auswertung der rheologischen und optischen untersuchungen ... · aus der literatur ist kein modell...
TRANSCRIPT

Auswertung der rheologischen und optischen Untersuchungen während der
Gelierung des Systems Gelatine / Wasser mit Hilfe der Perkolationstheorie
Dem Fachbereich 6 (Chemie-Geographie)
der
Gerhard-Mercator-Universität-Gesamthochschule Duisburg
zur Erlangung des akademischen Grades eines
Doktors der Naturwissenschaften (Dr. rer. nat.)
eingereichte Dissertation
von
Markus Lechtenfeld
aus
Duisburg

Die vorliegende Arbeit wurde im Fachgebiet
Angewandte Physikalische Chemie
der Gerhard-Mercator-Universität-GH Duisburg angefertigt.
Berichterstatter: Prof. Dr. Werner Borchard
Prof. Dr. Wiebren S. Veeman
Eingereicht am: 13.03.2001
Tag der mündlichen Prüfung: 13.06.2001

Ich danke Herrn Prof. Dr. W. BORCHARD für die interessante Aufgabenstellung,
seine fachliche Betreuung, Hilfestellung, die gewährte Freiheit bei der
Durchführung dieser Arbeit sowie seine ständige und unermüdliche Bereitschaft
zur fruchtbaren Diskussion.
Herrn Prof. Dr. W. S. VEEMAN danke ich für die freundliche Übernahme des
Korreferats.
Mein Dank gilt der DEUTSCHEN FORSCHUNGSGEMEINSCHAFT (DFG) für die
finanzielle Unterstützung.
Ich danke allen Mitarbeitern des Fachgebietes Angewandte Physikalische
Chemie, die zum Gelingen dieser Arbeit beigetragen haben. In
wissenschaftlicher Hinsicht danke ich MICHAEL KISCHEL für seine ständige
Hilfsbereitschaft in Hard- und Softwarefragen, die insbesondere bei der
Realisierung der neuen Steuereinrichtung des Rheometers auftraten. In diesem
Zusammenhang danke ich ebenfalls VOLKER FISCHER für seine unerlässlichen
Hilfestellungen beim Umgang mit dem Atari Computer sowie VOLKER
KÖRSTGENS für die zeitsparende Hilfe bei der Programmierung der neuen
Software. In verwaltungstechnischer Hinsicht danke ich Frau CHRISTEL VON DER
WARTH, die nicht selten unter Aufwendung von Mehrarbeit alle
personaltechnischen Fragen stets zuverlässig koordiniert hat.
Ich danke der Urbesetzung der Kaffeerunde, Dr. DIRK KISTERS und PASCAL
JABLONSKI für die wissenschaftlichen Diskussionen, Unterstützung und prompte
Hilfsbereitschaft bei auftretenden Problemen. Herrn JABLONSKI danke ich
weiterhin für die Übersicht des Manuskriptes und die Hilfestellungen bei der
Gestaltung dieser Arbeit.
Ganz besonders möchte ich mich bei zwei Lehrern bedanken, die mir aus
meiner Schul- bzw. Lehrzeit in bester Erinnerung geblieben sind. Frau BREXEL
danke ich für ihre Toleranz und Geduld, die ich während meiner Realschulzeit
sehr strapaziert habe. Herrn HAIER, der die Fähigkeit besitzt, Theorie und Praxis
der Chemie so zu vermitteln, dass daraus eine lebendige Wissenschaft wird,
die man sich nach seiner Ausbildung gerne zum Beruf macht, sei ebenfalls
gedankt.

If you are right – you are happy,if you are wrong – you learn something.
Sir Harold Kroto
(Winner of the 1996 Nobel Prize in Chemistry)
Justina,
Luca, dem Kikimann
und meiner lieben Mutter gewidmet.

INHALT
INHALT
EINLEITUNG ................................................................................... 1
THEORETISCHER TEIL ........................................................... 3
1 Dreidimensionale Netzwerke .............................................................. 3
1.1 Was ist ein Gel? ............................................................................ 3
1.2 Die Klassifizierung der Gele .......................................................... 4
1.3 Das System Gelatine / Wasser...................................................... 6
1.3.1 Struktur, Aufbau und Herstellung von Gelatine................. 7
1.3.2 Physikalische Eigenschaften ............................................ 9
2 Grundlagen der Rheologie................................................................ 11
2.1 Viskoses Verhalten von Flüssigkeiten ......................................... 11
2.2 Elastisches Verhalten von Festkörpern ....................................... 14
2.3 Viskoelastisches Verhalten von Polymerschmelzen.................... 16
2.4 Der Kriech- und Spannungsrelaxationsversuch .......................... 17
2.5 Das BOLTZMANNsche Superpositionsprinzip ................................ 18
2.6 Das dynamische Experiment....................................................... 20
3 Polarimetrie ........................................................................................ 23
4 Gelierung aus der Sicht der Perkolationstheorie............................ 27
4.1 Was ist Perkolation?.................................................................... 27
4.2 Perkolation und Sol-Gel-Umwandlung ........................................ 29
4.3 Kritische Exponenten .................................................................. 32
EXPERIMENTELLER TEIL...................................................... 35
5 Probenmaterial und Vorbereitung.................................................... 35
6 Versuchsaufbau zur simultanen Bestimmung der optischen
und rheologischen Kenngrößen....................................................... 36
6.1 Das dynamische Schwingungsviskosimeter................................ 37
6.1.1 Messsystematik .............................................................. 39

INHALT
6.1.2 Mathematische Beschreibung des dynamischen
Experiments.................................................................... 42
6.2 Das Polarimeter........................................................................... 47
6.2.1 Messsystematik .............................................................. 49
6.2.2 Berechnung der Messgrößen ......................................... 50
ERGEBNISSE UND DISKUSSION ............................................ 51
7 Übersicht der durchgeführten Messungen...................................... 51
8 Die Bestimmung des Gelpunkts und der kritischen Exponenten . 52
8.1 Frequenzabhängigkeit des komplexen Schubmoduls ................. 52
8.2 Auswertung nach der Perkolationstheorie................................... 54
8.2.1 Die normierten Perkolationsansätze............................... 56
8.2.2 Der modellierte Verlauf der Gelierkurven........................ 61
8.2.3 Die kombinierten Perkolationsansätze............................ 63
8.3 Diskussion der kritischen Exponenten......................................... 68
8.3.1 Der kritische Exponent ν................................................. 68
8.3.2 Der kritische Exponent µ................................................. 69
8.3.3 Das Skalenverhalten der kritischen Exponenten ............ 74
8.4 Zusammenhang zwischen dem komplexen Schubmodul
und der optischen Drehung ......................................................... 77
9 Die mathematische Beschreibung der G'(t) Funktion
- Das Aggregationsmodell - .............................................................. 83
ZUSAMMENFASSUNG........................................................... 89
ANHANG ............................................................................. 91
A-1 Ergebnisse der Auswertung nach der Perkolationstheorie. ................. 91
A-2 Ableitung weiterer CF. ......................................................................... 94
A-3 Messkurven der untersuchten Systeme............................................... 97
LITERATURVERZEICHNIS.................................................... 108

EINLEITUNG 1
EINLEITUNG
Gelatine ist ein Polypeptid und wird vornehmlich aus dem im Stützgewebe
(Haut, Knochen) von Rindern und Schweinen enthaltenen Kollagen gewonnen.
Sie findet mannigfaltigen Einsatz in der Kosmetik-, Photo-, Pharma- und
Lebensmittelchemie. Man bedient sich hierbei der besonderen Eigenschaft der
Gelatine, in wässerigem Milieu thermoreversible Gele auszubilden. Hierunter
versteht man, dass alleine durch die Veränderung der Temperatur der Über-
gang vom flüssigen Zustand (Sol) in den festen Zustand (Gel) beliebig oft wie-
derholt werden kann. Dieser Vorgang ist mit einer deutlichen Änderung der
rheologischen Eigenschaften verbunden, was mit Hilfe der Rheologie, die sich
mit den mechanischen Eigenschaften von Flüssigkeiten bzw. Festkörpern
beschäftigt, beobachtet werden kann. Grundvoraussetzung für die Gelbildung
im System Gelatine ist die Fähigkeit der Polypeptide, aufgrund von Wasserstoff-
brückenbindung Ein- oder Mehrfachhelices zu bilden, die letzten Endes die Bil-
dung der Netzwerkpunkte ausmachen. Der Charakter des Netzwerkes ist daher
physikalischer Natur, was die thermoreversiblen Eigenschaften des Systems
Gelatine / Wasser erklärt. Die Bildung der Helices lässt sich wiederum mit Hilfe
optischer Methoden verfolgen, da die Helices ähnlich der optisch aktiven Sub-
stanzen die Schwingungsebene des Lichtes drehen.
Der Vorgang der Sol-Gel-Umwandlung bzw. der Gelierung wird als Phasen-
umwandlung bezeichnet und stellt für Wissenschaftler, die sich mit derartigen
Problemen befassen, ein sogenanntes kritisches Phänomen dar. Für derartige
kritische Phänomene wurden Theorien entwickelt, die Aufschluss über das Ver-
halten der untersuchten Systeme in diesem kritischen Bereich geben und dabei
den Übergang - in diesem Fall die Umwandlung vom flüssigen in den festen
Zustand - genau bestimmen. Eine Theorie, die sich mit diesen kritischen Phä-
nomenen beschäftigt, ist die Perkolationstheorie. Sie sagt ein Potenzverhalten
für den zeitlichen Verlauf der rheologischen Kenngrößen in der Nähe der Sol-
Gel-Umwandlung voraus.
Ein Ziel der vorliegenden Arbeit ist es, mit Hilfe des in der Arbeitsgruppe Ange-
wandte Physikalische Chemie der Gerhard-Mercator-Universität um Prof. Dr.
W. BORCHARD konstruierten dynamischen Schwingungsviskosimeters den
Punkt der Sol-Gel-Umwandlung genau zu lokalisieren, sprich die Gelierzeit zu
bestimmen. Dies soll anhand der Auswertung der rheologischen Kenngrößen
nach der Perkolationstheorie geschehen. Vorab soll die Steuereinrichtung des
Schwingungsviskosimeters vom veralteten Atari Betrieb auf PC Betrieb umge-

EINLEITUNG 2
stellt werden, sodass die Messzeit zwischen zwei Messpunkten von jetzt 18 s
deutlich kleiner wird.
Die oben erwähnte Perkolationstheorie besitzt nur Gültigkeit in der Nähe des
Umwandlungspunktes. Das im späteren Verlauf der Arbeit als Perkolationsbe-
reich bezeichnete Gebiet kann bis heute nicht scharf eingegrenzt werden. Im
Zusammenhang mit der Auswertung nach der Perkolationstheorie soll hier der
Versuch unternommen werden, diesen Perkolationsbereich unter Berücksichti-
gung der in der Theorie gemachten Annahmen besser zu definieren.
Aus der Literatur ist kein Modell bekannt, das die Gelierung sowohl qualitativ als
auch quantitativ beschreibt. Basierend auf der Perkolationstheorie soll ein
Aggregationsmodell diskutiert werden, das die Kinetik in der Nähe des Um-
wandlungspunktes sowie in weiterer Entfernung dazu mathematisch und physi-
kalisch sinnvoll beschreibt.
Wie oben erwähnt lässt sich mit Hilfe rheologischer sowie optischer Kenngrö-
ßen die Sol-Gel-Umwandlung beobachten. Durch Konstruktion eines Ver-
suchsaufbaus, in dem das dynamische Schwingungsviskosimeter sowie ein
Polarimeter integriert sind, sollen diese Größen miteinander korreliert werden.
Es soll dabei die Frage geklärt werden, ob eine Proportionalität zwischen den
optischen und rheologischen Kenngrößen in der Nähe des Gelpunkts existiert.
Des Weiteren soll mit Hilfe der simultanen Messungen ein vor vielen Jahren
gefundenes Potenzverhalten zwischen den oben erwähnten Kenngrößen über-
prüft werden.

THEORETISCHER TEIL 3
THEORETISCHER TEIL
1 Dreidimensionale Netzwerke.
1.1 Was ist ein Gel?
"The colloidal state, the gel, is one which is easier to recognize than to define".1
Dieser Kommentar von JORDAN LLOYD aus dem Jahre 1926 trifft 75 Jahre später
die Sache deutlicher denn je. Die wissenschaftlichen Arbeiten auf dem Gebiet
der Gele halten weiterhin an und somit auch die Diskussion über die Definition
des Zustandes eines Gels.2 Dass die Wissenschaftler mit der Definitionsfrage
stellenweise sehr humorvoll umgehen, zeigt die Äußerung von KLAAS TE
NIJENHUIS in seinem Buch Thermoreversible Networks.3
"A gel is a gel, as long as one cannot prove that it is not a gel".
Eine allgemein anerkannte Definition stammt aus dem Jahre 1949 von P.H.
HERMANS: 4
• Gele sind kohärente, kolloide, disperse Systeme, die sich aus mindestens
zwei Komponenten zusammensetzen,
• sie verfügen über die für Festkörper charakteristischen mechanischen
Eigenschaften,
• sowohl die dispergierte Komponente als auch das Dispersionsmedium
erstrecken sich kontinuierlich über das gesamte System.
Etwas anschaulicher lässt sich der Zustand des Gels wie folgt beschreiben: Bil-
det ein makromolekularer Stoff bei der Synthese in einem Lösemittel unter der
Aufnahme desselben ein dreidimensionales Netzwerk aus oder quillt ein bereits
vorhandenes Netzwerk durch Aufnahme des Lösemittels, so spricht man von
einem Gel. Ein Gel ist dadurch charakterisiert, dass das Netzwerk seine äußere
Gestalt beibehält und elastische Eigenschaften besitzt. Im engeren physikali-
schen Sinne besitzt das Netzwerk jetzt viskoelastische Eigenschaften
(s. Kap. 2).5 Der Vorgang einer kontinuierlichen Phasenumwandlung einer
1 Lloyd JD (1926) In: Alexander J (ed) Colloid chemistry I. Chemical Catalog Company, NewYork, p. 7672 Almdal K, Dyre J, Hvidt S, Kramer O (1993) Polymer Gels Networks 1:53 te Nijenhuis K (1997) Thermoreversible Networks, Springer, Berlin Heidelberg New York, p. 34 Hermans PH (1949) In: Kruyt HR (ed) Colloid Science II, Elsevier, Amsterdam, p. 4835 Rehage G (1977) Berichte der Bunsengesellschaft Bd. 81, Nr.10

THEORETISCHER TEIL 4
polymeren Lösung (Sol) zu einem viskoelastischen Festkörper (Gel) wird als
Prozess der "Gelierung" bezeichnet.
Die kürzeste Definition eines Gels stammt von BORCHARD. Er definiert ein Gel
als eine aus dreidimensionalen, polymeren Netzwerken bestehende flüssige
Mischphase, die elastische Eigenschaften besitzt.6
1.2 Die Klassifizierung der Gele.
Anhand ihrer strukturellen Eigenschaften teilt FLORY die Gele in vier Gruppen
ein:7
1. Geordnete lamellare Strukturen, die Gelmesophasen enthalten.
2. Kovalente polymere Netzwerke, die vollständig ungeordnet vorliegen.
3. Durch physikalische Aggregation gebildete physikalische Netzwerke, die
überwiegend ungeordnete und teilweise geordnete Bereiche aufweisen.
4. Kleinste Teilchen mit ungeordneten Strukturen.
Beispiele für die in Gruppe 1 aufgeführten Gele sind Seifengele sowie wässrige
Dispersionen verschiedener Tonmaterialien (Kaolinit). Dabei können die Kräfte
zwischen den Lamellen polarer oder elektrostatischer Natur sein.8
Unter kovalenten polymeren Netzwerken (Gruppe 2) versteht man Makromole-
küle, die nach einer chemischen Reaktion (wie z.B. Polykondensation, radikali-
sche Polymerisation, Polyaddition) hauptvalenzmäßig, d.h. chemisch miteinan-
der verknüpft sind (s. Abb. 1.1.). Für die Ausbildung eines dreidimensionalen
Netzwerkes bedeutet dies, dass eine Verknüpfungsstelle im Netzwerk minde-
stens trifunktionell sein muss, d.h. mindestens drei von ihr ausgehende kova-
lente Bindungen aufweisen muss. Chemisch vernetzte Polymersysteme werden
außerdem als permanente oder auch irreversible Gele bezeichnet, da sie nur
durch einen Bindungsbruch wieder in einzelne Polymerketten (bzw. Solzustand)
überführt werden können.9 Beispiele für diese Art von Gelen sind vulkanisierter
Kautschuk (Gummi) oder wassersensitive Polyurethannetzwerke.
6 Borchard W (1998) Ber Bunsenges Phys Chem 102:15807 Flory PJ (1974) Disc Faraday Soc 57:18 Borchard W (1983) In: Finch CA (ed) Thermoreversible Gelation, Cambridge ResidentalSchool of Chemistry, Plenum Press9 Rehage G (1977) Berichte der Bunsengesellschaft Bd. 81, Nr.10

