regulatorische betrachtung* von cad-cam-verfahren in der ... · regulatorische betrachtung* von...
TRANSCRIPT

Regulatorische Betrachtung* von
CAD-CAM-Verfahren in der Medizintechnik
• Deutsche Medizinproduktegesetz & Verordnungen
• Europäische Medizinprodukterichtlinie 93/42/EWG (bis 25. Mai 2020)
• Europäische Medizinprodukteverordnung 2017/745 (ab 26. Mai 2020)Seite 1

Zertifizierte Beratungen zur Unternehmenszertifizierung
– ISO 9001 Qualitätsmanagementsysteme - www.isologisch.de -
– ISO 13485 Qualitätsmanagement Medizinprodukte
– MDD 93/42/EWG und EU-MDR 2017/745
– CMDCAS (Kanada), PAL/JGMP (Japan), 21CFR (USA), TGA
Beratungen zur Produktzulassung
– Gestaltung und Ausarbeitung der Technischen Dokumentation
• Risikoanalysen, Grundlegende Anforderungen, Verträge
• Klinische Bewertungen, Prozessvalidierungen, Marktbeobachtungen
• Lieferantenaudits und Interne Audits
• Projektmanagement bei Start-Ups, Spin-Offs und Fusionen
Referent für o.g. Bereichen (Medical Mountains, MDC und In-House)
….. und Ersteller der „EU-MDR Checklisten zur Orientierung“ >> Am Stand in der Halle
Seite 2

Attraktiver Medizinproduktemarkt
• Die Medizintechnik ist die Branche mit aktuell den meisten
Patentanmeldungen aus der Forschung
>> Patente brauchen 2-5 Jahre zum Produkt!
• Die Gesundheitswirtschaft gilt als Wachstumsmotor mit guten
Karrieremöglichkeiten in Pflege, Wellness und Industrie:
– über 4 Mil. Menschen arbeiten heute in diesem Bereich,
– bis zu 700 000 neue Arbeitsplätze sollen dazukommen.
• Die richtige Technologie und Qualifikation und deren Erhalt sind
entscheidende Faktoren für den unternehmerischen Erfolg

Definition „Hersteller“ (NB-MED 2.5.5/Rec 5)
• Jedes Unternehmen bzw. jede Person ist bereits dann legaler und haftbarer
„Hersteller“ wenn er die Verantwortlichkeit übernimmt ein Produkt als
Medizinprodukt unter seinem Namen in Verkehr zu bringen.
• Die gesetzliche Verantwortung für die Übereinstimmung des Produktes mit
der anwendbaren EU-Regularien ist unabhängig davon, ob der Hersteller
das Produkt selbst entwickelt und herstellt oder ob relevante Tätigkeiten in
seinem Auftrag bei Dritten (Lieferanten) stattfinden.
• Änderung der Zweckbestimmung = Risiken kreieren = Herstellen
3DDD Druck GmbH

Definition „Medizinprodukt“
Zu den Medizinprodukten gehören u.a.:
• medizinisch-technische Geräte (Beatmung, Röntgengeräte)
• Implantate, (Herzschrittmacher, Gelenke, Gefäße)
• Produkte zur Injektion, Dialyse (Katheter, Spritzen, Blutbeutel …),
• diagnostische Instrumente (Blutdruck, Temperatur, Sensorik)
• Orthopädie- und Dentalprodukte (worunter Sonderanfertigungen),
• Verbandstoffe, Heftpflaster, Sehhilfen,
• Software zur Therapie, Diagnostik, Steuerung

Pro
du
kth
aft
un
g g
ilt
Fü
r a
lle H
ers
telle
r !
Sicherheit < &/ > Qualität
• Produktsicherheit wird bei
• Lebensmitteln,
• Arzneimitteln und
• Medizinprodukten
von Regulierungsbehörden vorausgesetzt.
• Produktqualität kann unterschiedlich sein, Schäden
und Risiken dürfen aber nicht entstehen.
• Der Hersteller muss beweisen (Beweislastumkehr),
dass das Produkt/Leistung fehlerfrei war.

