobalamin)
Einzige Reaktion in Säugern die N5-THF benötigt
10=Methylmalonyl-CoA mutase 12=N5N10 methylene Tetrahydrofolate reductase
Adrenalin
Phosphatidyl- cholin
Bevorzugter Methylgruppen Überträger für Biochem. reaktionen
Ohne B12 -> Blockierung -> Weniger erythrocyten Werden produziert Homocysteinspiegel steigt
Folat Mangel kann Ähnliche Symptome Hervorrufen und zu Anämie führen. Nur Folat Behandlung würde hier nicht greifen
-> zuviele ungeradzahlige Fett- Säuren in neuronalen Membranen
Hoher homocystein Spiegel -> Kardiovaskuläre Probleme

6
The biosynthetic fates of the C1 units in the THF pool.
Pag
e 10
29
= adrenalin
= membranlipid
Anemies v anemie hemolytique autoimmune : - Anticorps contre erythrocytes - Etiologie: idiopathique (primaire), maladie lymphoproliferative, troubles autoimmunes systemiques (SLE), médicaments, infections (viral; bacterial) - Diagnostique: test antiglobuline direct - Therapie: steroides

7
B-Anémies hémolytiques non corpusculaires: maladies acquises, hémolyse induite par agression externe
B.1-Anémies hémolytiques d’origine immunologique
-post-transfusionnelles, MHNN, par incompatibilité
-AHAI: Auto-Ac (« chauds ou froids ») anti-Ag de gps sanguins apparaissant ds infections virales,MAI,médicaments, tumeurs…
-AH immuno-allergiques origine médicamenteuse
B.2-Anémies hémolytiques d’origine toxique: médicaments, venins, champignons, dérivés benzéniques, arsenic, PLOMB Saturnisme: Pb conduit à inhibition ALA synthétase et Héme synthétase, accumulation ALA ds sang et urines. Diminution capacité fixation du fer, troubles de maturation des réticulocytes
B.3-Anémies hémolytiques d’origine infectieuse: C.perfringens, Plasmodium
B.4-Anémies hémolytiques d’origine mécanique: destruction intra-vasculaire, présence de schizocytes, cas des prothèses valvulaires cardiaques, ou microangiopathies (vaisseaux altérés) de HTA, cancers, purpura thrombotique et thrombocytopénique, et Syndrome hémolytique et urémique de l’enfant
Anémie inflammatoire
Tout syndrome inflammatoire (infectieux, auto-immun) chronique conduit à une séquestration du fer par les macrophages, avec livraison difficile du fer aux érythroblastes
Syndrome anémique peu intense souvent détecté après la cause inflammatoire.
Anémie modérée (Hb entre 9 et 11g/dl) longtemps normocytaire et normochrome puis discrètement microcytaire (rarement VGM < 75fl) et faiblement hypochrome (rarement TCMH < 24pg).
Anémie arégénérative, avec anomalies légères sur frottis.
Souvent Thrombocytose, et Polynucléose neutrophile.
Anémie hyposidérémique (Fer rarement < 300µg/l).
Transferrine faiblement diminuée
Coefficient de Saturation normal à faiblement diminué (rarement < 20%).
Ferritine plasmatique normale, parfois augmentée.
Syndrome biologique inflammatoire : VS augmentée, Protéines plasmatiques de l'inflammation élevées (CRP, Fibrinogène, Haptoglobine …), γglobulines sériques et α2globulines augmentées…

8
Anemie anemie pernicieuse : - maladie autoimmune - implication hématologique et neurologique - traitement à l’acide folique corrige l’anémie,mais pas les troubles neurologiques
Anemies v Sickle-cell anemie: Drépanocytose (HbS)
mutation du gene β-globine état héterozygote– protection de la malaria? état homozygous – forme grave: - hémolyse - crises douloureuses - crises respiratoires - priapisme - déficience multiorganes

9
U.Albrecht BC1
Über 400 Hämoglobin mutanten bekannt. Bei 95% nur eine einzige AS ausgetauscht. Solche Mutationen werden Hämoglobinopathien genannt. Dagegen werden Störungen in der Hämoglobinsynthese als Thalassämien bezeichent.
A. Molekulare Pathologie des Hämoglobins
1. Austausch von AS an der Oberfläche Änderungen von oberflächlich gelegenen AS sind oft unauffällig, da Oberflächen AS keine spezielle Funktion haben. Ausnahme: Sichelzellenanämie (HbS, siehe später). HbE ist die häufigste Mutante nach HbS. Glu B8(26)ß -> Lys, d.h. saure AS wird durch basische er- setzt. Da aber an Oberfläche und keine spezifische Funktion -> keine Folgen 2. Austausch intern gelegener AS Austausch interner AS führt meist zu Destabilisierung von Hämoglobin -> Abbauprodukte, speziell zer- fallenes Häm kleben an Zellmembran der Erythrocyten -> vorzeitige Lyse. Träger instabiler Hämoglobine haben hämolytische Anämie
Abnormale Hämoglobine