THEORETISCHER TEIL 5
Physikalisch vernetzte Makromoleküle (Gruppe 3) werden in den Vernetzungs-
stellen durch schwächere Kräfte wie z.B. durch inter- und intramolekulare H-
Brückenbindungen oder durch mechanische Verschlaufungen der Polymer-
ketten (entanglements) zusammengehalten.
Abb. 1.1. Chemisch (hauptvalenzmäßig) vernetzte Makromoleküle mit einer Funktio-nalität (f) von f = 4 an den Verknüpfungstellen des Netzwerkes
Weiterhin können polymere Ketten über kristalline sowie glasige Bereiche mit-
einander verknüpft sein.10 (s. Abb. 1.2.). Bei den physikalisch vernetzten Gelen
ist es möglich, die Struktur des Netzwerkes (Gel) durch Änderung der Tempe-
ratur oder der Konzentration zu zerstören und wieder in den Zustand der Poly-
merlösung (Sol) zu überführen. Diese Gel-Sol-Umwandlung kann rückgängig
gemacht werden, wenn die ursprünglichen äußeren Bedingungen wieder her-
gestellt werden. Aus diesem Grund werden diese physikalisch vernetzten Gele
auch als reversible Gele bezeichnet 11 Vernetzungen durch mechanische Ver-
schlaufungen (entanglements) treten bei Molekülen mit großen molekularen
Massen, langen Ketten und ausreichender Kettenflexibilität auf. Diese Ver-
schlaufungen oder Verhakungen der Polymerketten werden durch hohe Kon-
zentrationen und niedrige Temperaturen begünstigt. Vernetzungen über glasige
Bereiche können bei aus zwei Komponenten bestehenden Blockcopolymeren
auftreten. Dabei liegt eine der beiden Komponenten bei der Versuchstempera-
tur glasig erstarrt, die andere im gummielastischen Zustand vor. Bei der Ver-
netzung über kristalline Bereiche liegt ein teilkristallines Polymer vor, dessen
amorphe Bereiche über kristalline Bereiche vernetzt sind. Die Ausbildung von
H-Brückenbindungen wird häufig bei aminofunktionellen Makromolekülen beob-
achtet. Es können sich hier Assoziate oder Mikrokristallite in den Netzwerkbe-
10 Rehage G (1977) Berichte der Bunsengesellschaft Bd. 81, Nr.1011 Borchard W (1994) In: Water Based Polymers, Gels and Mesophases, The Centre ofProfessional Advancement 5, Chicago

THEORETISCHER TEIL 6
reichen bilden.12 Diese Art der Vernetzung wird ebenfalls bei der Gelierung von
wässriger Gelatine-Lösung beobachtet. Hinsichtlich der Thematik der vorlie-
genden Arbeit soll der Gelatine und deren physikalischem Verhalten in Wasser
ein gesondertes Kapitel gewidmet werden (s. Kap. 1.3).
Abb. 1.2. Nebenvalenzmäßige Vernetzung über a) mechanische Verschlaufungen(entanglements), b) kristalline Bereiche und c) glasige Bereiche
Bei den in der Gruppe 4 definierten Gelen handelt es sich im Allgemeinen um
lockere Ausflockungen, die gewöhnlich aus Partikeln mit großer geometrischer
Anisotropie bestehen, wie z.B. Protein-Aggregate oder das natürliche Kolla-
gen.13
1.3 Das System Gelatine / Wasser.
Die wässrigen Lösungen der Gelatine sind die klassischen Vertreter der ther-
moreversiblen Gele. Sie gelten als die in der Vergangenheit am häufigsten
untersuchten reversibel gelierenden Systeme. In den Forschungsarbeiten des
20. Jahrhunderts erfreuen sie sich deshalb hoher Beliebtheit, da noch kein
zuverlässiges Modell gefunden wurde, das eine qualitative und quantitative
Beschreibung der Kinetik der Netzwerkbildung zulässt. Im Folgenden soll kurz
auf die Struktur, die Gewinnung bzw. Herstellung, die Anwendungsgebiete der
Gelatine sowie auf ihr physikalisches Verhalten im wässrigen Medium einge-
gangen werden.
12 Rehage G (1977) Berichte der Bunsengesellschaft Bd. 81, Nr.1013 Borchard W (1983) In: Finch CA (ed) Thermoreversible Gelation, Cambridge ResidentalSchool of Chemistry, Plenum Press
a b c

THEORETISCHER TEIL 7
1.3.1 Struktur, Aufbau und Herstellung von Gelatine.
Bei der Gelatine handelt es sich um ein hochmolekulares Polypeptidgemisch,
welches aus dem im Stützgewebe (Knochen, Haut) von Schweinen oder Rin-
dern enthaltenen Kollagen gewonnen wird. Bis heute sind 17 verschiedene
Kollagen-Typen identifiziert, die sich nur durch die Zusammensetzungen der
Aminosäuren unterscheiden.14
Abb. 1.3. Feinstruktur und molekularer Aufbau von Kollagen15
Der Aufbau des Kollagens ergibt sich aus einer gestaffelten Anordnung der
Kollagenmoleküle, dem sogenannten fibrillären Aufbau. Jedes dieser einzelnen
Kettenmoleküle, der Tropokollagene, wird durch die Ausbildung einer rechts-
gängigen Superhelix aufgebaut. Diese Superhelix besteht wiederum aus drei
Polypeptidketten, von denen sich jede aus ca. 1050 Aminosäuren zusammen-
setzt. Die Polypeptidketten unterscheiden sich ihrerseits durch die Anordnung
der Aminosäuren in den Ketten. Das Mittelstück der Peptidketten weist einen
regelmäßigen Aufbau der Aminosäuresequenzen auf, wobei die folgenden
Anordnungen häufig beobachtet werden:
14 Babel W (1996) Chemie in unserer Zeit 30:8615 Pezron I, Djabourov J (1990) Polym Sci B, Polym Phys 28:1823
- X - Y - GLY - X - Y
300 nm
300 nm35 nm
67 nm
Kollagen-Fibrille(Quartärstruktur)
stäbchenförmigesTropokollagen
rechtsgängige Superhelix(Tertiärstruktur)
Aminosäuresequenz(Primärstruktur)
linksgängige α-Helix(Sekundärstruktur)

THEORETISCHER TEIL 8
gly - pro - hyp
gly - pro - ala
gly - ala - hyp
(gly: Glycin; pro: Prolin; ala: Alanin; hyp: Hydroxyprolin).
Die Realisierung der Helicierung sowie die Bildung der Superhelix ist vermutlich
durch die kleine, räumlich günstige Aminosäure Glycin gegeben.16,17
Herstellung. Bei den Herstellungsverfahren zur Gewinnung der Gelatine aus
Kollagen unterscheidet man zwei Aufschlussverfahren:
Beim alkalischen Verfahren werden Knochen oder Rinderspalt als Rohstoff
verwendet, welcher in einem mehrwöchigen, als Äscherung bezeichneten Ver-
fahren, mit konzentrierter Calciumhydroxid-Lösung behandelt wird.
Durch die Verwendung von Schweineschwarten von Jungtieren, die einen
geringen Anteil an quervernetztem Kollagen enthalten, ist dieser langwierige
Prozess der Äscherung nicht erforderlich. Im sogenannten sauren Verfahren
genügt eine kurze Behandlungszeit von drei Tagen mit verdünnter Salzsäure,
um das Kollagen aufzuschließen.
Beiden Verfahren schließt sich derselbe Extrahierungsprozess an. Dieser
gestaltet sich derart, dass, nach einer ausreichenden Neutralisation, die Gela-
tine mit heißem Wasser in mehreren Schritten zwischen 40 und 90°C behandelt
wird. Die Extrakte unterscheiden sich anschließend in ihren chemischen und
physikalischen Eigenschaften. So weisen die ersten Extrakte höhere molare
Massen, eine engere Molmassenverteilung und eine höhere Gelfestigkeit auf.18
Anwendungsgebiete. Aufgrund der Fähigkeit der Gelatine, thermoreversible
Gele zu bilden, sowie als Schutzkolloid zu fungieren, findet sie in der Industrie
ein weitgefächertes Anwendungsgebiet. In der Lebensmittelchemie wird sie als
Gelierungsmittel, Emulgator, Film- oder Schaumbildner verwendet. In der Pho-
toindustrie zum Beschichten von Filmmaterial, in der Pharmaindustrie hingegen
in hydrolisierter Form als Blutplasmaersatz. In der Kosmetik verwendet man
16 Pezron I, Djabourov J (1990) Polym Sci B, Polym Phys 28:182317 Piez KA (1985) In: Encyclopedia of Polymer Science and Engeneering, Collagen, 2nd Ed. Vol.3, Wiley and Sons, London, p.69918 Internetseite der Firma DGF Stoess AG http://www.gelita.com

THEORETISCHER TEIL 9
Gelatine als Emulgator in Salben und Cremes. Technische Gelatine dient z.B.
zur Mikroverkapselung von Farbstoffen.19
1.3.2 Physikalische Eigenschaften.
Durch den chemischen Aufbau eines Gelatinemoleküls sind drei Prozesse
denkbar, die zur Netzwerkbildung führen können: Die Bildung von H-Brücken-
bindungen, die Ausbildung kristalliner Bereiche sowie die Verschlaufungen der
langen in der Solphase flexiblen Ketten (entanglements).
Betrachtet man nun eine wässrige Gelatine-Lösung bei einer Temperatur von
40°C, so liegen die Polymerketten als statistische Knäule vor. Senkt man die
Temperatur, nimmt die Beweglichkeit der Polymerketten ab und ermöglicht den
entlang der Ketten befindlichen Carbonylsauerstoffatomen mit den in nächster
Nachbarschaft liegenden Wasserstoffatomen der NH-Gruppen "intramoleku-
lare" H-Brückenbindungen auszubilden. Dies hat die Bildung einer Helix bzw.
eines helikalen Bereiches in der Kette zur Folge - man sagt die Polymerkette ist
jetzt partiell heliciert. Das ursprüngliche Gelatinemolekül weist jetzt mehrere
steife helikale Bereiche auf, ist aber weiterhin durch die nicht helicierten Ketten-
abschnitte weitestgehend flexibel. Diese helikalen Bereiche unterschiedlicher,
vorzugsweise benachbarter Polymerketten, sind nun in der Lage, über "inter-
molekulare" H-Brückenbindungen zu aggregieren. Bei genügend hoher Poly-
merkonzentration findet so eine räumliche Vernetzung der Gelatineketten statt,
deren Netzwerkpunkte die aggregierten Helices darstellen.
Es sei erwähnt, dass in der Literatur weitere Modelle zur Klärung des Gelier-
mechanismus diskutiert werden, wobei es unterschiedliche Auffassungen von
der Ausbildung bzw. Gestalt der Netzwerkpunkte gibt. Diese interessanten,
nach Studium der Arbeiten von BORCHARD und MAIBAUM20,21,22 jedoch als sehr
fraglich erscheinenden Modelle, finden weitverbreitet enormen Zuspruch und
sollen daher hier nicht unerwähnt bleiben.
Ein Modell geht davon aus, dass die Netzwerkpunkte durch die diffusionskon-
trollierte Aggregation bereits vorhandener Helices gebildet werden. Bei günsti-
19 Internetseite der Firma DGF Stoess AG http://www.gelita.com - siehe hierzu auch Venohr H(1999) Dissertation Duisburg.20 Borchard W, Lechtenfeld M (1999) Macromolecules eingereicht21 Borchard W (1998) Ber Bunsenges Phys Chem 102:158022 Maibaum R (1999) Dissertation Duisburg

THEORETISCHER TEIL 10
ger räumlicher Anordnung der Ketten wird hier die Bildung von Mehrfachhelices
nicht ausgeschlossen, die als solche ebenfalls an der Aggregation teilneh-
men.23,24
Ein zweites Modell schließt eine Aggregation von Einfachhelices vollständig
aus. Die Netzwerkpunkte werden hier durch die Verdrillung von anfänglich
gebildeten Einfachhelices zu Dreifachhelices gebildet.25,26
Ein weiteres Modell geht nicht von einer anfänglichen Einfachhelicierung aus,
sondern von einer direkten Bildung von Dreifachhelices. Dies kann entweder
durch die Verdrillung dreier nahezu parallel verlaufender Ketten erfolgen 27,28
oder über die Verdrillung zweier Ketten, indem die eine Kette einen 180°-Knick
aufweist (hairpine-model) und somit über einen zweiten Bereich in der Kette die
Bildung der Dreifachhelix ermöglicht.29,30
MAIBAUMs Kritik richtet sich vor allem gegen die Renaturierung des Kollagens,
d.h. die Entstehung von Tripelhelices, die in solch einer Form im Kollagen als
Superhelix vorliegen. Seiner Ansicht nach bedarf es einer zu komplexen Kon-
formationsänderung bei der Verdrillung der Einfachhelices, die darüber hinaus
noch simultan ablaufen müsste.31 Zur Verdrillung ist weiterhin erforderlich, dass
der Rest der jeweiligen Einzelketten frei beweglich sein muss und keinesfalls in
anderen Netzwerkpunkten fixiert vorliet. Ein Zustand, der mit voranschreitender
Reaktion immer unwahrscheinlicher wird.
MAIBAUMs Ansicht nach spricht der schlagartige Abbau des Netzwerks bei
Temperaturerhöhung für ein Modell, das hauptsächlich aus aggregierten Ein-
fachhelices aufgebaut ist. Die für die Entwirrung der Dreifachhelices erforder-
liche konformative Umlagerung dürfte dahingegen weit mehr Zeit in Anspruch
nehmen.
Unumstritten ist aber, dass die Knäuel-Helix-Umwandlung mit einem erheb-
lichen Anstieg des End-zu-End-Abstandes der vorher geknäuelten Kette ver-
23 v. Hippel PH, Harrington WF (1959) Biochem Biophys Acta 37:42724 Engel J (1962) Arch Biochem Biophysics 97:15025 te Nijenhuis K (1981) Colloid Polym Sci 259(5):52226 Bohidar HB, Jena SS (1993) J Chem Phys 8(11):897027 Djabourov M (1988) Contemp Phys 29(3):27328 Pezron I, Djabourov M (1990) J Polm Sci B, Polym Phys 28:182329 Benguigui L, Busnel JP, Durand D (1991) Polymer 32(14):268030 Ross-Murphy SB (1992) Polymer 33(12):262231 Maibaum R (1999) Dissertation Duisburg