3D Druck in der Medizintechnik
• Bionische Assistenzsysteme und Exoprothesen
• Bohr- und Frässchablonen zur Positionierung
• Zahnimplantate, Zahnersatz
• Epithesen
• Prothesen
• Orthesen
• Knochen, Gefäße oder Gewebe (Organisches Material)
• Gelenksimplantate (Metall/Kunststoff/Keramik)
• Pumpen, Antriebe, Gehäuse, Halterungen, Verbindungen
• Chirurgische Instrumente
• Wer Materialien im Auftrag eines Arztes verarbeitet im
Rahmen einer einmaligen Sonderanfertigung oder Teile für ein
Medizinprodukt in Serie produziert ist Hersteller im Sinne des
Medizinproduktgesetzes.
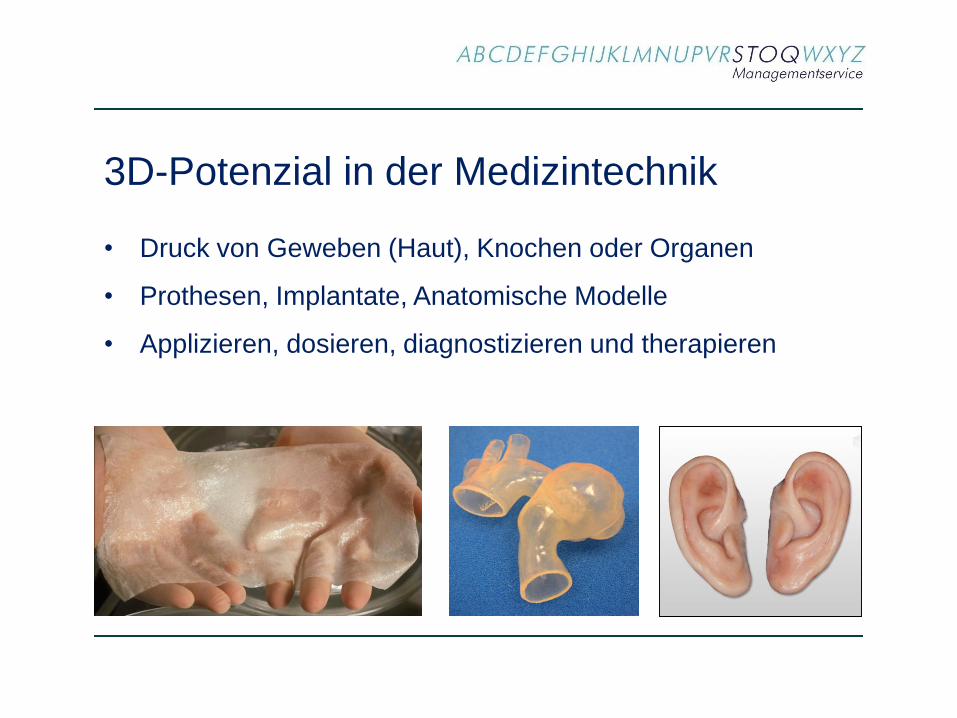
3D-Potenzial in der Medizintechnik
• Druck von Geweben (Haut), Knochen oder Organen
• Prothesen, Implantate, Anatomische Modelle
• Applizieren, dosieren, diagnostizieren und therapieren
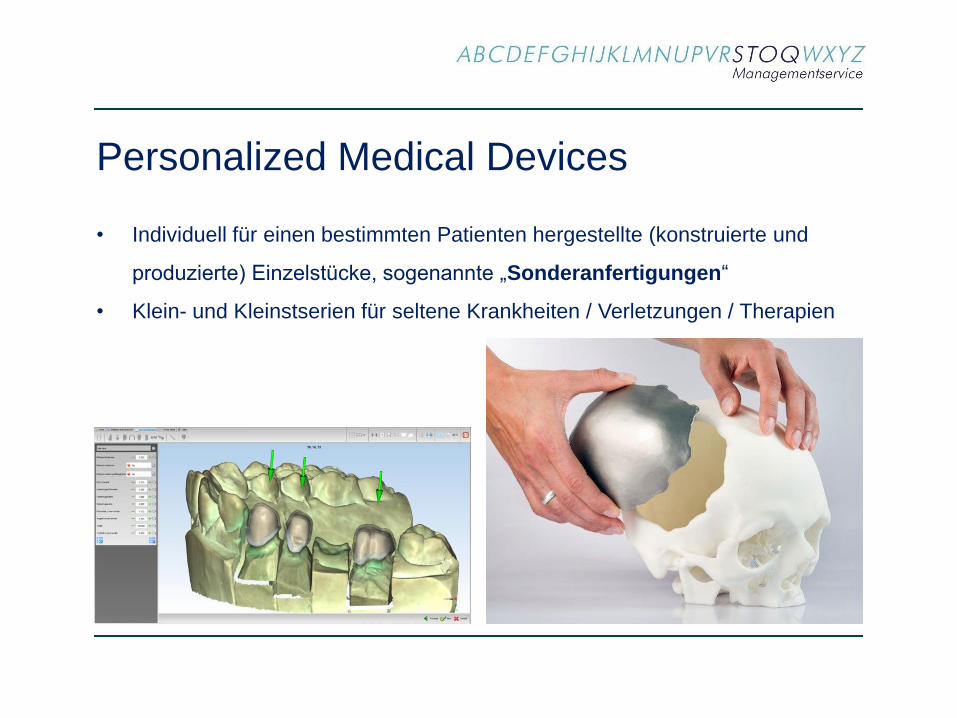
Personalized Medical Devices
• Individuell für einen bestimmten Patienten hergestellte (konstruierte und
produzierte) Einzelstücke, sogenannte „Sonderanfertigungen“
• Klein- und Kleinstserien für seltene Krankheiten / Verletzungen / Therapien