10
kleinste Konformationsänderung im Zentrum kann zum Verlust der Bindung von Häm führen -> Funktionslosigkeit Hb Hammersmith Phe CD1(42)ß -> Ser Häm wird herausgelöst Hb Bristol Val E11(67)ß -> Asp Häm wird herausgelöst Hb Bibba Leu H19(136)α -> Pro Helix wird gebrochen -> instabil Hb Savanna Gly B6(24)ß -> Val Überkreuzung B und E Helix gestört
Hb Philly Tyr C1(35) α->Phe α1-ß1 Kontakt gestört
U.Albrecht BC1
Sichelzellenanämie
HbS GluA3(6)ß -> Val
Molekularer Hintergrund der Sichelzell-Anämie U.Albrecht BC1
Erythrocyten haben Sichelähnliche Form -> weniger Flexibel -> führt zu fatalen Blockaden des Blutflusses. Durchblutung in Peripherie verschlechtert -> gestörte Blutzirkulation. Mutation führt zur Verklumpung der deformierten Erythrocyten -> Homozygote Träger leiden an hämolytischer Anämie. In Afrika 25% der Bevölkerung heterozygote Träger von HbS GluA3(6)ß -> Val. Bietet Schutz gegen Malaria. Grund: Malaria Parasit benötigt eine bestimmte Kalium-Ionen Konzentration für seine Entwicklung die in normalen EC vorhanden ist. Die HbS Mutation senkt die Kalium-Ionen Konzentration in den Sichel- Zellen und der Parasit kann nicht überleben.
HbA
HbS
Normale EC
Sichelförmige EC

11
Drépanocytose (HbS)
U.Albrecht BC1 Die HbS-Fasern werden durch intermolekulare Kontakte stabilisiert
HbS GluA3(6)ß -> Val Glu macht keinen Kontakt OxyHb keine hydrophobe Tasche
Kontakte zwischen Asp73 und Glu 23 auch in HbA -> genügt alleine nicht um Fasern zu Bilden. Jedoch ist auch dieser Kontakt wichtig. Weitere Kontakte -> mischen von Hb verschiedener Mutanten
Hb Harlem Glu3 -> Val,Asp73 -> Asn -> höhere Konz. von HbH/S nötig für Polymerisation als für HbA/S Hb Kole-Bu Asp 73 -> Asn Polymeri- sation auch verzögert. Asp73 wichtig für intermolekulare Kontakte von HbS. Hb Memphis Glu23 -> Gln α-UE von HbMemphis und ß-UE von HbS gelieren langsamer.
Anordnung der DesoxyHb
12
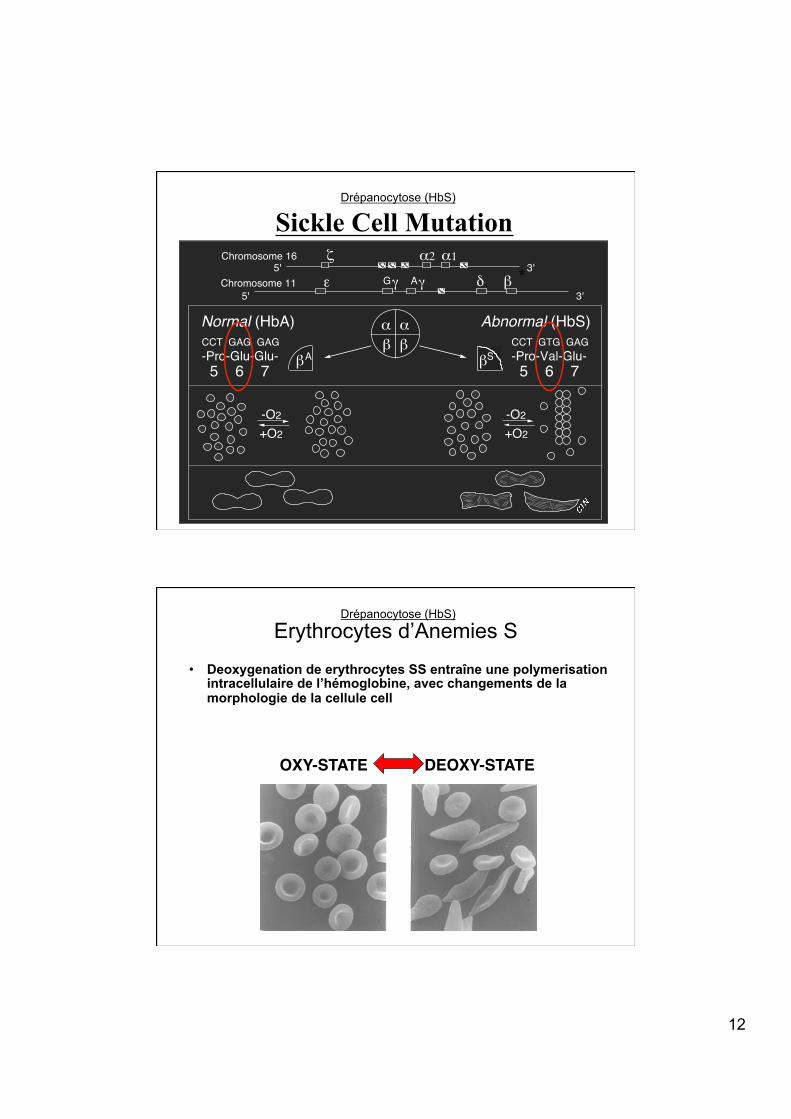
Sickle Cell Mutation
+O2
-O2
+O2
-O2
5'α1α2ζ
3'Chromosome 16
5' 3'Chromosome 11 ε γ γ δ βG A
α αβ βCCT GAG GAG
-Pro-Glu-Glu-5 6 7
CCT GTG GAG-Pro-Val-Glu-5 6 7 βA βS
Normal (HbA) Abnormal (HbS)
CTN
*
Drépanocytose (HbS)
Erythrocytes d’Anemies S
OXY-STATE! DEOXY-STATE!
• Deoxygenation de erythrocytes SS entraîne une polymerisation intracellulaire de l’hémoglobine, avec changements de la morphologie de la cellule cell
Drépanocytose (HbS)