THEORETISCHER TEIL 11
bunden ist.32 Diese Erkenntnis ermöglicht es erst, die Gelierung von O / W -
Emulsionen zu erklären, in denen die Konzentration der Gelatine in der konti-
nuierlichen Phase, in Bezug auf das gesamte System, weit unterhalb der erfor-
derlichen sogenannten kritischen Konzentration zur Netzwerkbildung liegt.33
Zur Untersuchung der Gelierung bieten sich ganz besonders optische und
rheologische Methoden an. Optische Untersuchungen erfolgen u.a. mit Hilfe der
Polarimetrie. Hier nutzt man den Effekt aus, dass die während der Gelierung
gebildeten Helices die Schwingungsebene linear polarisierten Lichtes drehen
(s. Kap. 3). Die Helicierung bzw. deren Aggregation zu immer größer werden-
den Clustern bewirkt einen Anstieg der Viskosität. Die Viskosität divergiert am
Gelpunkt (GP) - damit meint man den Übergangspunkt vom Sol- in den Gelzu-
stand - liegt die Polymerkonzentration oberhalb der kritischen Konzentration.
Signifikant ändert sich aber im Bereich dieser Phasenumwandlung die für ein
Gel charakteristische elastische Verformbarkeit. Diese Effekte lassen sich im
rheologischen Experiment beobachten.
2 Grundlagen der Rheologie.
Unter Rheologie versteht man die Wissenschaft von der Deformation und dem
Fließen der Körper. Fließvorgänge werden vornehmlich in Flüssigkeiten beob-
achtet, Deformationen hingegen in Festkörpern. Der idealisierte Zustand einer
Flüssigkeit und der eines Festkörpers gehen auf NEWTON und HOOKE zurück.
NEWTON bezeichnet eine Flüssigkeit, die ausschließlich über viskose Anteile
verfügt, als ideal-viskose Flüssigkeit, HOOKE bezeichnet einen Festkörper, der
ausschließlich über elastische Anteile verfügt, als einen ideal-elastischen Fest-
körper. In der Literatur spricht man von der sogenannten NEWTONschen Flüs-
sigkeit bzw. dem HOOKEschen Körper.34
2.1 Viskoses Verhalten von Flüssigkeiten.
Das Fließverhalten einer NEWTONschen Flüssigkeit lässt sich am Modell der
einfachen Laminarströmung (Scherströmung) erklären.
32 Hinsken H, Borchard W (1995) Colloid Polym Sci 273:91333 Lechtenfeld M, Borchard W (1999) Phys Chem Chem Phys 1:312934 Pahl MH (1983) Praktische Rheologie der Kunststoffschmelzen und Lösungen, VDI-Verlag
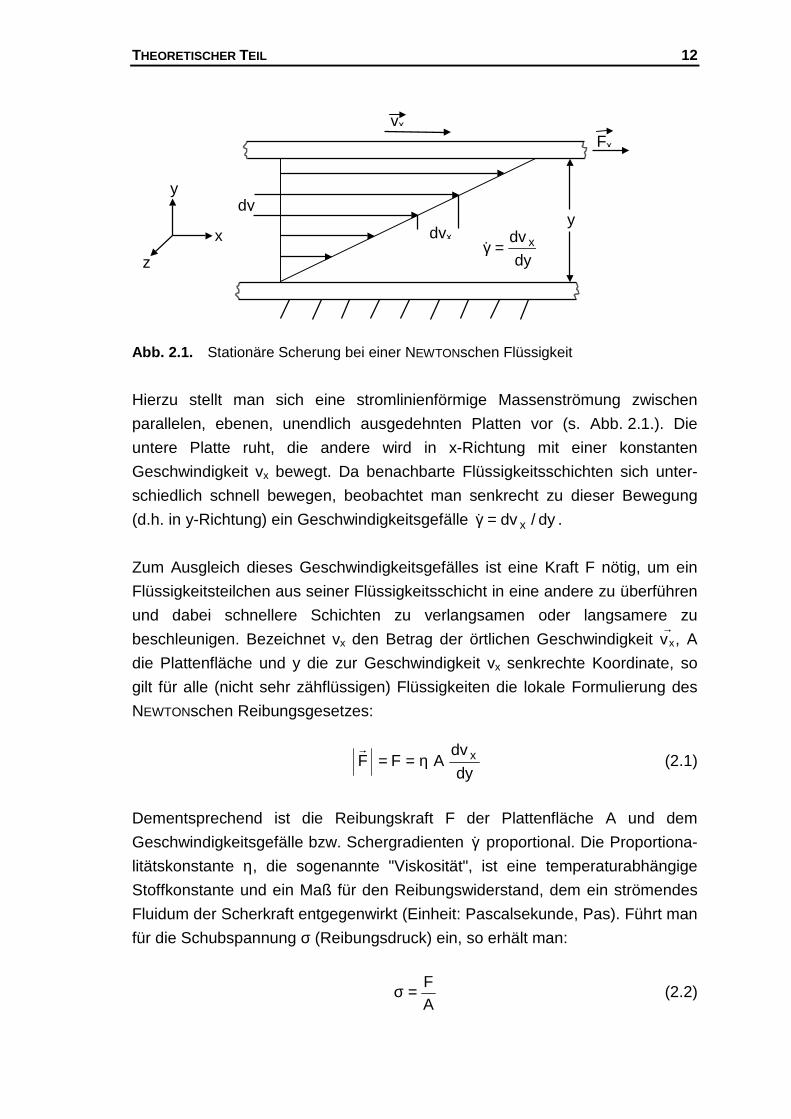
THEORETISCHER TEIL 12
Abb. 2.1. Stationäre Scherung bei einer NEWTONschen Flüssigkeit
Hierzu stellt man sich eine stromlinienförmige Massenströmung zwischen
parallelen, ebenen, unendlich ausgedehnten Platten vor (s. Abb. 2.1.). Die
untere Platte ruht, die andere wird in x-Richtung mit einer konstanten
Geschwindigkeit vx bewegt. Da benachbarte Flüssigkeitsschichten sich unter-
schiedlich schnell bewegen, beobachtet man senkrecht zu dieser Bewegung
(d.h. in y-Richtung) ein Geschwindigkeitsgefälle dy/dv x=γ� .
Zum Ausgleich dieses Geschwindigkeitsgefälles ist eine Kraft F nötig, um ein
Flüssigkeitsteilchen aus seiner Flüssigkeitsschicht in eine andere zu überführen
und dabei schnellere Schichten zu verlangsamen oder langsamere zu
beschleunigen. Bezeichnet vx den Betrag der örtlichen Geschwindigkeit vx→
, A
die Plattenfläche und y die zur Geschwindigkeit vx senkrechte Koordinate, so
gilt für alle (nicht sehr zähflüssigen) Flüssigkeiten die lokale Formulierung des
NEWTONschen Reibungsgesetzes:
dy
dvAFF xη==
&(2.1)
Dementsprechend ist die Reibungskraft F der Plattenfläche A und dem
Geschwindigkeitsgefälle bzw. Schergradienten γ� proportional. Die Proportiona-
litätskonstante η, die sogenannte "Viskosität", ist eine temperaturabhängige
Stoffkonstante und ein Maß für den Reibungswiderstand, dem ein strömendes
Fluidum der Scherkraft entgegenwirkt (Einheit: Pascalsekunde, Pas). Führt man
für die Schubspannung σ (Reibungsdruck) ein, so erhält man:
AF=σ (2.2)
dy
x
z
y
vx
dvx
Fx
dy
dv x=γ� y

THEORETISCHER TEIL 13
.γ⋅η=σ � (2.3)
Aus Gl. (2.3) geht hervor, dass η unabhängig vom Geschwindigkeitsgefälle γ�ist. Flüssigkeiten, die dieser Gesetzmäßigkeit gehorchen, bezeichnet man dem-
zufolge auch als NEWTONsche Flüssigkeiten.
Bei einer Vielzahl von Flüssigkeiten gibt es jedoch keinen linearen Zusammen-
hang zwischen der Schubspannung σ und dem Schergradienten γ� , sodass ηselbst bei einwandfrei laminarem Fließen eine Funktion von γ� ist. Dieses Ver-
halten findet man bei den sogenannten "nicht-NEWTONschen" Flüssigkeiten, wie
z.B. Polymerlösungen, deren Teilchen durch die Strömung orientiert, verformt
oder zerkleinert werden und so im Scherexperiment zu "scheinbaren Viskositä-
ten" und nicht zu Viskositäten im Sinne von NEWTONschen Flüssigkeiten führen.
Zu nicht-NEWTONschem Verhalten zählt man u.a. "Dilatanz" und "Strukturvisko-
sität", wobei im ersten Fall die Viskosität mit zunehmender Scherbeanspru-
chung zunimmt, im letzteren Fall abnimmt. Dilatantes Verhalten beobachtet
man in der Regel bei Schmelzen und Lösungen von Makromolekülen (z.B.
Stärke / Wasser), selten dagegen bei Dispersionen. Dieses erklärt man durch
die Immobilisierung des Lösemittels im System. Strukturviskoses Verhalten tritt
dagegen sowohl bei asymmetrischen, starren Teilchen als auch bei flexiblen
Knäueln in Erscheinung, wobei die Platzwechselvorgänge durch parallele Aus-
richtung der Polymerketten bzw. Deformation der Knäuels im Schergefälle mit
einem geringeren Energieaufwand verbunden sind als bei NEWTONschen Flüs-
sigkeiten. Ein besonderes strukturviskoses Verhalten zeigen die sogenannten
plastischen Flüssigkeiten (Bingham Körper). Bei ihnen stellt sich das Fließen
erst oberhalb einer bestimmten Schubspannung ein. Man spricht hier von einer
Fließgrenze. Beobachtet man außerdem noch eine zeitabhängige Änderung der
Viskosität, so spricht man im Fall einer Zunahme von "Rheopexie", im Fall einer
Abnahme von "Thixotropie". Beide Phänomene können reversibler oder irrever-
sibler Art sein. Anhand von sogenannten Fließkurven [ )(f γ=σ � ] sollen diese
unterschiedlichen rheologischen Verhalten deutlich gemacht werden.35
35 Pahl MH (1983) Praktische Rheologie der Kunststoffschmelzen und Lösungen, VDI-Verlag

THEORETISCHER TEIL 14
Abb. 2.2. Fließkurven (a) und (b) für verschiedene rheologische Verhalten: A =NEWTONsch, B = dilatant, C = strukturviskos, D = plastisch, E = thixotrop
2.2 Elastisches Verhalten von Festkörpern.
Wirken auf einen festen Körper äußere Kräfte, die im Gleichgewicht sind, so tritt
eine Änderung des Volumens und der Form ein. Gehen Volumen- und Formän-
derung nach Beendigung der äußeren Krafteinwirkung vollständig zurück, so
finden reversible Verformungsprozesse statt, der Körper ist ideal elastisch. Der
Zustand eines solchen Körpers unter Spannung und Deformation kann durch
die korrespondierenden Tensoren beschrieben werden. Die Komponenten des
Spannungs- und Deformationstensors beschreiben die an einem kubischen
Volumenelement angreifenden Kräfte sowie dessen Änderung bzw. äußeren
Abmessungen. Der Spannungszustand lässt sich durch die drei Normalspan-
nungen σxx, σyy, σzz und sechs Tangentialspannungen (auch Scher- oder
Schubspannungen genannt) σxy, σxz, σyx, σyz, σzx, σzy beschreiben (s. Abb. 2.3.).
Die ursprünglichen neun Komponenten des Spannungstenors reduzieren sich
auf sechs, schließt man eine Rotation des Körpers während der Beanspru-
chung aus, d.h σxz = σzx, σyz = σzy, und σxy = σyx.36,37,38
Im allgemeinsten Fall ist jede unabhängige Komponente des Deformations-
tensors eine Funktion aller 6 unabhängigen Komponenten des Spannungsten-
sors. Nimmt man eine lineare Abhängigkeit an, so erhält man ein Gleichungs-
system mit 36 Komponenten.
36 Tschoegel NW (1989) The phenomenological theory of linear viscoelastic behaviour,Springer, Berlin Heidelberg New York37 Borchard W (1994) In: Water Based Polymers, Rheology I: Dilute Polymer-Water Systems,The Centre of Professional Advancement 5, Chicago38 Ferry JD (1970) Viscoelastic Properties of Polymers, John Wiley & Sons Inc., New York
γ�γ�
σ
a
A
B
C
Dσ
b
E

THEORETISCHER TEIL 15
Abb. 2.3. Nomenklatur der Spannungen im würfelförmigen Körper
Diese Zahl reduziert sich deutlich, geht man von einem isotropen,
inkompressiblen Körper aus, der, wie oben erwähnt, bei Beanspruchung nicht
zu rotieren beginnt. Legt man weiterhin Volumenkonstanz des Körpers im
Experiment zu Grunde, so lässt sich das Deformationsverhalten aus der
einfachen Scherung ableiten (s. Abb. 2.4.).39
Abb. 2.4. Einfache Scherung am HOOKEschen Körper
Hierzu wird ein Würfel (in der Zeichnung stark vereinfacht) mit der Seitenfläche
A zu einem Parallelepiped gleicher Höhe und Breite durch eine in x-Richtung
angreifende Kraft Fx verzerrt, wonach die ursprünglich zur y-Richtung parallelen
39 Borchard W (1994) In: Water Based Polymers, Rheology I: Dilute Polymer-Water Systems,The Centre of Professional Advancement 5, Chicago
σyx
σyy
σyz
z
x
y
σxx
σxy
σxz
σzx
σzy
σzz
x
z
y
Fx
dx
ϕ
y

THEORETISCHER TEIL 16
Kanten einen Winkel ϕ mit der y-Richtung einschließen. Die Scherung wird
definiert als:
ϕ==γ tany
dxxy (2.4)
Die Schubspannung σxy ist dann proportional der Scherung γxy. Da man nur
Deformationen bezüglich der indizierten Koordinaten zulässt, kann für die wei-
teren Behandlungen auf diese Indizierung verzichtet werden. In Analogie zum
NEWTONschen Gesetz besteht für einen HOOKEschen Körper eine direkte Pro-
portionalität zwischen der Schubspannung σ und der Scherung γ.
γ⋅=σ G (2.5)
In Gl. (2.5) nennt man die Proportionalitätskonstante G den Schubmodul.
Unter einem HOOKEschen Körper versteht man einen Körper, bei dem bei Ein-
wirken einer Kraft die gesamte Deformation eintritt und nach der Entlastung
direkt auf ihren Ausgangswert wieder zurückgeht.
2.3 Viskoelastisches Verhalten von Polymerschmelzen.
Das viskoelastische Verhalten von Stoffen ergibt sich aus der Überlagerung von
viskosem und elastischem Verhalten und wird bei Polymerschmelzen und
Polymer – Lösemittel - Systemen beobachtet. Bei der Behandlung dieser
Systeme ist also das Zusammenspiel von elastischer Verformung, zeitabhängi-
ger elastischer Deformation und viskosem Fließen zu erwarten. Es kann nicht
mehr davon ausgegangen werden, dass die in Kap. 2.1 und Kap. 2.2 einge-
führten Größen G und η zeitunabhängige Materialkonstanten, sondern vor-
nehmlich frequenzabhängige Größen sind. Dass Spannung und Dehnung in
viskoelastischen Mischphasen frequenz- bzw. zeitabhängige Größen sind, zei-
gen die nun folgenden zwei klassischen Versuche.

THEORETISCHER TEIL 17
2.4 Der Kriech und Spannungsrelaxationsversuch.
Zur Beobachtung des zeitlichen
mechanischen Verhaltens eines
viskoelastischen Systems wird die
Probe im Kriechversuch in einem
bestimmten Zeitraum einer kon-
stanten Spannung σ0 ausgesetzt
(s. Abb. 2.5.oben).
Im anschließenden Erholungsver-
such (s. Abb. 2.5.unten) verfolgt
man den Deformationsverlauf γ(t)
während und nach der Beanspru-
chung.
AAbb. 2.5. Zeitprofil des Kriech (oben)- undErholungsversuches (unten)
Der Versuch ist für viskoelastische Systeme wie folgt zu deuten. Mit einsetzen-
der konstanter Spannung σo zum Zeitpunkt t = t0 tritt ein Teil der Deformation γ0
sofort auf, ein anderer bildet sich erst mit zunehmender Dauer der Beanspru-
chung aus. Im Erholungsversuch (t > t1) federt ein Teil der Deformation teil-
weise um den Betrag zurück, der durch die zu Beginn des Kriechexperiments
aufgebrachten Spannung σo hervorgerufen wurde. Ein weiterer Teil kann blei-
bend sein und ist auf das Fließen der Probe zurückzuführen. Für ideal elasti-
sche Körper setzt eine der aufgebrachten Spannung proportionale Deformation
spontan ein, welche im Erholungsversuch ebenfalls spontan zurückfedert. Für
ideal viskose Flüssigkeiten setzt eine zeitverzögerte Deformation ein, die im
Erholungsexperiment vollständig erhalten bleibt.40
40 Es muss erwähnt werden, dass es sich bei diesem Deformationsverlauf um eine Näherunghandelt, da die Träge der Masse nicht berücksichtigt wurde.
σ
tt0 t1
σ0
0
γ
t0 t0
viskoelastisch
t1
elastisch
viskos