Bei Sonderanfertigungen:
Sonderanfertigungen sind Medizinprodukte, welche;
• nach schriftlicher Verordnung und spezifischen
Vorgaben vom Chirurg/in oder Arzt/Ärztin,
• meist von einem Gesundheitshandwerker für einen
namentlich benannten Patienten angefertigt werden.
• Zulieferer (CAD-CAM) von Gesundheitshandwerker
müssen in der Nachweiskette eingebunden werden!
• Serienmäßige Medizinprodukte ohne Patientenbezug
sind keine Sonderanfertigungen, tragen dann aber das
CE-Zeichen für die jeweilige Medizinprodukteklasse!

Typ
isch
er
Pro
ze
ss in
de
r M
ed
izin
tech
nik
3D-Profil (Scan/MRT/Röntgen) des Patienten
Daten Transferieren und Matching in CAD
Material für Anwendung definieren
Lösung in CAD konstruieren
Transferieren in CAM-/STL-File
Übertragung an 3D-Print oder CAM-Anlage
Herstellung Prothese
Abtrennen von Halterungen/Stegen
Sintern/Verdichten/Tempern
Manuelle/Maschinelle Nachbearbeitung
Vorreinigung
(Sterilisation)
Verpackung
Anwendung am Patienten

Fragen an Materialien in der 3D Druck
• Bei Hautkontakt ist die Biokompatibilität der verwendeten Werkstoffe nach EN
ISO 10993-Reihe als auch die Entfernung von Betriebshilfsstoffen (z.B. Kühl-
oder Trennmittel, Bisphenol A- und Phthalate-Freiheit) nachzuweisen.
• Bei mechanisch belastbaren Teilen ist die Statik unter Worst-Case Betrachtung
zu prüfen und belegen. Normen, worunter die EN ISO 62366 dienen als Basis.
• Bei implantierbaren Werkstoffen ist die Sinterdichte sowie Stabilität und
Oberflächenqualität (z.B. Abrieb bei Gelenkprothesen vermeiden) nachzuweisen.
• Bei Produkten zum Einsatz an mehreren Patienten ist die Wiederverwendbarkeit
und die Reinigung/Sterilisation nachzuweisen nach EN ISO 17664