13
HbS bildet Fasern die ver- Antwortlich sind für die Sichelfrom der EC. Die EC können platzen und die Fasern treten aus.
Fasern des HbS die aus einem Erythrocyten austreten.
U.Albrecht BC1 Der Beginn der HbS - Gelbildung ist ein komplexer Prozess
Faserbildung tritt mit einer Zeitlichen Verzögerung auf. Abhängig von: Sauerstoffpartialdruch (T->R Übergang) HbS Konzentration Temperatur 1 / td = k (ct / cs ) td = Verzögerungszeit
ct = total HbS Konzentration cs = Löslichkeit von HbS k,n = Konstanten
Zweistufenprozess: 1. Bildung eines Nucleus von HbS Molekülen
homogener Nukleationsprozess hohe Konzentrationsabhängigkeit
2. Gebildete Fasern = Nukleationskeime heterogener Nukleationsprozess rasche Beendigung der Gelierung
= kinetische Hypothese episodenhaftes Auftreten von Krisen -> Stocken Blutstrom Oxygenierung -> Fasern aufgelöst -> keine Fasern in arter- iellem Blut -> Kapillargebiet in 0.5-2s durchströmt -> HbS kann ausfallen wenn aber td > Transitzeit keine Unterbre- chung des Blutstroms. td kann verkürzt werden bei Fieber, O2 Mangel oder Dehydratisierung -> Krisen.
n
Zeitverlauf der Gelbildung von HbS
Initiation der Gelbildung durch Temperatursprung von 0 auf 20°C
Konz. Abhängigkeit der Gelbildung von DesoxyHb bei 30°C

14
erhöht Anteil an HbF (wie ist unbekannt) -> andere dynamik in der Faserbildung dauert länger als Aufenthaltsdauer im peripheren Gewebe.