THEORETISCHER TEIL 18
Beim Spannungsrelaxationsversuch wird dem
viskoelastischen System von einem Zeitpunkt
t0 an für eine unbestimmte Zeit eine definierte
Deformation γ0 aufgezwungen (s. Abb. 2.6.
oben).
Die für den Deformationsablauf erforderliche
Spannung wird als Funktion der Zeit σ(t)
gemessen (s. Abb. 2.6.unten).
Die zu beobachtende Abnahme der Spannung nach der Deformation ist auf
Fließ- und Platzwechselvorgänge in der viskoelastischen Probe zurückzufüh-
ren. Mit diesen Erkenntnissen lässt sich eine Beziehung zwischen dem in
Kap. 2.2 eingeführten Schubmodul G und der Zeit herstellen. Bildet man gemäß
Gl. (2.6) den Quotienten aus der Spannung σ(t) und der konstanten Dehnung
γ0, so erhält man den Spannugsrelaxationsmodul, G(t).41,42
o
)t()t(G
γσ= (2.6)
2.5 Das BOLTZMANNsche Superpositionsprinzip.
Möchte man das viskoelastische Verhalten eines Systems bei zeitlich variabler
Belastungsfolge beschreiben, dann genügen die oben erläuterten Kriech- und
Spannungsrelaxationsversuche nicht, da es sich hier um zwei statische Versu-
che unter Vorgabe einer "konstanten" Spannung bzw. Dehnung handelt. Eine
Lösung dieses Problems bietet das von BOLTZMANN im Jahre 1874 aufgestellte
stoffunabhängige Superpositionsprinzip. Es besagt: Die Wirkung einer Summe
41 Ferry JD (1970) Viscoelastic Properties of Polymers, John Wiley & Sons Inc., New York42 Schwarzl FR (1990) Polymermechanik, Springer, Berlin Heidelberg New York
γ(t)
tt0
γ0
σ(t)
tt0
γ0
γ0 G(t)
Abb. 2.6. Zeitprofil des Span-nungsrelaxationsversuches

THEORETISCHER TEIL 19
von Ursachen ist gleich der Summe der Wirkungen der einzelnen Ursachen.43
Man kann auch sagen: Wenn eine Spannung σ1(t) erforderlich ist, um die
Deformation γ1(t) hervorzurufen und σ2(t) nötig ist, um γ2(t) zu bewirken, so
muss für die Deformation γ1(t) + γ2(t) die Summe der Spannungen wirksam sein.
In symbolischer Schreibweise liest sich:44,45
( ) ( )∑∑ γ∝σ tt ii (2.7)
Mit Hilfe der mathematischen Formulierung des Superpositionsprinzips ist man
nun in der Lage, die Deformation zum Zeitpunkt t zu bestimmen, wenn die Vor-
geschichte des Spannungsablaufes bekannt ist. Aufgrund der dualen Fassung
des Superpositionsprinzips gilt gleiches auch für die Berechnung der Span-
nung, wenn die Vorgeschichte des Deformationsverlaufes bekannt ist.
Die Vorgeschichte einer Deformation berücksichtigt man, indem man alle
Deformationen im Zeitbereich von -∞ bis t betrachtet. Es gilt:
( ) tfür ≤ξ<∞−ξγ (2.8)
Durch eine Graphik veranschaulicht sucht man die Fläche unter der Deforma-
tions – Zeitkurve, die sich durch Aufsummieren der horizontalen Streifen ergibt.
Der Flächeninhalt eines Streifens ist gegeben durch die zu einem Zeitpunkt ξeinsetzende konstante Deformation ( ) ξ∆ξγ� (s. Abb. 2.7.).46
Wählt man die Stufenbreite ∆ξ infinitisemal klein (∆ξ→0), dann ergibt sich unter
Berücksichtigung der Gl. (2.6) für die Summierung über alle Beiträge:
( ) ( ) ( ) ξξγξ−=σ ∫∞−
dtGtt � (2.9)
Für den Fall der einfachen Scherung bezeichnet man Gl. (2.9) als die lineare
rheologische Zustandsgleichung eines isotropen, viskoelastischen Körpers.
43 Boltzmann L (1874) Sitzber KGl Akad Wiss Wien 2. Abt. 70:22544 Ferry JD (1970) Viscoelastic Properties of Polymers, John Wiley & Sons Inc., New York45 Tschoegel NW (1989) The phenomenological theory of linear viscoelastic behaviour,Springer, Berlin Heidelberg New York46 Schwarzl FR (1990) Polymermechanik, Springer, Berlin Heidelberg New York

THEORETISCHER TEIL 20
Abb. 2.7. Zerlegung des Deformationsverlaufes in horizontale Streifen47
2.6 Das dynamische Experiment.
Mit Hilfe des dynamischen Experiments lassen sich die viskosen und die elasti-
schen Anteile eines viskoelastischen Systems getrennt voneinander bestim-
men. Hierzu wird die Probe einer sinusförmigen oszillierenden Scherbeanspru-
chung unterworfen. Für die vorgegebene Deformation, bzw. für die Ableitung
nach der Zeit gilt folgender Ausdruck:
( ) ( )tsint 0 ωγ=γ (2.10)
( ) ( ).tcost 0 ωωγ=γ� (2.11)
Ersetzt man in Gl. (2.11) t durch ξ und bringt den daraus resultierenden Aus-
druck für ( )ξγ� in die lineare rheologische Zustandsgleichung Gl. (2.9) ein, dann
erhält man:
( ) ( ) ( ) ξωξξ−ωγ=σ ∫∞−
dcostGtt
0 . (2.12)
Zur Vereinfachung substituiert man (t - ξ) durch s und ändert die Integrations-
variablen auf das Zeitintervall [0 bis ∞]:48
47 Schwarzl FR (1990) Polymermechanik, Springer, Berlin Heidelberg New York48 Goodwin JW, Hughes RW (2000) Rheology for chemists, Royal Society of Chemistry
( ) ξ∆ξγ�
tξ
∆ξ = t - ξ
γ(ξ)
laufende Zeit
)(ξγ�

THEORETISCHER TEIL 21
( ) ( ) ( )[ ]dsstcossGt0
0 −ωωγ=σ ∫∞
(2.13)
mit Hilfe des Additionstheorems Gl. (2.14) ergibt sich:
cos(α−β) = cosα cosβ + sinα sinβ (2.14)
( ) ( ) ( ) ( ) ds)scos()s(GtcosdsssinsGtsin)t(0
00
0
ωωωγ+
ωωωγ=σ ∫∫
∞∞(2.15)
Der linke Teil der Gleichung schwingt im Experiment in Phase mit der aufge-
brachten sinusförmigen Anregung, er wird ausgedrückt durch den frequenzab-
hängigen Speichermodul G'. Der rechte Teil der Gleichung schwingt um den
Betrag der Differenz von Sinus zu Cosinus außer Phase, dies wird durch den
ebenfalls frequenzabhängigen Verlustmodul G'', ausgedrückt.
( ) s)dssin()s(G'G0
ωω=ω ∫∞
(2.16)
( ) ds)scos()s(G''G0
ωω=ω ∫∞
(2.17)
Entsprechend kann Gl. (2.15) wie folgt formuliert werden:
( ) ( ) )(''Gtcos)(G'tsin)t( 00 ωωγ+ωωγ=σ (2.18)
Der Spannungs- und Deformationsverlauf der Probe lässt sich im dynamischen
Experiment (s. Abb. 2.8.) veranschaulichen, wobei im Regelfall die aufge-
brachte Spannung der Deformation um den Winkel δ vorauseilt.
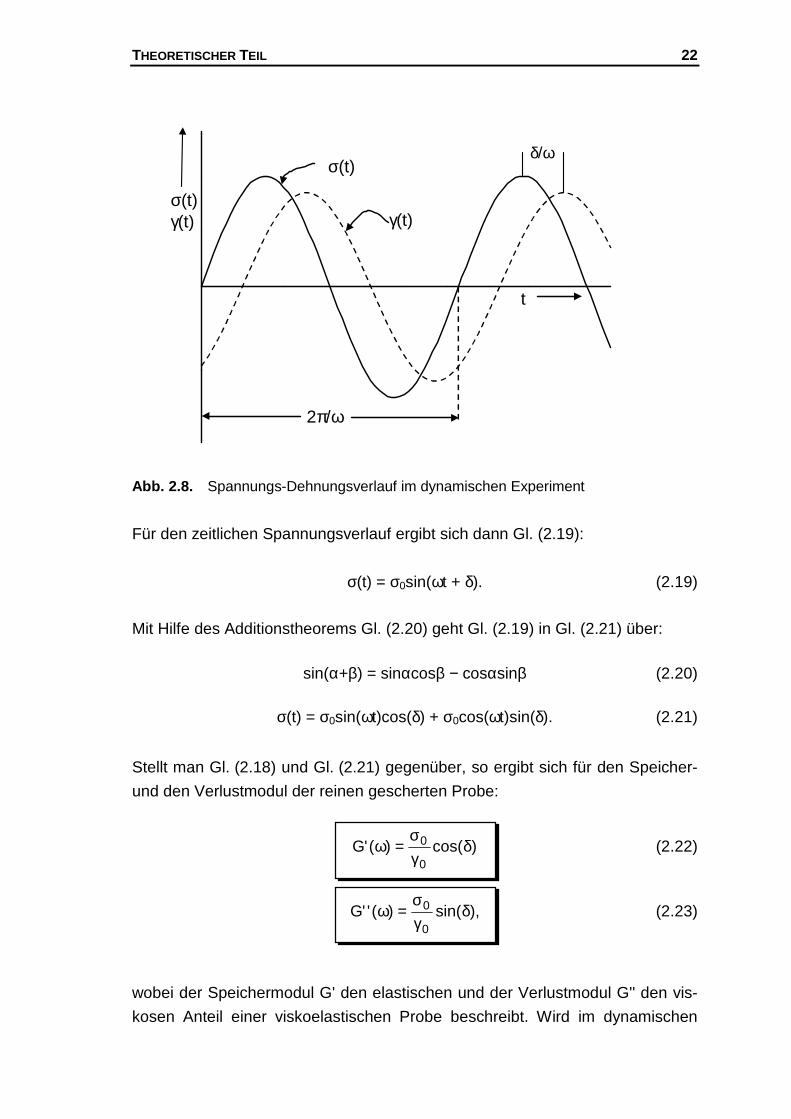
THEORETISCHER TEIL 22
Abb. 2.8. Spannungs-Dehnungsverlauf im dynamischen Experiment
Für den zeitlichen Spannungsverlauf ergibt sich dann Gl. (2.19):
σ(t) = σ0sin(ωt + δ). (2.19)
Mit Hilfe des Additionstheorems Gl. (2.20) geht Gl. (2.19) in Gl. (2.21) über:
sin(α+β) = sinαcosβ − cosαsinβ (2.20)
σ(t) = σ0sin(ωt)cos(δ) + σ0cos(ωt)sin(δ). (2.21)
Stellt man Gl. (2.18) und Gl. (2.21) gegenüber, so ergibt sich für den Speicher-
und den Verlustmodul der reinen gescherten Probe:
)cos()('G0
0 δγσ
=ω (2.22)
),sin()(''G0
0 δγσ
=ω (2.23)
wobei der Speichermodul G' den elastischen und der Verlustmodul G'' den vis-
kosen Anteil einer viskoelastischen Probe beschreibt. Wird im dynamischen
σ(t)γ(t)
σ(t)
γ(t)
2π/ω
δ/ω
t

THEORETISCHER TEIL 23
Experiment keine Phasenverschiebung beobachtet (d.h. δ = 0°), so ergibt sich
nach Gl. (2.23) für G'' der Wert Null, d.h. die Probe verfügt nur über elastische
Anteile und kann als ein ideal elastischer Körper angesehen werden. Beob-
achtet man eine Phasenverschiebung von δ = 90°, so ergibt sich nach Gl. (2.22)
für G' der Wert Null, d.h. die Probe verfügt ausschließlich über viskose Anteile
und ist demzufolge als ideal viskos anzusehen. An dieser Stelle sollte erwähnt
werden, dass die Gl. (2.22) und Gl. (2.23) nur dann als Auswertegleichung für
die Moduli herangezogen werden können, wenn es gelingt, den Einfluss der
Messapparatur gering zu halten.
Der Speicher- und der Verlustmodul lassen sich in einer komplexen Schreib-
weise darstellen:
( ) ( ) ( ).''iG'G*G ω+ω=ω (2.24)
Der Speichermodul stellt hier den Realteil und der Verlustmodul den Imaginä-
rteil des sogenannten komplexen Schubmoduls dar.49,50,51
Der Realteil in Gl. (2.24) repräsentiert die während einer Schwingung im Netz-
werk reversibel gespeicherte Arbeit, der Imaginärteil repräsentiert hingegen die
während einer Schwingung durch Reibung im Netzwerk dissipierte Arbeit.
3 Polarimetrie.
Optisch aktive Verbindungen drehen die Schwingungsebene von Licht. Je
höher die Konzentration der optisch aktiven Verbindung, desto größer der
Betrag, um den die Schwingungsebene gedreht wird. Eine gewöhnliche Licht-
quelle hat aber beliebig viele Schwingungsebenen, d.h., es kann unter diesen
Bedingungen keine Aussage über den Betrag, um den die Schwingungsebene
gedreht wurde, gemacht werden. Letzten Endes wird man auch so keine Aus-
sage darüber machen können, ob überhaupt eine Verbindung optisch aktiv ist.
Aus diesem Grunde verwendet man bei den polarimetrischen Untersuchungen
Licht mit "einer" definierten Schwingungsebene. Diese Bedingung erfüllt das
sogenannte linear polarisierte Licht.
49 Schwarzl FR (1990) Polymermechanik, Springer, Berlin Heidelberg New York50 Tschoegel NW (1989) The phenomenological theory of linear viscoelastic behaviour,Springer, Berlin Heidelberg New York51 Ferry JD (1970) Viscoelastic Properties of Polymers, John Wiley & Sons Inc., New York