Medizinprodukteklassen* nach EU-Kriterien
• Klasse I: Produkte mit einem CE-Zeichen ohne Kenn-Nummer einer
Benannten Stelle
• Klasse I Steril, Klasse I mit Messfunktion, Klasse I wiederverwendbare
chirurgische Instrumente (ab 26. Mai 2020) mit CE-Zeichen und Kenn-
Nummer einer Benannten Stelle
• Klassen IIa, IIb und III mit CE-Zeichen und Kenn-Nummer einer
Benannten Stelle
* Die Klassifizierung erfolgt nach MDD 93/42/EWG Anhang IX oder ab 2020 nach EU-MDR 2017/745 Anhang VIII

Klassifizierung &
Kennzeichnung
Medizinprodukte Klasse I
Medizinprodukte Klasse Is, Im, Ir (neu), IIa, IIb und III
0483
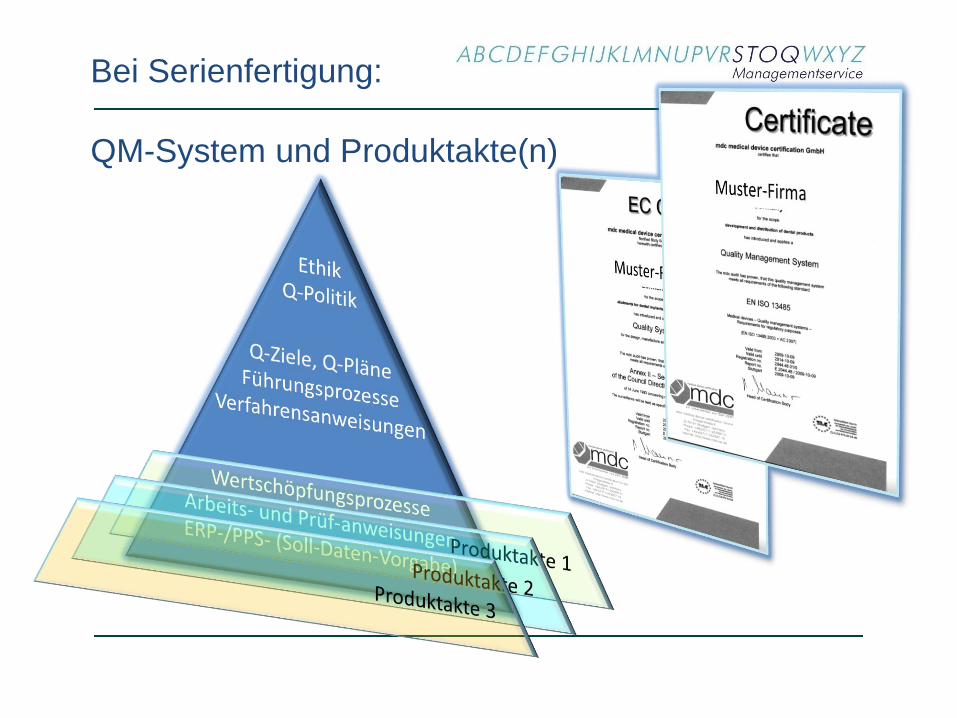
Bei Serienfertigung:
QM-System und Produktakte(n)