15
ANEMIES liées à des MALADIES CHRONIQUES
• Trouble de la Thyroide • Maladie vasculaire du Collagène
– Arthritis rheumatoid – Lupus Erythematosus Systemique – Polymyositis – Polyarteritis Nodosa
• Maladie Inflammatoire intestinale – Colitis Ulcerative – Maladie de Crohn’s
• Tumeur • Maladie infectieuse chronique
– Osteomyelitis – Tuberculosis
• Insuffisance rénale
• Synthèse de la chaine α: par 2 genes α: α1 et α2, sur chromosome 16. • Les chaînes β & δ: proviennent d’un seul gene sur chromosome 11. • Chaine γ: dirigée par 2 genes, γG & γA, sur le chromosome 11.
Thalassémies Anomalies quantitatives constitutionnelles de la synthèse de globine
• Syndromes Thalassémiques: diminution ou absence de synthèse d'une ou plusieurs chaînes de globine : α-Thalassémies et β-Thalassémies.

16
Globinketten sind auf 2 verschiedenen Chromosomen codiert
β
β
α
α
- Hémoglobinose H : délétions de 3 gènes (--/-α). Anémie microcytaire hypochrome, régénérative, et hémolytique, avec corps de Heinz. HbA 70%, HbH 10 à 30 %, traces Hb Bart's. Croissance normale, retard pubertaire.
- α-thalassémies mineures :
délétion de 2 gènes en cis (--/αα) : α0-thalassémie hétérozygote
délétion de 2 gènes en trans (-α/-α) : α+-thalassémie homozygote
Microcytose sans anémie, ou pseudopolyglobulie microcytaire et hypochrome. Electrophorèse de l'Hb normale chez l'adulte, parfois HbA2 < 2,5%. Naissance Hb Bart's 5%. Cliniquement asymptomatique.
- α-thalassémie silencieuse: délétion d'un gène (-α/αα), α+-thalassémie hétérozygote. Electrophorèse de l'Hb: à la naissance Hb Bart's 1%, normale chez l'adulte. Cliniquement et biologiquement asymptomatique.
• Les α-Thalassémies
Altérations génétiques: délétions, (rarement mutations), d'un ou plusieurs des 4 gènes α.
Populations: Sud-Est asiatique (Thaïlande, Laos, Vietnam), Afrique noire, Bassin méditerranéen, Moyen-Orient.
- Délétions des 4 gènes : létale, mort in utero ou à la naissance
- Anémie sévère macrocytaire, Hb Bart's (γ4)80%, HbH (β4)10%, Hb Portland (ζ2γ2) 10%
Thalassémies

17
Erscheinungsformen der α-Thalassämie
α-Thalassämien weniger dramatisch im Verlauf als β-Thalassämien, da 4 Allele vorhanden sind. Bart = γ4-tetramere (Hbγ4) Nach Geburt γ durch β ersetzt -> β4-tetramere Zeigen keinen Bohr Effekt, sind unstabil Und neigen zur Verklumpung -> Lebensdauer der Erthrozyten reduziert und haben bizarre Formen
Anomalies quantitatives constitutionnelles de la synthèse de globine
• Syndromes Thalassémiques: diminution ou absence de synthèse d'une ou plusieurs chaînes de globine : α-Thalassémies et β-Thalassémies.
• Les β-Thalassémies
Populations : Bassin méditerranéen (Grèce, Italie du Sud, Sicile, Sardaigne, Afrique du Nord), Asie du Sud-Est, Moyen Orient, Afrique noire, Antilles, Noirs américains.
Altérations génétiques : 150 mutations ponctuelles connues (délétions plus rares), affectant le promoteur des gènes β, les mécanismes d'excision-épissage, ou le signal de terminaison de traduction.
∅β thalassémie majeure ou maladie de Cooley
Anémie n'apparaissant pas à la naissance car synthèse HbF restant majoritaire, donc diagnostic établi entre 3 mois et 18 mois.
Anémie sévère (Hb entre 4 et 7g/dl), microcytaire et hypochrome, peu régénérative.
Anomalies sur le frottis caractéristiques : microcytose, poïkylocytose, elliptocytes, hypochromie, annulocytes et hématies cibles, ponctuations basophiles, et érythroblastose sanguine (imposant la correction de la numération leucocytaire).
Thalassémies

18
β-Thalassémie hétérozygote
Forme mineure : Anémie modérée microcytaire et hypochrome, peu régénérative. Hb A2 > 3%, et HbF 1 à 5%. Forme clinique très atténuée de la maladie de Cooley.
Forme minime : Pseudopolyglobulie microcytaire et hypochrome sans anémie, HbA2 > 3%. Porteur inapparent de la maladie, cliniquement asymptomatique, diagnostic au hasard à un âge avancé, parfois au cours d'une enquête familiale après dépistage d'un enfant atteint de la maladie de Cooley
β-Thalassémies homozygotes
Mutation portant sur les 2 gènes β : β0-Thalassémie si aucune chaînes β produites ou β+-Thalassémie si seulement diminution de production des chaînes β.
β-Thalassémie intermédiaire
Forme atténuée de la maladie de Cooley, définition strictement clinique représentant 10% des β-Thalassémies homozygotes.
Anémie modérée, bien supportée, sans déglobulisation rapide, HbF augmentée. Croissance normale, mais retard pubertaire.
Thalassémies
Globinketten sind auf 2 verschiedenen Chromosomen codiert
β
β
α
α