THEORETISCHER TEIL 24
Linear polarisiertes Licht. Licht breitet sich als transversale elektromagne-
tische Welle aus. Das elektrische- (Ê) und magnetische Feld (V� stehen dabei
senkrecht zur Ausbreitungsrichtung und selbst in einem rechten Winkel zuein-
ander (s. Abb. 3.1.).52
Abb. 3.1. Elektromagnetische Welle
Bei gewöhnlichen Lichtquellen schwingen die Feldvektoren in alle Raumrich-
tungen mit verschiedenen Wellenlängen, das Licht ist unpolarisiert. Setzt sich
aber Licht aus Strahlen einer Wellenlänge zusammen, deren elektrisches Feld
in einer Ebenen liegt, dann spricht man von linear polarisiertem Licht.
Dieses Licht stellt man sich jetzt als die Überlagerung von zwei entgegenge-
setzt rotierenden zirkular polarisierten Lichtkomponenten vor (s. Abb. 3.2.).
Blickt ein Beobachter dem Lichtstrahl entgegen, und der elektrische Feldvektor
rotiert im Uhrzeigersinn, spricht man von rechts zirkular polarisiertem Licht,
rotiert der elektrische Feldvektor entgegengesetzt dem Uhrzeigersinn, spricht
man von links zirkular polarisiertem Licht. Man betrachtet demnach eine links-
drehende L-Komponente und eine rechtsdrehende R-Komponente. Tritt dieses
Licht nun in ein optisch aktives Medium ein, dann findet eine Wechselwirkung
der Elektronen der verschiedenen chemischen Spezies mit dem elektrischen
Feld einer der Komponenten statt.
Dies hat eine Polarisation der Materie, verbunden mit einer Abnahme der Fort-
pflanzungsgeschwindigkeit v und damit des Brechungsindex n53 der Kompo-
52 Fa. LOT Oriel Instruments (2000) The book of photon tools53 Der Brechungsindex (Brechungsquotient, Brechzahl) ist das Verhältnis der Lichtgeschwindig-keit im Vakuum zur Lichtgeschwindigkeit in dem betroffenen Medium.
ÇV
ElektrischesFeld
MagnetischesFeld
Strahlungsrichtung
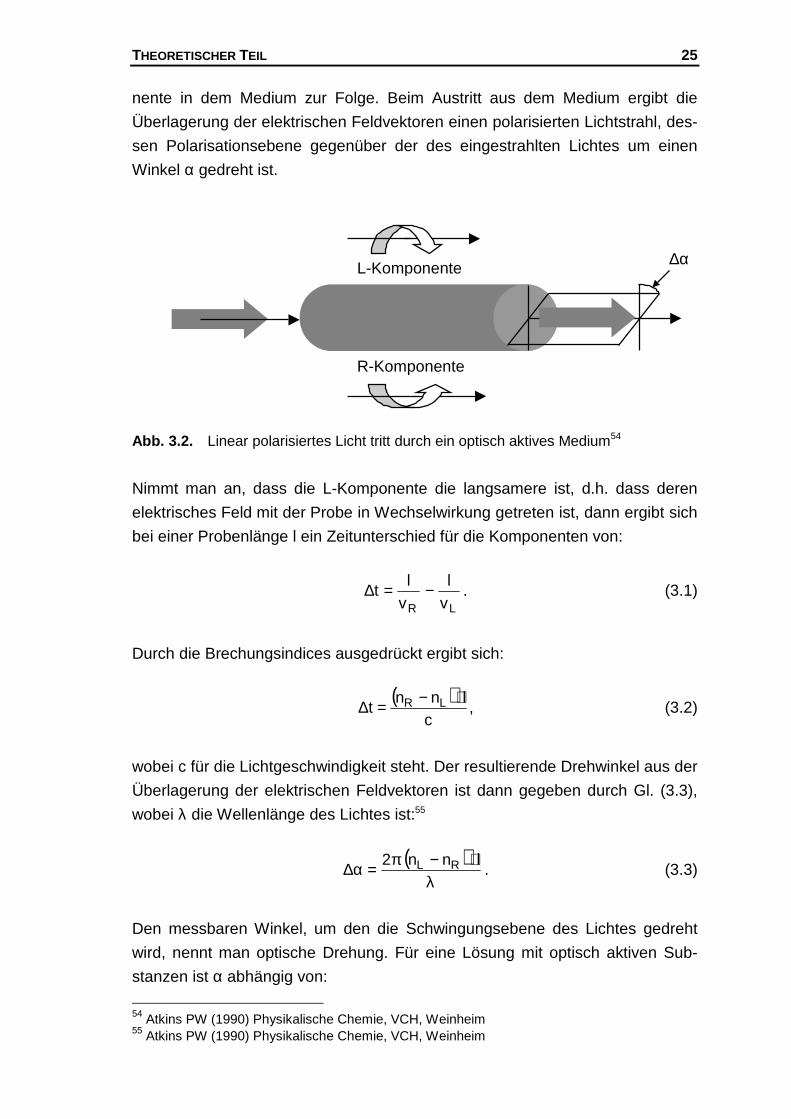
THEORETISCHER TEIL 25
nente in dem Medium zur Folge. Beim Austritt aus dem Medium ergibt die
Überlagerung der elektrischen Feldvektoren einen polarisierten Lichtstrahl, des-
sen Polarisationsebene gegenüber der des eingestrahlten Lichtes um einen
Winkel α gedreht ist.
Abb. 3.2. Linear polarisiertes Licht tritt durch ein optisch aktives Medium54
Nimmt man an, dass die L-Komponente die langsamere ist, d.h. dass deren
elektrisches Feld mit der Probe in Wechselwirkung getreten ist, dann ergibt sich
bei einer Probenlänge l ein Zeitunterschied für die Komponenten von:
LR vl
vl
t −=∆ . (3.1)
Durch die Brechungsindices ausgedrückt ergibt sich:
( )c
lnnt LR ⋅−=∆ , (3.2)
wobei c für die Lichtgeschwindigkeit steht. Der resultierende Drehwinkel aus der
Überlagerung der elektrischen Feldvektoren ist dann gegeben durch Gl. (3.3),
wobei λ die Wellenlänge des Lichtes ist:55
( )λ
⋅−π=α∆ lnn2 RL . (3.3)
Den messbaren Winkel, um den die Schwingungsebene des Lichtes gedreht
wird, nennt man optische Drehung. Für eine Lösung mit optisch aktiven Sub-
stanzen ist α abhängig von:
54 Atkins PW (1990) Physikalische Chemie, VCH, Weinheim55 Atkins PW (1990) Physikalische Chemie, VCH, Weinheim
∆αL-Komponente
R-Komponente

THEORETISCHER TEIL 26
• der Temperatur
• der Wellenlänge des Lichtes
• der Konzentration
• der Länge der Messstrecke
• der räumlichen Anordnung der Liganden, Unsymmetrien.
Häufig wird eine stoffspezifische Größe, eine sogenannte spezifische optische
Drehung [α] angegeben, in welcher die oben angegebenen Abhängigkeiten
berücksichtigt werden:
[ ]cl ⋅
α=α ϑλ . (3.4)
In Gl. (3.4) bedeuten:
α = optischer Drehwinkel
l = Länge der Messstrecke
c = Konzentration des gelösten Stoffes
ϑ = Temperatur.
Worin unterschieden sich optisch aktive von optisch inaktiven Substanzen
molekular? Optisch aktiv sind Moleküle, die ein asymmetrisches C-Atom haben,
d.h. ein Kohlenstoffatom mit vier unterschiedlichen Substituenten. Die Spiegel-
bilder von Molekülen, die solche asymmetrischen C-Atome enthalten, können
nicht mit dem Molekül zur Deckung gebracht werden. Man bezeichnet solche
Moleküle als chiral. Diese Moleküle haben kein Symmetriezentrum. Grob for-
muliert: Aufgrund der Symmetrie eines optisch inaktiven Moleküls wird jeder
Effekt, den der Teil des Moleküls bzw. der Elektronen auf den elektrischen
Feldvektor eines zirkular polarisierten Lichtes hat, durch den Effekt des spiegel-
bildlichen Teils des Moleküls auf den anderen Teil des zirkular polarisierten
Lichtes aufgehoben. Die Schraubenstruktur eines Moleküls (Helixstruktur s.
Kap. 1.3.2) trägt ebenfalls zur Drehung der Schwingungsebene von Licht bei.56
Man erwartet demnach bei den in dieser Arbeit untersuchten Gelatine / Wasser
Systemen, dass ein Teil des ermittelten Drehwinkels durch die in der Gelatine
enthaltenen asymmetrischen Kohlenstoffatome hervorgerufen wird, ein weiterer
Teil durch die Helicierung der Ketten während der Sol-Gel-Umwandlung.
56 Vollhardt KPC (1990) Organische Chemie, VCH Weinheim

THEORETISCHER TEIL 27
4 Gelierung aus der Sicht der Perkolationstheorie.
Eine Infektionskrankheit breitet sich in der Bevölkerung aus, Erdöl sickert durch
Gestein, ein Waldbrand breitet sich aus, ein Ei wird beim Kochen hart, Quitten-
gelee wird im Marmeladenglas fest. All diese Prozesse lassen sich mit den
Mitteln der Perkolationstheorie mathematisch beschreiben. Durch die Anwen-
dung der Perkolationstheorie auf derartige Beispiele kann z.B. folgende Frage
beantwortet werden. Wieviel Prozent der Bevölkerung muss geimpft sein, damit
keine Epidemie ausbricht? Weitere, wissenschaftlicher abgehandelte Beispiele
findet man in der Literatur.57,58
Mit Hilfe der Perkolationstheorie lassen sich auch Phasenübergänge beschrei-
ben, was für die in dieser Arbeit untersuchten Sol-Gel-Umwandlung während
der thermoreversiblen Gelierung des Systems Gelatine / Wasser von beson-
derer Bedeutung ist. Anhand dieses Prozesses soll die Perkolationstheorie
näher beschrieben werden.
4.1 Was ist Perkolation?
Hierzu stellt man sich eine große Anordnung von Quadraten vor, die zusammen
in idealer Weise ein unendlich großes Gitter darstellen sollen (s. Abb. 4.1.).
Einige Quadrate sind dabei mit einem Punkt versehen, andere Quadrate blei-
ben leer. Quadrate, die eine gemeinsame Seite haben bezeichnet man als
nächste Nachbarn. Sind solche Quadrate mit einem Punkt versehen, dann bil-
den sie gemeinsam einen sogenannten Cluster. Die Perkolationstheorie handelt
nun von der Anzahl und den Eigenschaften solcher Cluster.
Die Belegung der Quadrate mit Punkten findet dabei zufällig statt, so als ob die
Punkte untereinander nichts voneinander wüßten bzw. sich ignorierten. Man
nimmt an, dass die Quadrate mit der Wahrscheinlichkeit p mit einem Punkt
belegt sind, bzw. mit der Wahrscheinlichkeit (1-p) frei sind. Nimmt p einen kriti-
schen Wert pc an, beobachtet man einen Cluster, der sich von einer Seite des
Gitters zur anderen Seite erstreckt.
57 Sahimi M (1994) Applications of Percolation Theory, Taylor & Francis58 Stauffer D, Aharony A (1995) Perkolationstheorie, VCH Weinheim

THEORETISCHER TEIL 28
Abb. 4.1. Definition von Perkolation und seinen Clustern. a) Teile eines quadrati-schen Gitters, b) Besetzung einzelner Quadrate durch Punkte angedeutet, c) Zusam-menfassung nächstbenachbarter besetzter Quadrate zu Clustern was, durch Kreiseangedeutet ist59
Man spricht von einem Cluster, der durch das System perkoliert, wie Wasser
durch den mit Kaffee gefüllten Filter in einer Kaffeemaschine, die deshalb auch
"percolator" genannt wird (lat. percolare = durchsickern). Durchquert man den
Bereich knapp unterhalb bis knapp oberhalb dieser kritischen Konzentration,
dann erfahren die Systeme eine starke Änderung ihrer Eigenschaften.
Unterhalb von pc kann ein System nichtleitend sein, oberhalb dagegen leitend,
oder das System liegt unterhalb von pc als Sol vor, oberhalb hingegen als Gel.
Diese Änderungen während der Perkolation bezeichnet man als kritische
Phänomene und die Theorie, die versucht, diese Phänomene zu beschreiben
als Skalentheorie.60
Der Beginn der Perkolationstheorie wird mit einer Veröffentlichung von
BROADBENT und HAMMERSLEY aus dem Jahre 1957 in Verbindung gebracht. Hier
wurde der Name Perkolationstheorie eingeführt und das oben erläuterte geo-
metrische und wahrscheinlichkeitstheoretische Konzept mathematisch behan-
delt. Die damals aufkommenden Computer waren nach Angaben von
HAMMERSLEY ausschlaggebend für die Entwicklung dieser Theorie.61 Eine aus-
führliche und sehr anschauliche Erklärung, wie mit Hilfe der Monte - Carlo
Simulation die Perkolation auf einem Gitter abläuft, geben STAUFFER und
AHARONY in ihrem Buch Perkolationstheorie.62
59 Stauffer D, Aharony A (1995) Perkolationstheorie, VCH Weinheim60 Stauffer D, Aharony A (1995) Perkolationstheorie, VCH Weinheim61 Broadbent SR, Hammersley JM (1954) Proc Camp Phil Soc 53:62962 Stauffer D, Aharony A (1995) Perkolationstheorie, VCH Weinheim
a b c

THEORETISCHER TEIL 29
4.2 Perkolation und Sol–Gel-Umwandlung.
Bereits im Zweiten Weltkrieg entwickelten FLORY und STOCKMAYER die erste
Theorie der Gelierung für die Sol – Gel Phasenumwandlung.63,64 Grundlage
ihres Modells stellt das baumartige Wachstum f-funktioneller Monomerer auf
einem Bethe – Gitter (Cayley – Baum) dar. Diese Theorie bezeichnet man
heute als die Perkolationstheorie auf dem Bethe – Gitter.65 In der Literatur
spricht man in der Regel von der klassischen Theorie (mean field theory) sie
soll im weiteren Verlauf der Arbeit auch als solche bezeichnet werden. Die für
die Bildung eines vollständigen Netzwerkes erforderliche Konzentration an f-
funktionellen Gruppen pc ist gegeben durch:
1f1
pc −= . (4.1)
Hauptkritikpunkt an dieser Theorie ist, dass sie keine Ringschlüsse der Makro-
moleküle zulässt und den Einfluss des ausgeschlossenen Volumens
(excluded volume effect) nicht berücksichtigt.66,67,68 STEPTO behandelt diese
Probleme in seinen neueren Arbeiten ausführlich.69
Wie in Kap. 4.1 erwähnt behandelt die Perkolationstheorie die kritischen Phä-
nomene in der Nähe von pc, d.h. knapp unter- bzw. knapp oberhalb von pc. Im
Falle der Sol–Gel-Umwandlung liegt ein System im Bereich p < pc als Sol vor -
hier schließen sich multifunktionelle Monomere zu endlichen Clustern zusam-
men. Im Bereich p > pc liegt es als Gel vor - hier existiert ein unendlich großer
Cluster (unendliches Netzwerk), das eine Seite der Probe mit der anderen ver-
bindet. Diese Phasenumwandlung vollzieht sich am sogenannten Gelpunkt
(GP). Das besondere an diesem Punkt ist, dass bestimmte Messgrößen an die-
sem Punkt gegen Null gehen oder gegen unendlich.
63 Flory PJ (1941) J Amer Chem Soc 63:3083,3091,309664 Stockmayer WH (1944) J Chem Phys 11:45 ibid 12:12565 Letztlich perkoliert auch hier ein Cluster durch ein Gitter, wobei nur nächste Nachbarn eineBindung ausbilden können. Bei dieser Theorie ist jedoch die Richtung durch den baumartigenWachstum vorgegeben, nicht zuletzt erfolgen die Berechnungen nicht überComputersimulationen. Aus diesen Gründen ist die Bezeichnung Perkolationstheorie eherirreführend.66 Stauffer D (1979) Physics Reports 54:167 de Gennes PG (1979) Scalling Concepts in Polymer Physics, Cornell University Press, IthacaNew York68 Stauffer D, Coniglio A, Adam M (1982) Advances in Polymer Science 44:103, SpringerVerlag, Berlin69 Stepto RFT persönliche Mitteilung, World Polymer Congress 2000, Warschau, Polen

THEORETISCHER TEIL 30
Abb. 4.2. Schematischer Verlauf der dynamischen Viskosität ηdyn und des Speicher-moduls G’ am Gelpunkt (GP) in einer logarithmischen Darstellung
Eines dieser Verhalten zeigt die dynamische Viskosität ηdyn. Sie ist im wesent-
lichen über den Verlustmodul über folgende Beziehung verknüpft
gel
22
dyn tt;''G)''G'G( 2
1
<ω
≈ω
+=η (4.2)
und gilt nur in dem Zeitbereich vor dem Gelpunkt. Am Gelpunkt zeigt das
System kein Fließverhalten mehr, d.h. die Viskosität muss am Gelpunkt diver-
gieren. Das entgegengestzte Verhalten zeigt der Speichermodul G'. Er geht am
Gelpunkt gegen Null (s. Abb. 4.2.).
Für diesen Fall finden STAUFFER und DE GENNES folgende sogenannte Potenz-
gesetze:70,71
( ) 0undppfürppK''G
ccdyn >ν<−=ω
=η ν−ν (4.3)
( ) 0undppfürppK'G cc >µ>−= µµ . (4.4)
In diesen Formeln bedeuten die Konstanten Kν und Kµ die kritischen Amplitu-
den, p den Anteil bereits geschlossener Bindungen während einer Reaktion,
demnach pc den kritischen Anteil an bereits geschlossenen Bindungen, die zur
70 de Gennes PG (1979) Scalling Concepts in Polymer Physics, Cornell University Press, IthacaNew York71 Stauffer D, Coniglio A, Adam M (1982) Advances in Polymer Science 44:103, SpringerVerlag, Berlin
G‘
ηdyn
GP
ηdyn, G‘
p