Medizinprodukte-Regularien-Fazit:
G.M.P. (Good Manufacturing Practice)
1. Die Technische Dokumentation mit allen verifizierten Angaben zur
Herstellung klinisch sicheren Produkten (DIN EN ISO 13485:2016
Kap. 7.3.8 und MDD 93/42/EWG Anhang II / V / VII)
2. Die Entwicklungsakte (DHF) mit allen Vorgaben und im Projekt
gesammelten Test-Protokolle, Erkenntnisse, Qualifizierungen und
Entscheidungen (DIN EN ISO 13485:2016 Kap. 4.2.3)
3. Die Beschreibung aller Prozesse und beteiligten (potenziell
kritischen) Lieferanten von Materialien und Leistungen in einem
zertifizierten QM-System nach EN ISO 13485:2016
4. Die Anzeige als Hersteller und Produkt-GMDN beim DIMDI
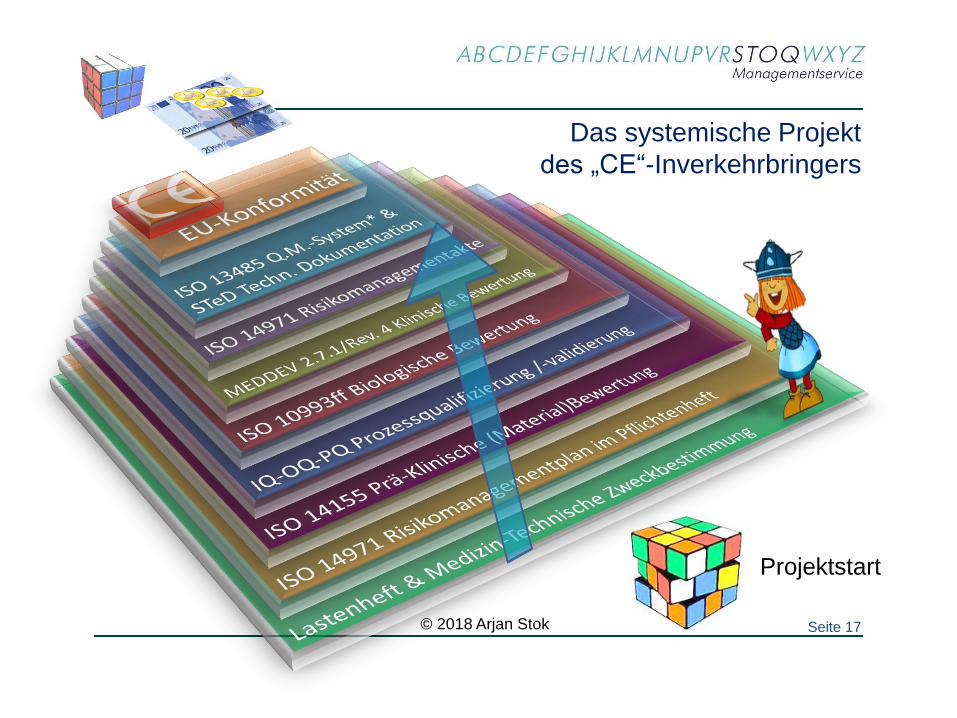
Das systemische Projekt
des „CE“-Inverkehrbringers
Projektstart
Seite 17© 2018 Arjan Stok

Kennen Sie für jede Vertragsbeziehung Ihre Rolle?
• Materialhersteller (von Vorprodukten für 3D-Drucker) mit CE-Zeichen
• Medizinproduktehersteller (Serien-Verarbeiter von 3D-Vorprodukten)
Besitzt eine T.D. und ein QM-System nach CE-Richtlinie / CE-Verordnung
Arbeitet mit einem validierten System für alle CAD-CAM-Komponenten
• Sonderanfertiger (Einzel-Verarbeiter von 3D-Vorprodukten)
Verwendet Materialien mit CE-Kennzeichen im Rahmen der Zweckbestimmung
• Kritischer Zulieferer
Bekommt detaillierte Angaben zur Herstellung von wesentlichen Teilen vom MP-Hersteller
und ist im Rahmen des Zertifizierungsverfahrens der Benannten Stelle zu auditieren
Seite 18

Guten Appetit und gutes Verdauen der Inhalte!
Arjan J. H. Stok
Zertifizierte Unternehmensberatung
und (In-House) Schulungen für
Medizinproduktehersteller und -zulieferer
www.stoq.de
Der Vortrag stellt keine Rechtsberatung dar. Gesetze, Richtlinien, Verordnungen und Norminhalte wurden nicht wortwörtlich übernommen.
Vollständigkeit und Richtigkeit wird nicht gewährleistet, hierzu Verweisen wir auf die Originaldokumente und behördliche Interpretationen.
Seite 19