19
β-Thalassémies
Das klinische Bild ist abhängig von der genetischen Konstellation: Thalassämia minor: Heterozygotie: nur ein Allel (Genkopie) ist mutiert, dies ist meist symptomlos. Die Erythrozyten sind klein (MCV und MCH sind vermindert). Thalassämia intermedia: Meist compound Heterozygotie: die beiden Allele sind an unterschiedlichen Stellen mutiert, es entsteht eine mittelschwere Anämie mit Ikterus („Gelbsucht“) und Milzvergrößerung. Je nach Ausprägung kann eine Eisenüberladung entstehen. Thalassämia major: Meist Homozygotie: beide Allele sind identisch mutiert, es entsteht eine schwere, im Kindesalter auftretende schwere Anämie mit zunehmender Milzvergrößerung und Wachstumsverzögerung. Früher war die Haupttodesursache eine Eisenüberladung mit Organschädigung
Krankheitszeichen
20
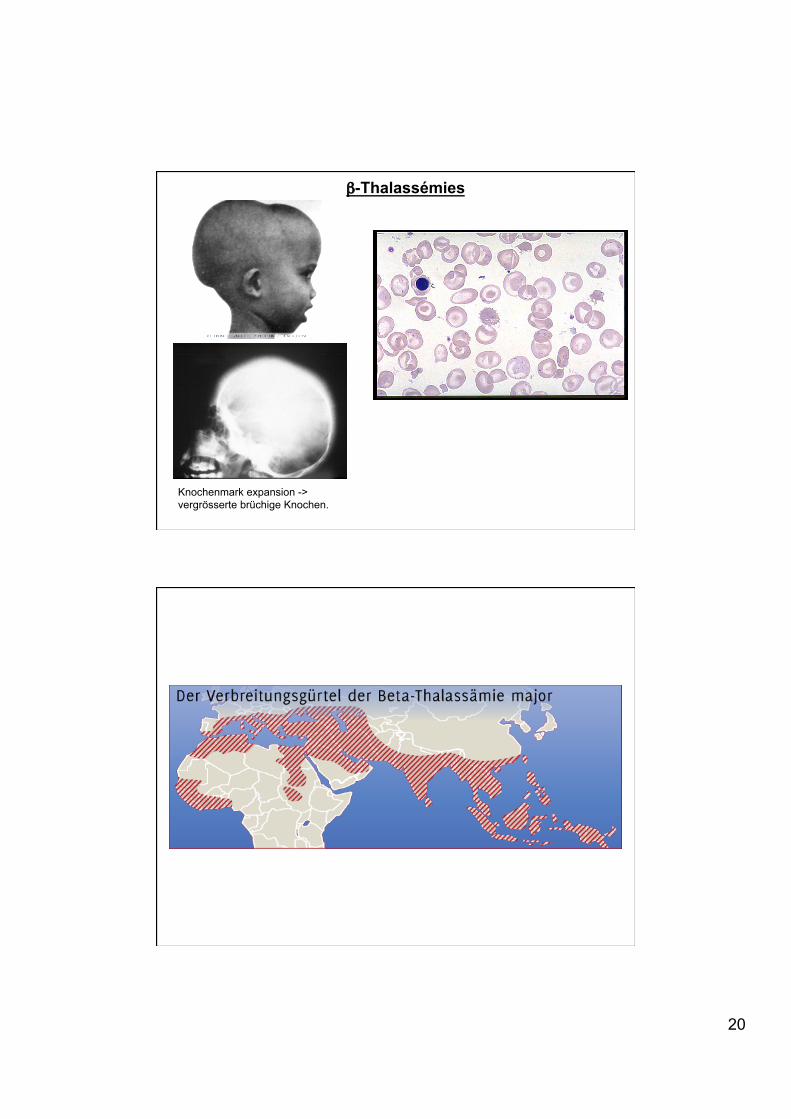
β-Thalassémies
Knochenmark expansion -> vergrösserte brüchige Knochen.