THEORETISCHER TEIL 31
Netzwerkbildung erforderlich sind und ν sowie µ die kritischen Exponenten.72
Den kritischen Exponenten kommt dabei eine besondere Bedeutung zu. Sie
sollen in Kap. 4.3 gesondert behandelt werden.
Beim Umgang mit den Gln. (4.3) und (4.4) stellt sich dem experimentierenden
Wissenschaftler ein Problem: Welche durch Experimente zugängliche Größe
verwendet man anstelle der durch Computersimulationen erhaltene Größe p,
um anhand des im Experiment ermittelten komplexen Schubmoduls kritische
Phänomene mit Hilfe der Perkolationstheorie zu beschreiben?
Dieses Problem wird von verschiedenen Wissenschaftlern unterschiedlich
angegangen. Im Falle der Sol–Gel-Umwandlung des Systems Gela-
tine / Wasser bestimmen DJABOUROV und Mitarbeiter eine Konvertierungsvarible
Φ73, die sich auf den Helixanteil in einer Probe bezieht. Zum Aufbau eines
Netzwerkes muss ein kritischer Helixanteil im System vorliegen, der durch die
kritische Konvertierungsvariable Φc angegeben wird. Bei einer bestimmten
Temperatur beobachten DJABOUROV und Mitarbeiter das Verhalten der rheolo-
gischen Kenngrößen im Bereich Φ < Φc und Φ > Φc.74 Für das gleiche System
ermittelten KUMAGAI und Mitarbeiter eine kritische Konzentration an Gelatine φc
und ermittelt für eine Konzentrationsreihe im Bereich φ < φc und φ > φc die rheo-
logischen Größen.75
MICHALCZYK76 und später VENOHR77 folgten der Empfehlung STAUFFERs, die
Wahrscheinlichkeiten p in den Gln. (4.3) und (4.4) durch die Zeit zu substituie-
ren. Dass in der Nähe des Gelpunkts eine Proportionalität zwischen diesen
Größen existiert, ist in verschiedenen Veröffentlichungen niedergeschrieben.78,79,80,81,82 Für eine konstante Frequenz gehen damit die Gln. (4.3) und (4.4) in
folgende Ausdrücke über:
72 Je nach dem welche kritische Phänomene mit der Perkolationstheorie behandelt werden,werden in der Literatur unterschiedliche Symbole für die kritischen Exponenten benutzt. Die indieser Arbeit verwendeten Symbolen wurden von Stauffer vorgeschlagen.73 siehe hierzu Gl. (8.30) in Kap. 8.474 Djabourov M, Leblond J, Papon P (1988) J Phys (France) 49:33375 Kumagai H, Fujii T, Inukai, T, Yano T (1993) Biosci Biotech Biochem 57(4):53276 Michalczyk A (1993) Dissertation Duisburg77 Venohr H (1999) Dissertation Duisburg78 Penich-Covas C, Dev SB, Gordon M, Judd M, Kajiwara K (1974) Discussion of the FaradayDivision on Gels and Gelling Processes 57:16579 Parker TG, Dalgleish DG (1977) J Dairy Res 44:8580 Adam M, Delsanti M, Okasha R, Hild G (1979) J Phys Lett (Paris) 40:L 53981 Gauthier-Manuel B, Guyon E (1980) J Phys Lett (Paris) 41:L50382 Borchard W (1998) Ber Bunsenges Phys Chem 102:1580

THEORETISCHER TEIL 32
( ) ην−
ηη <−= ,gel,gel ttfürttK''G (4.5)
( ) G,gelG,gelG ttfürttK'G >−= µ (4.6)
Durch die Anwendung der Gln. (4.4) und (4.5) auf die rheologischen Experi-
mente ist nun die Möglichkeit gegeben, den Gelpunkt bzw. die Gelierzeit, (tgel)
d.h. die Zeit zu bestimmen, die vergeht bis sich ein Gel gebildet hat.83 Für die
industrielle Anwendung gelierender Systeme ist eine genaue Bestimmung der
Gelierzeit von großer Bedeutung.
4.3 Kritische Exponenten.
Die kritischen Exponenten sind entscheidend vom Verhalten der untersuchten
Systeme am kritischen Punkt abhängig. Anhand von unterschiedlichen Simula-
tionen und Modellrechnungen versuchen verschiedene Wissenschaftler für die
kritischen Phänomene, wie z.B. die Gelierung, die kritischen Exponenten vor-
herzusagen.
DE GENNES simuliert die Gelierung mit Hilfe eines Widerstandsnetzwerkes.84
Später bemühte sich SAHIMI, die Bindung in einem Netzwerk als vektorielle Grö-
ßen zu berücksichtigen und nicht als skalare Größen wie durch das Wider-
standsnetzwerk vorgegeben. SAHIMI trägt damit dem Dehn- und Biegevermögen
(bond bending) eines Netzwerkes Rechnung.85,86,87 Im Folgenden soll eine von
VENOHR angefertigte Zusammenstellung der in diesen und weiteren Fällen vor-
hergesagten kritischen Exponenten wiedergegeben werden.88
83 Die Auswertung zweier experimentell ermittelter Größen ηdyn und G‘ nach derPerkolationstheorie in der Form Gln. (4.5) und (4.6) hat zwei Lösungen für die Gelierzeit zurFolge tgel,η und tgel,G. Auf dieses "Problem" wird in Kap. 8 genauer eingegangen.84 de Gennes PG (1979) Scalling Concepts in Polymer Physics, Cornell University Press, IthacaNew York85 Arbabi S, Sahimi M (1990) Phys Review Lett 65:72586 Sahimi M (1992) Mod Phys Lett B 6:50787 Sahimi M (1994) Applications of Percolation Theory, Taylor & Francis88 Venohr H (1999) Dissertation Duisburg

THEORETISCHER TEIL 33
Vorhersage für den kritischen Exponenten ν:
ν = 0: Ergebnis der klassischen Theorie.89,90 Demnach ergibt sich am
Gelpunkt für die Viskosität ein endlicher Wert.
ν ≈ 0.65: ARBABI und SAHIMI nehmen hier an, dass starke hydrodynamische
Wechselwirkungen zwischen den Polymeren am Gelpunkt beste-
hen und keine oder wenig Diffusion stattfindet (ZIMM –
Regime).91,92
ν = 0.7: DE GENNES vergleicht hierbei die Viskosität mit der Leitfähigkeit
einer Mischung aus Leitern und Supraleitern.93,94
ν = 1.3: Ergebnis der ROUSE Approximation. Der Beitrag eines Clusters zur
Viskosität ist hierbei proportional zum Quadrat seines Radius. Die
Polymere werden hierbei als lange inflexible Ketten angenommen.
Geringe Polymer - Polymer und Polymer - LM Wechselwirkun-
gen.95,96
ν ≈ 1.35: ARBABI und SAHIMI nehmen an, dass in der Nähe des Gelpunkts
keine hydrodynamische Wechselwirkung zwischen den Polymeren
verschiedener Größen besteht (ROUSE – Regime).97,98
0 ≤ν ≤ 1.35: Resultate der Berechnungen von MARTIN und Mitarbeitern, abhän-
gig vom Ausmaß der hydrodynamischen Wechselwirkungen der
Polymere.99
Vorhersage für den kritischen Exponenten µ:
µ = 1.7: Annahme der Analogie von Elastizität zur Leitfähigkeit bei einem
Netzwerk aus Leitern und Isolatoren.100
89 Flory PJ (1941) J Amer Chem Soc 63:3083,3091,309690 Stockmayer WH (1944) J Chem Phys 11:45 ibid 12:12591 Arbabi S, Sahimi M (1990) Phys Review Lett 65:72592 Sahimi M (1992) Mod Phys Lett B 6:50793 de Gennes PG (1979) Scalling Concepts in Polymer Physics, Cornell University Press, IthacaNew York94 de Gennes PG (1979) J Physique (Paris) Lett 40:19795 de Gennes PG (1979) J Physique (Paris) Lett 40:19796 de Gennes PG (1980) Comp Rendus Acad Sci (Paris) 286B:13197 Arbabi S, Sahimi M (1990) Phys Review Lett 65:72598 Sahimi M (1992) Mod Phys Lett B 6:50799 Martin JE, Adolf D, Wilcoxon JP (1989) Phys Rev A 39:1325

THEORETISCHER TEIL 34
µ = 2.1: Die Bindungen auf einem Perkolationsnetzwerk werden durch
elastische Elemente dargestellt, die gedehnt werden können
(stretching forces).101
µ = 2.67: Diesen Wert berechnete Martin für ein Sol aus verzweigten Poly-
meren.102
µ = 3: Ergebnis der klassischen Theorie.103,104,105
µ = 3.75: Die Bindungen auf einem Perkolationsnetzwerk werden durch
elastische Elemente dargestellt, die sowohl gedehnt als auch
gebogen werden können (bond bending).106
µ ≤ 3.78: Diesen Wert erhalten ROUx und GUYON unter der Annahme, dass
sich Drehmomente wie die elektrische Leitung ausbreiten.107
µ = 2.85
bzw. 3.55: KANTOR und WEBMAN ermittelten diese Werte unter Berücksichti-
gung der Änderung von Bindungswinkeln, der Gestalt der Poly-
merketten und der Richtung der angreifenden Kraft.108
Behandelt man ein kritisches Phänomen mit Hilfe der Skalentheorie und erhält
identische oder zumindest ähnliche kritische Exponenten, dann spricht man von
einer Universalität der kritischen Exponenten. Systemen, die einer Universali-
tätsklasse zugeordnet werden können, kann man unterstellen, dass sie sich,
bezüglich ihres Verhaltens am kritischen Punkt, sehr ähnlich sind. Im Falle der
Polymerisation kann man daraus schließen, dass ähnliche molekulare Abläufe
stattfinden.
100 de Gennes PG (1979) Scalling Concepts in Polymer Physics, Cornell University Press,Ithaca New York101 Sahimi M (1992) Mod Phys Lett B 6:507102 Martin JE, Adolf D, Wilcoxon JP (1989) Phys Rev A 39:1325103 Dobson GR, Gordon M (1965) J Chem Phys 43:705104 de Gennes PG (1976) J Phys (Paris) 37:L1105 Gordon M, Ross-Murphy SB (1979) J. Phys A 12:L155106 Sahimi M (1992) Mod Phys Lett B 6:507107 Roux S, Guyon E (1986) In: Stanley HE, Ostrowski N (eds) On Growth and Form, MartinusNijhoff Boston108 Kantor Y, Webman I (1984) Phys Rev Lett 52:1891

EXPERIMENTELLER TEIL 35
EXPERIMENTELLER TEIL
5 Probenmaterial und Vorbereitung.
In dieser Arbeit wurden zwei verschiedene Gelatine - Typen verwendet. Bei der
einen Sorte handelte es sich um eine sauer aufbereitete Schweineschwarten -
Gelatine der Firma DEUTSCHE-GELATINE-FABRIKEN STOESS AG (kurz DGF
STOESS AG),109 bei der anderen handelt es sich um eine basisch geäscherte
Rinderknochen - Gelatine vom Typ M92 der Firma ROUSSELOT S.A..110
Das Gelatinegranulat wurde mit Hilfe einer Analysenwaage unter Berücksichti-
gung des in einem Trocknungsversuch bestimmten Wassergehaltes der Gela-
tine von 14.00 Gew.-% (STOESS Gelatine) und 11.97% (ROUSSELOT Gelatine) in
mit Schwefelsäure gereinigte Hochdruckflaschen eingewogen. Die Proben wur-
den durch Zusatz von 0.15mL Raschitlösung111 pro 1g Gelatine gegen den
bakteriellen Befall geschützt. Im Anschluss daran wurden die Proben über
Nacht bei 5-7°C quellen gelassen, um beim späteren Lösen des Polymeren ein
Verklumpung des Gelatinegranulats (gel-blocking) zu verhindern. Hierdurch lie-
ßen sich die Proben problemlos im 45°C temperierten Wasserbad homogenisie-
ren. Das längere Behandeln von Gelatinelösungen oberhalb von 45°C führt zu
irreversiblen Zerstörungen der Gelatinemoleküle. Dies wurde durch die Ver-
wendung eines auf 45°C eingestellten Kontaktthermometers, welches in das
Wasserbad ragt, verhindert. Das Wasserbad verhindert gleichzeitig, dass die
Hochdruckflaschen direkt mit der Heizplatte in Verbindung stehen und somit die
Temperatur in der Probe lokal über 45°C ansteigt. Die Gelatine-Lösungen wur-
den mit Hilfe einer Plastikspritze aus den Hochdruckflaschen entnommen und
direkt, wie oben beschrieben in die Messzellen eingebracht. Diese wässrigen
Proben sind sowohl als Sol als auch als Gel äußerst transparent und sind somit
bestens für die optischen Untersuchungen geeignet. Die Verwahrung der Pro-
ben erfolgte im Kühlschrank bei 5-7°C.
109 Herrn Dipl. Ing. Pflaumbaum von der Firma DGF Stoess AG danke ich für die Überlassungder Gelatine Proben.110 Der Firma Dupont de Nemours danke ich für die Überlassung einiger Gelatineproben.111 5%ige Lösung aus 4-Chlor-3-Methylphenol in Methanol

EXPERIMENTELLER TEIL 36
6 Versuchsaufbau zur simultanen Bestimmung der optischen und
rheologischen Kenngrößen.
Die in dieser Arbeit durchgeführten Bestimmungen des Drehwertes des Lichts
und des komplexen Schubmoduls während der isothermen Gelierung des
Systems Gelatine / Wasser erfolgten mit einem in der Arbeitsgruppe Ange-
wandte Physikalischen Chemie an der Universität Duisburg selbst konstruier-
ten, dynamischen Schwingungsviskosimeter112,113,114 und einem Präzisionspola-
rimeter vom Typ POL S-1 der Firma DRE – DR. RISS ELLIPSOMETERBAU GmbH.
Abb. 6.1. Blockschaltbild für den Messplatz zur simultanen Bestimmung der opti-schen und rheologischen Kenngrößen
Da sowohl die optische Drehung und der komplexe Schubmodul sehr stark
temperaturabhängig sind, wurden zur Gewährleistung identischer thermischer
Bedingungen in den Messzellen beide Messgeräte über einen gemeinsamen
Thermostaten (T1) temperiert (s. Abb. 6.1.). Zum Aufschmelzen der Probe in
den Messzellen kann ein zweiter Thermostat (T2) über die in den Temperier-
112 Michalczyk A (1993) Dissertation Duisburg113 Lechtenfeld M, Michalczyk A, Borchard W (2001) Rheol Acta angenommen114 im Verlauf der Arbeit wird das dynamische Schwingungsviskosimeter gelegentlich auch alsRheometer bezeichnet
PC PC
T1
T2
Rheometer Polarimeter

EXPERIMENTELLER TEIL 37
kreislauf eingebauten Drei-Wege-Hähne "kurzgeschlossen" werden. Der
eigentliche Thermostat des primären Kreislaufes kann dadurch bei einer Tem-
peratur eingestellt bleiben. Sowohl das dynamische Schwingungsviskosimeter
als auch das Polarimeter werden separat mit einem Personalcomputer (PC)
gesteuert. In den nächsten beiden Kapiteln sollen diese beiden Messgeräte
beschrieben werden.
6.1 Das dynamische Schwingungsviskosimeter.
Bei dem in dieser Arbeit verwendeten Rheometer handelt es sich wie oben
erwähnt um eine Eigenkonstruktion der Arbeitsgruppe Angewandte Physikali-
sche Chemie der Universität Duisburg. Es stellt die "zweite Generation" eines
von BORCHARD entwickelten und BURG konstruierten dynamischen Schwin-
gungsviskosimeters dar.115,116 Bei der Entwicklung des neuen Rheometers reali-
sierten BORCHARD und MICHALCZYK eine Messzelle mit wesentlich geringerem
Probenvolumen und damit deutlich geringerem Gewicht der schwingenden
Komponenten (s. Abb. 6.2.).117,118 Dies hat zum einen den Vorteil, dass die
Abkühlzeiten der Proben in der Messzelle kürzer sind, zum anderen erhöht sich
der Frequenzmessbereich. Die erheblich kürzeren Abkühlzeiten des neu kon-
struierten Rheometers gehen hauptsächlich auf die neu entwickelte Dop-
pelspaltanordnung zurück.119 Die Probe wird hierbei sowohl durch einen äuße-
ren Glaszylinder sowie durch einen inneren Stahlzylinder temperiert. Durch
diese Anordnung ergibt sich eine Rheometergeometrie, die sich aus einem
sogenannten Searl- und einem Couette-Typ zusammensetzt.120
Die zu untersuchende Probe in dem erwähnten Doppelspalt wird im Experiment
über einen einseitig offenen, schwingungsfähigen Hohlzylinder einer oszillie-
renden, sinusförmigen Scherdeformation ausgesetzt. In diesen Zylinder ragt ein
auf einer Metallplatte fixierter, etwas kleinerer Zylinder, der von der Temperier-
flüssigkeit durchströmt wird.
115 Burg B (1988) Dissertation Duisburg116 Borchard W, Burg B (1990) Progr Colloid Polym Sci 83:200117 Michalczyk A (1993) Dissertation Duisburg118 Lechtenfeld M, Michalczyk A, Borchard W (2001) Rheol Acta angenommen119 Eine von mir beim Europäischen Patentamt in Auftrag gegebene Patentrecherche hatergeben, dass diese Messzellenanordnung nicht bekannt ist.120 Searl – Typ: Der innere Zylinder wird angetrieben, der äußere Zylinder steht fest. Couette-Typ: Der innere Zylinder steht fest, der äußere wird angetrieben.

EXPERIMENTELLER TEIL 38
Abb. 6.2. Skizzierung des dynamischen Schwingungsviskosimeter mit Doppelspalt-anordnung121
Nach außen ist die Messzelle durch den temperierten Glaszylinder abgegrenzt.
Die Schwingungsanregung erfolgt elektrodynamisch durch ein induziertes
Magnetfeld, welches durch zwei Wechselstrom durchflossene Kupferspulen
erzeugt wird. Dieses Magnetfeld wirkt orthogonal auf einen Stabmagneten, der
starr mit dem beweglichen Innenzylinder verbunden ist. Die daraus resultie-
rende Auslenkung des schwingenden Systems wird berührungslos durch einen
induktiven Wegaufnehmer über eine Messfahne am Innenzylinder gemessen.
Der zur Schwingungsanregung erforderliche sinusförmige Wechselstrom wird
durch einen Frequenzganganalysator erzeugt, der seinerseits durch einen Lei-
stungsverstärker unterstützt wird. Das am Innenzylinder wirkende Drehmoment
ist abhängig von der momentanen Stromstärke in der Erregerspule, welcher
proportional einem Spannungsabfall UE über einem reinen Ohmschen Wider-
stand ist. Der Messverstärker des induktiven Wegaufnehmers liefert als Signal
eine der momentanen Auslenkung des schwingenden Systems proportionalen
Spannung UA. Der Frequenzganganalysator analysiert den Zusammenhang
zwischen aufgegebenem Drehmoment und resultierender Auslenkung des
Systems. Die Größen sind hier die aus dem Quotienten von UE(max) und UA(max)
erhaltene Amplitudenverhältnis und Phasenverschiebung zwischen Erregungs-
schwingung und der Schwingung der Auslenkung. Beide Größen werden im
121 Ich bedanke mich bei Dr. H. Venohr für die freundliche Überlassung eines Teils der Skizzedes dynamischen Schwingungsviskosimeters.
StabmagnetMetallmessfahne
temperierterGlaszylinder
äußeresanregendesMagnetfeld
gelierende Probe
oszillierenderStahlzylinder
innerer temperierterStahlzylinder
rostfreier Stahldraht
Einspritzvor-richtung

EXPERIMENTELLER TEIL 39
Display des Frequenzganganalysators angezeigt. Die zu bestimmenden Grö-
ßen G' und G'' werden anhand einer Kalibrierungsmessung von Ölen mit defi-
nierter Viskosität aus den ermittelten Messdaten berechnet.
Im Rahmen dieser Arbeit wurde die Steuerung des dynamischen Schwingungs-
viskosimeters von längst veraltetem Ataribetrieb auf PC umgestellt.122 Der oben
erwähnte Frequenzganganalysator wird hierbei über einen GPIB–Port mit einer
in den PC eingebauten IEEE 486 Schnittstelle der Firma KEITHLEY gesteuert.
Mit Hilfe des Programms Test PointTM wurde ein Programm zur Steuerung des
Rheometers geschrieben, das die Einstellungen aller relevanten Versuchs-
parameter über eine Programmmaske erlaubt. Das Softwareprogramm wurde
dabei so gestaltet, dass der Verlauf der Gelierung online verfolgt werden kann.
Der Vorteil gegenüber der Steuerung über Atari ist, dass Fehlmessungen sofort
erkannt und abgebrochen werden können. Durch den Ataribetrieb wurden
Fehlmessungen erst dann sichtbar, wenn nach Ende der Messungen die Daten
umgeformt und dann mit Hilfe einer geeigneten Software als Diagramm darge-
stellt wurden. Insbesondere bei Messungen, die über einen längeren Zeitraum
laufen (1 Woche), ist diese Neuerung sehr zeitsparend. Der alles entschei-
dende Vorteil ist aber, dass die wesentlich höhere Rechnerleistung Abtastraten
in Sekundenintervallen erlaubt. Durch den Ataribetrieb konnte maximal alle 18 s
ein Messwert ermittelt werden. Bei Untersuchungen schnell gelierender
Systeme stehen somit 18mal so viele Messwerte zur Verfügung, wodurch eine
Auswertung der Gelierkurven nach der Perkolationstheorie überhaupt erst mög-
lich wird.
Die Bestimmung der Temperatur während des Experiments geschieht mit Hilfe
eines Thermoelements. Über eine ebenfalls in den PC eingebaute Schnittstelle
der Firma KEITHLEY ist es möglich, die Temperaturmessung in das Softwarepro-
gramm zu implementieren, sodass die Kontrolle des Temperaturverlaufs eben-
falls online erfolgen kann.
6.1.1 Messsystematik.
Für die Durchführung der Experimente ist es unbedingt erforderlich im linear
viskoelastischen Bereich zu arbeiten. Dies ist gewährleistet, wenn unterschied-
lich große Schwingungsamplituden keinen Einfluss auf die Messgrößen, sprich
122 Herrn Michael Kischel, Herrn Volker Körstgens und Herrn Volker Fischer danke ich für dieHilfe bei der Hardwarekonfiguration sowie der Formulierung der Auswertesoftware.

EXPERIMENTELLER TEIL 40
Speicher- und Verlustmodul, haben. Dies wurde durch die Bestimmung des
Speichermoduls als Funktion der Anregungsspannug, ständig überprüft.
Da die Messungen sehr empfindlich auf äußere Einflüsse reagieren, wurden die
Beeinträchtigungen durch Gebäudeschwingung, welche insbesondere Messun-
gen bei kleinen Frequenzen sehr stören, weitestgehend verhindert, indem das
dynamische Schwingungsviskosimeter auf eine massive Stahlplatte gestellt
wurde, die ihrerseits von vier nach unten spitz zulaufende Messingkegeln
getragen wurde.123
Die vorliegenden Arbeit hat die Absicht, die Gelierung möglichst von Anbeginn
der Messung unter isothermen Bedingungen zu verfolgen. Dies wurde in den
früheren Arbeiten weitestgehend durch die Verwendung zweier Temperatur-
kreisläufe realisiert. Ein Temperaturkreislauf wurde auf die gewünschte Tempe-
ratur, bei der die Untersuchung durchgeführt werden sollte, eingestellt, der
zweite temperierte die Messzelle mit einer Temperatur oberhalb der Gelbil-
dungstemperatur. Über die Einspritzvorrichtung (s. Abb. 6.2.) wurde nun die wie
in Kap. 5 aufbereitete Probe in die Messzelle eingebracht. Durch das Verstellen
zweier Drei-Wege-Hähne wurde das höher temperierte Wasserreservoir vom
Kreislauf abgeklemmt und die Messzelle jetzt von dem auf Geliertemperatur
eingestellten Thermostaten temperiert. Die Abkühlzeiten in der Probe lagen bei
18 s.124,125,126
Zur Realisierung wirklicher isothermer Bedingungen von Anbeginn der Messung
an wurde in dieser Arbeit ein einfacher, aber sehr wirksamer "Kunstgriff" getä-
tigt. Das Probenmaterial wurde hier direkt in die bereits auf die gewünschte
Geliertemperatur eingestellte Messzelle eingespritzt. Da garantiert werden
sollte, dass die Temperatur des schwingenden Zylinders, der nicht an den
Temperierkreislauf angeschlossen ist, mit der Temperatur des Stahl- bzw.
Glaszylinders identisch ist (kein Temperaturgradient in der Probe), wurde vor
allen Messungen die Messzelle mit Aceton gefüllt und ausreichend lange
gewartet bis ein Temperaturausgleich mit dem schwingenden Stahlylinder statt-
finden konnte. Im Anschluss wurde das Aceton durch die Spritzeinrichtung
abgesaugt und die Messzelle zügig mit Pressluft vom Restlösemittel befreit.
Unmittelbar danach wurde das Probenmaterial blasenfrei mit Hilfe einer Plastik-
123 Trotz aller Vorkehrungen konnte der Einfluss der Straßenbahnlinie 901 der DuisburgerVerkehrsgesellschaft, die unmittelbar am Gebäude vorbeiführt, auf die Gebäudeschwingungnicht vollständig eliminiert werden.124 Michalczyk A (1993) Dissertation Duisburg125 Venohr H (1999) Dissertation Duisburg126 Lechtenfeld M, Michalczyk A, Borchard W (2001) Rheol Acta angenommen
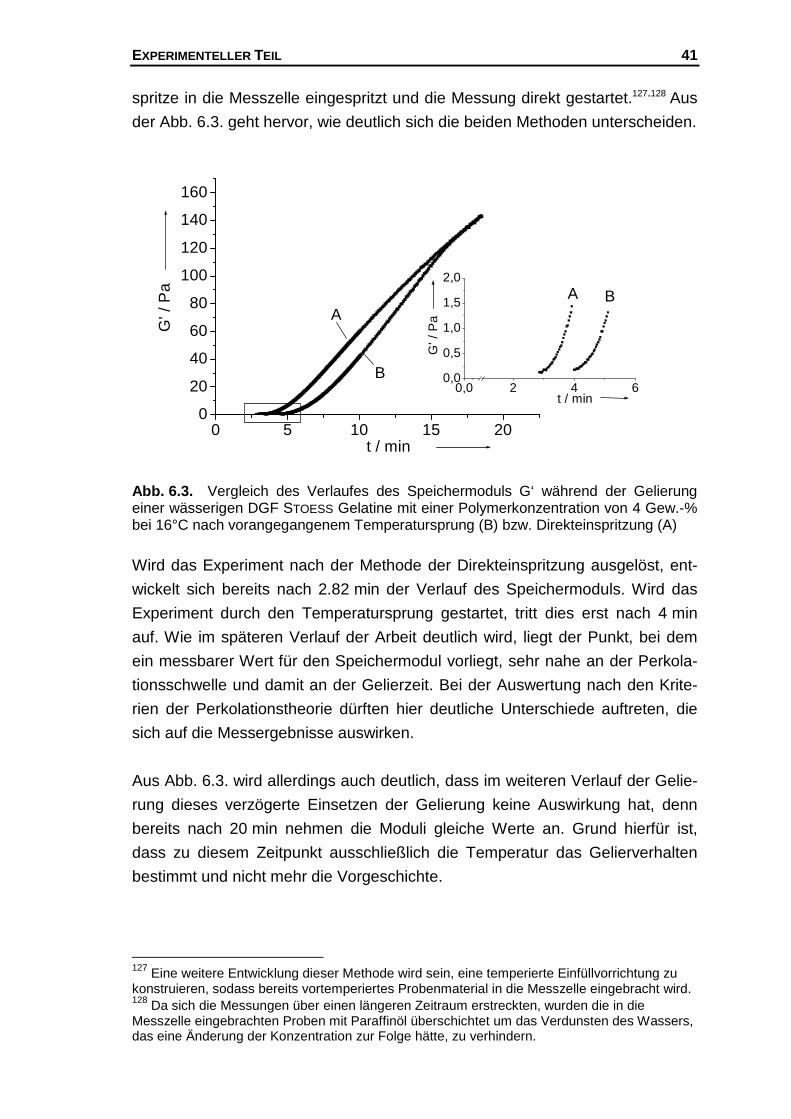
EXPERIMENTELLER TEIL 41
spritze in die Messzelle eingespritzt und die Messung direkt gestartet.127,128 Aus
der Abb. 6.3. geht hervor, wie deutlich sich die beiden Methoden unterscheiden.
Abb. 6.3. Vergleich des Verlaufes des Speichermoduls G‘ während der Gelierungeiner wässerigen DGF STOESS Gelatine mit einer Polymerkonzentration von 4 Gew.-%bei 16°C nach vorangegangenem Temperatursprung (B) bzw. Direkteinspritzung (A)
Wird das Experiment nach der Methode der Direkteinspritzung ausgelöst, ent-
wickelt sich bereits nach 2.82 min der Verlauf des Speichermoduls. Wird das
Experiment durch den Temperatursprung gestartet, tritt dies erst nach 4 min
auf. Wie im späteren Verlauf der Arbeit deutlich wird, liegt der Punkt, bei dem
ein messbarer Wert für den Speichermodul vorliegt, sehr nahe an der Perkola-
tionsschwelle und damit an der Gelierzeit. Bei der Auswertung nach den Krite-
rien der Perkolationstheorie dürften hier deutliche Unterschiede auftreten, die
sich auf die Messergebnisse auswirken.
Aus Abb. 6.3. wird allerdings auch deutlich, dass im weiteren Verlauf der Gelie-
rung dieses verzögerte Einsetzen der Gelierung keine Auswirkung hat, denn
bereits nach 20 min nehmen die Moduli gleiche Werte an. Grund hierfür ist,
dass zu diesem Zeitpunkt ausschließlich die Temperatur das Gelierverhalten
bestimmt und nicht mehr die Vorgeschichte.
127 Eine weitere Entwicklung dieser Methode wird sein, eine temperierte Einfüllvorrichtung zukonstruieren, sodass bereits vortemperiertes Probenmaterial in die Messzelle eingebracht wird.128 Da sich die Messungen über einen längeren Zeitraum erstreckten, wurden die in dieMesszelle eingebrachten Proben mit Paraffinöl überschichtet um das Verdunsten des Wassers,das eine Änderung der Konzentration zur Folge hätte, zu verhindern.
0 5 10 15 200
20
40
60
80
100
120
140
160
B
A
G' /
Pa
t / min
0,0 2 4 60,0
0,5
1,0
1,5
2,0A B
t / min
G' /
Pa

EXPERIMENTELLER TEIL 42
6.1.2 Mathematische Beschreibung des dynamischen Experiments.
Die erzwungene harmonische Schwingung des oszillierenden Zylinders kann
durch Gl. (6.1) ausgedrückt werden:129
( ) ( ) ( ) ( ) ti0eMtMtDt*t ω==ϕ+ϕη+ϕ ���I . (6.1)
Auf der rechten Seite der Gl. (6.1) steht der Ausdruck für den durch das äußere
Feld hervorgerufene Drehmoment M(t), das auf den Zylinder wirkt - M0 ist dabei
die maximale Amplitude. Auf der linken Seite stehen alle Beiträge, die auf die
Messzelle zurückzuführen sind. Dies sind der oszillierende Zylinder, der
Magnet, der Stab, an dem die beiden Komponenten befestigt sind, der Draht
und die viskoelastische Probe, die sich im Verlauf des Experiments am Zylinder
anlagert.130 ( )tϕ��I repräsentiert den Beitrag der Massenträgheit mit dem Mas-
senträgheitsmoment I des oszillierenden Zylinders inklusive aller damit verbun-
denen Massen und der Winkelgeschwindigkeit ( )tϕ�� . Der Ausdruck ( )t* ϕη � steht
für die Reibungsverluste bei der Winkelgeschwindigkeit ϕ(t) und der komplexen
Viskosität η*.131 Dϕ(t) gibt den Beitrag des Rückstellmoments mit der Direktions-
konstanten D des Systems und dem Winkel ϕ(t) wieder.
η* und D setzten sich aus einem apparativen Anteil (Index ap) und einem
Anteil, verursacht durch die viskoelastische Probe (Index pr), zusammen.132
prprap i* η′′−η′+η′=η (6.2)
prap DDD += . (6.3)
apη′ wird durch die Verwendung von nicht rein elastischem Stahldraht verur-
sacht, an dem der oszillierende Zylinder aufgehängt ist anstelle von reinem
aber korrodierenden Stahl.
Dpr in Gl. (6.3) berücksichtigt das Rückstellmoment der Probe nach dem Gel-
punkt. Der Rückstellmoment ist proportional zum Winkel ϕ.
129 Borchard W, Burg B (1990) Progr Colloid Polym Sci 83:200130 Dieser Term kann bei schwachen Gelen und niedrigen Frequenzen unberücksichtigt bleiben.131 In Analogie zu dem in Kap. (2.5) eingeführten komplexen Schubmodul Gl. (2.25), lässt sichauch eine komplexe Viskosität formulieren. η* = η' - iη''. Hierbei ist η' die dynamische Viskosität(im Verlauf der Arbeit nur als ηdyn bezeichnet), die die wirklichen rein - viskosen Anteile einerProbe berücksichtigt, η'' repräsentiert hingegen mögliche elastische Anteile eines Fluids.132 Borchard W, Burg B (1990) Progr Colloid Polym Sci 83:200

EXPERIMENTELLER TEIL 43
Gl. (6.4) liefert die Lösung der Gl. (6.1):
( ) ( )δ−ωϕ=ϕ ti0et . (6.4)
ϕ0 ist die maximale Auslenkungsamplitude und δ der Phasenwinkel zwischen
dem äußeren Drehmoment und dem des schwingenden Zylinders.
Die erste und zweite Ableitung des Winkels ϕ nach der Zeit sind gegeben
durch:
( ) ( ) ( )tieit ti0 ωϕ=ωϕ=ϕ δ−ω� ( ) ( ) )t(iet ti
02 ϕω=ϕω−=ϕ δ−ω ��� . (6.5)
Aus den Gln. (6.1), (6.4) und (6.5) ergibt sich:
( )( )
δ
ωϕ=
ω+η+ω=
ϕi
0
0 eiM
iD
*ittM
I� . (6.6)
Der Quotient aus M(t) und der Winkelgeschwindigkeit ( )tϕ� wird die komplexe
mechanische Impedanz genannt.133,134 Gl. (6.6) lautet dann:
( )δ+δωϕ
=ωϕ
≡ δ sinicosiM
eiM
Z0
0i
0
0 . (6.7)
Unter Berücksichtigung der Gln. (6.1), (6.2) und (6.7) können zwei Gleichungen
für Z formuliert werden, eine für die leere (Index l) und eine für die mit Probe
gefüllte (Index g) Messzelle:135
( )prapprprapg DDi1
iiZ +ω
+η′′−η′+η′+ω= I (6.8)
apapl Di1
iZω
+η′+ω= I . (6.9)
Aus der Differenz der Gln. (6.8) und (6.9) ergibt sich die Komplexe mechani-
sche Impedanz der viskoelastischen Probe Zpr:
133 Schwarzl F, Staverman AJ (1956) In: Stuart HA (ed) Physik der Hochpolymere IV, SpringerVerlag, Berlin Göttingen Heidelberg134 Ferry JD (1970) Viscoelastic Properties of Polymers, John Wiley & Sons Inc. New York135 Borchard W, Burg B (1990) Progr Colloid Polym Sci 83:200

EXPERIMENTELLER TEIL 44
ω
+η′′−η′=ω
+η′′−η′= prprprprprprpr
DiD
i1
iZ (6.10)
Unter Verwendung der Abkürzungen Gl. (6.11): geht Gl. (6.7) in Gl. (6.12) über,
ein Ausdruck nach dem sich Zpr aus der Zeitanalyse der Erregerschwingung
bestimmen lässt.136
l,0
0l
MK
ϕ= und
g,0
0g
MK
ϕ= (6.11)
( ) ( )[ ]llggllggpr sinKsinKicosKcosKi1
Z δ−δ+δ−δω
= . (6.12)
Der komplexe Schubmodul G* hängt über die Beziehung:
Zb1
i*G ω= (6.13)
mit der komplexen mechanischen Impedanz Z zusammen und lässt sich unter
Berücksichtigung der Gl. (6.10) bestimmen, wenn b der Formfaktor in Gl. (6.13)
richtig angesetzt ist.
Der Formfaktor berücksichtigt die Geometrie der verwendeten Messzelle. In
geometrischer Hinsicht stellt sich das neu entwickelte dynamische Schwin-
gungsviskosimeter, wie oben erwähnt, aus einem kombinierten Searl und
Couette Typ zusammen. Bei der Berechnung des Formfaktors sind daher 4
Radien zu berücksichtigen. Der Radius R1 des inneren feststehenden Zylinders,
der "innere" Radius R2 des oszillierenden Zylinders, der "äußere" Radius R3 des
oszillierenden Zylinders und R4, der Radius des äußeren Glaszylinders. In
Anbetracht der dünnen Wandstärke des oszillierenden Zylinders (R3 –
R2 = 0.2mm) kann folgende Näherung eingeführt werden:
(R2 + R3) / 2 = Rm≈ R2 ≈ R3. Aus den Berechnungen ergibt sich für den kombi-
nierten Searl – Couette Typ folgender Formfaktor:137,138,139
( ) ( )[ ]( )( )2
m21
2m
24
2m
24
21
2m
21
24
2m
RRRR
RRRRRRLR4b
−−
−+−π= (6.14)
136 Borchard W, Burg B (1990) Progr Colloid Polym Sci 83:200137 Ferry JD (1970) Viscoelastic Properties of Polymers, John Wiley & Sons Inc., New York138 Michalczyk A (1993) Dissertation Duisburg139 Lechtenfeld M, Michalczyk A, Borchard W (2001) Rheol Acta angenommen

EXPERIMENTELLER TEIL 45
Unter Berücksichtigung dieses Terms lässt sich für den wie in Gl. (2.23) kom-
plex angesetzten Schubmodul folgender Ausdruck formulieren:
[ ]b1
iDZb
iGiGG prprprprprprpr η′′ω+η′′ω+=ω=′′+′=∗ . (6.15)
Die separaten Ausdrücke für den frequenzabhängigen Real- bzw. Imaginärteil
des komplexen Schubmoduls sind gegeben durch:
( ) [ ]b1
DG prprpr η′′ω+=ω′ (6.16a)
.b
)(G prpr η′ω=ω′′ (6.16b)
Berücksichtigt man Gl. (6.12) ergibt sich entsprechend für den Real- und Imagi-
närteil:
llggpr cosKcosKG δ′−δ′=′ (6.17a)
( ) llggpr sinKsinKG δ′−δ′=ω′′ (6.17b)
mit bK
K,b
KK l
lg
g =′=′ .
Experimentell lassen sich gK′ und lK′ indirekt aus dem Verhältnis zweier Span-
nungen Um und Uϕ bestimmen. Führt man km und kϕ als Proportionalitätskon-
stanten ein, dann erhält man:140
g0g,
0M
0g,
0MM
g,0
0g EA
U
UE
Ubk
UkbM
K ===ϕ
=′ϕϕϕ
(6.18)
.EAU
UE
Ubk
Uk
b
MK l0
l,
0M
0l,
0MM
l,0
0l ===
ϕ=′
ϕϕϕ(6.19)
Die Größe E ist hierbei die elektro – mechanische Apparatekonstante, deren
Bestimmung weiter unten beschrieben wird. Durch die Einführung der Verhält-
nisse der Spannungsamplituden Ag bzw. Al können schließlich der Speicher-
und Verlustmodul nach folgender Gleichung berechnet werden:
140 Borchard W, Burg B (1990) Progr Colloid Polym Sci 83:200

EXPERIMENTELLER TEIL 46
( ) [ ]llggpr cosAcosAEG δ−δ=ω′ (6.20)
( ) [ ].sinAsinAEG llggpr δ−δ=ω′′ (6.21)
Wie bereits in Kap. 2.6 erwähnt, können die Gln. (2.22) und (2.23) nur dann zur
Auswertung herangezogen werden, wenn der Einfluss der Messapparatur ver-
nachlässigt werden kann. Da dies, wie gerade gezeigt, nicht der Fall ist, muss
die oben erwähnte elektro - mechanische Apparatekonstante ermittelt werden.
Dies geschieht, indem die Apparatur mit einem Mineralöl kalibriert wird, dessen
Viskosität sehr genau bestimmt und der NEWTONsche Bereich kontrolliert wurde.
Dies wurde bei unterschiedlichen Temperaturen durchgeführt, um der Tem-
peraturabhängigkeit der Viskosität Rechnung zu tragen.
Für die späteren frequenzabhängigen Messungen ist es erforderlich, eine Appa-
ratekonstante ebenfalls frequenzabhängig zu bestimmen.141 Die Apparatekon-
stante lässt sich nach folgender Gleichung berechnen:
( )( ) ( )2211
21
sinAsinAE
δ−δ
η−η⋅ω= (6.22)
In Gl. (6.22) bedeuten:
η1 = dynamische Viskosität des Öls bei der Temperatur T1
η2 = dynamische Viskosität des Öls bei der Temperatur T2
δ1 = Phasenverschiebung bei der Messung mit Öl bei der Temperatur T1
δ2 = Phasenverschiebung bei der Messung mit Öl bei der Temperatur T2
A1 = Amplitudenverhältnis bei der Messung mit Öl bei der Temperatur T1
A2 = Amplitudenverhältnis bei der Messung mit Öl bei der Temperatur T2
6.2 Das Polarimeter.
Die in dieser Arbeit durchgeführten Bestimmungen der optischen Drehung wäh-
rend der thermoreversiblen Gelierung des Systems Gelatine / Wasser wurden
mit einem Präzisionspolarimeter vom Typ Pol S-1 der Firma DRE DR. RISS
ELLIPSOMETERBAU GmbH durchgeführt (s. Abb. 6.4.). Die Steuerung des Mess-
platzes erfolgte durch eine vom Hersteller programmierte Software.
141 Während der Durchführung der vorliegenden Arbeit reichte es aus, die Kalibrierung alle 6Monate zu wiederholen.

EXPERIMENTELLER TEIL 47
Das Polarimeter arbeitet nach dem Prinzip des automatischen, optischen Null-
abgleichs. Lichtquelle ist eine Laserdiode mit einer Wellenlänge von 670nm. Die
Auswertung des von der zu messenden Substanz gedrehten Winkel erfolgt über
einen fehlerkorrigierten Schrittmotor höchster Genauigkeit (360 000 Schritte je
Umdrehung), der mit einem Polarisationsprisma verbunden ist. Ausgewertet
wird das Licht mit einem Lichtdetektionssystem, das sich automatisch an die
detektierte Lichtintensität anpasst. In Abb. 6.5. ist das Blockschaltbild des Pola-
rimeters dargestellt.
Abb. 6.4. Schematische Darstellung des Polarimetermessplatzes142
Ein- bzw. Ausgangfür Thermostaten
Steuerelektronik

EXPERIMENTELLER TEIL 48
Abb. 6.5. Blockschaltbild des Polarimeters143
Bei höchster Auflösung beträgt die Messgenauigkeit 0.002° bei einem Drehwert
zwischen –90° und +90°.144 Je nach Wahl der vier möglichen Genauigkeiten
kann alle 2 – 15 s ein Messwert aufgenommen werden.
Für die Verwendung dieses Polarimeters sprechen mehrere Gründe. Im
Gegensatz zu herkömmlichen Polarimetern mit mechanischem Getriebe und
Faraday-Modulator, kann im Pol S-1 kein fehlerproduzierender Verschleiß eines
Zahnradgetriebes auftreten, da kein Getriebe vorhanden ist. Weiterhin ist kein
Glasstab aus Schwerflintglas vorhanden, da der Faraday-Modulator ebenfalls
entfällt. Schwerflintglas hat die negative Eigenschaft einer sehr großen Rest-
anisotropie, die einen Langzeitdrift aufweist und damit die Linearität verändert.
Die Spule eines Farady-Modulators unterliegt der Wärmeausdehnung und ver-
ändert in Abhängigkeit von der Temperatur den Stelleffekt. Ein weiterer negati-
ver Effekt ist der Einfluss des Magnetfeldes des Faraday-Modulators auf die
Endfenster der Küvetten. Eine mögliche Torsion des Chassis zwischen den
beiden Polarisationsfiltern wird kompensiert, indem nach dem Anschalten des
Gerätes bzw. nach der Aufwärmphase der Nullpunkt durch eine im Steuerpro-
gramm integrierte "Zero orientation" neu gesetzt wird. Diese Funktion dient
ebenfalls zur Kalibrierung des Polarimeters. Nach dieser Prozedur wird ein
Drehwinkel ermittelt und abgespeichert, der automatisch von den im Experi-
ment ermittelten Werten subtrahiert wird.
Die Hersteller bieten für diesen Typ Polarimeter eine Reihe thermostatisierbarer
Quarzküvetten mit einem Probenvolumen von 0.8 bis 16 mL an. Die Rotation
einer leeren Küvette kann durch einen im Steurprogramm integrierten Menü-
142 Übernommen aus der Bedienungsanleitung für das Präzisionspolarimeter POL S-1 derFirma DRE DR. RISS ELLIPSOMETERBAU GmbH.143 Werbeprospekt für das Polarimeter POL S-1 der Firma LOT ORIEL.144 Genauigkeit bezogen auf den Mittelwert einer Mehrfachmessung.
Interferenzfilter undPolarisator auf
Schrittmotor montiert
Detektorfest orientierterPolarisator
ProbeLaser

EXPERIMENTELLER TEIL 49
punkt ermittelt und somit bei den Untersuchungen der Proben berücksichtigt
werden.145,146
6.2.1 Messsystematik.
Bei den in dieser Arbeit durchgeführten Experimenten wurde ausschließlich
eine zylindrische, thermostatisierbare Polarimeterküvette aus Quarzglas ver-
wendet. Die Länge der Küvette betrug 200 +/- 0.1 mm und hatte einen Innen-
durchmesser von 10mm, das Probenvolumen betrug 16 mL. Die Küvette verfügt
über einen Ein- und Auslass für die Temperierflüssigkeit. Diese sind über zwei
Silikonschläuche mit zwei Schnellkupplungen, inklusive Ventil, mit dem Tempe-
rierkreislauf verbunden. Durch die Betätigung der Schnellkupplung konnte die
Küvette problemlos aus dem Polarimeter entnommen werden.
Die wie in Kap. 5 aufbereiteten Proben wurden als Sol blasenfrei in die bereits
auf die gewünschte Messtemperatur eingestellte Messzelle mit Hilfe einer Pla-
stikspritze in die Küvette eingebracht. Hierdurch ergeben sich konform zur Mes-
sung mit dem Rheometer Abkühlzeiten, die innerhalb der Messraten von 2 s
liegen. Von Anbeginn der Messung können auch hier isotherme Bedingungen
zu Grunde gelegt werden.147
Während der Untersuchungen bei tiefen Temperaturen bildete sich an den
Küvettenfenstern Kondenswasser, das den ein- bzw. ausfallenden Laserstrahl
so stark streute, dass die Intensität des Lichtes am Detektor derart gering war,
was einen automatischen Abbruch der Messung zur Folge hatte. Zur Verhinde-
rung der Bildung von Kondenswasser, wurde das Polarimeter in einer Glove-
Box aufgestellt. Sämtliche Zuleitungen (Strom, Kühlung) wurden mit Hilfe von
Silikonstopfen und Silikonkleber so in die Glove-Box geführt, dass diese luft-
dicht abgeschlossen werden konnte. Nachdem die Probe in die Küvette einge-
bracht, und die Glove-Box geschlossen wurde, sorgte das darin ausreichend
verteilte wasserfreie Kieselgel dafür, dass störende Kodensationen an den
Küvettenfenstern ausblieben.
145 Riss (DRE DR. RISS ELLIPSOMETERBAU GmbH), persönliche Mitteilung146 Bedienungsanleitung vom 28.7.1997 für das Präzisionspolarimeter POL S-1 der Firma DREDR. RISS ELLIPSOMETERBAU GmbH.147 An dieser Stelle sei erwähnt, dass es dem Experimentator alleine nicht möglich ist, einesimultane Messung zu starten. Da die Messungen über die PC's ohne Zeitunterschied gestartetwerden müssen, bedarf es zweier Personen, die in gemeinsamer Absprache die Messzellenzeitgleich befüllen und die Messungen starten. In einer weiteren Entwicklung könnte dasBefüllen der Probenmesszellen automatisiert werden, sodass die Hilfe einer weiteren Personzum Befüllen der Probenmesszellen nicht mehr erforderlich wäre.

EXPERIMENTELLER TEIL 50
6.2.2 Berechnung der Messgrößen.
Das Steuerprogramm des Polarimeters ermöglicht es, wahlweise den Wert der
optischen Drehung bzw. den in Kap. 3 beschrieben Wert der spezifischen opti-
schen Drehung, automatisch zu bestimmen bzw. die Entwicklung dieser Werte
während der Gelierung online zu verfolgen. Die Messwerte wurden auf entspre-
chenden Datenträgern gespeichert und standen in dieser Form für die weiteren
Auswertearbeiten bzw. graphischen Darstellungen mit den geläufigen Mathe-
matikprogrammen zur Verfügung.

ERGEBNISSE UND DISKUSSION 51
ERGEBNISSE UND DISKUSSION
7 Übersicht der durchgeführten Messungen.
In der vorliegenden Arbeit wurden am System Gelatine / Wasser in einem Kon-
zentrationsbereich von 2 bis 8 Gew.-% der komplexe Schubmodul und die opti-
sche Drehung während der isothermen Gelierung bei verschiedenen Tempera-
turen simultan bestimmt. Die Geliertemperaturen wurden dabei so gewählt,
dass eine schnelle Gelierung zu erwarten war, und somit Messzeiten von
120 min ausreichten, um die Sol – Gel – Umwandlung zu beobachten.
Vor der Umstellung auf den PC gesteuerten Rheometermessplatz wurden am
System Gelatine / Wasser Messungen über einen Zeitbereich von 4000 min
durchgeführt. Anhand dieser Langzeitmessungen sollte ein Modell überprüft
werden, das den Verlauf des Speichermoduls in der Nähe und in weiter Entfer-
nung des Gelpunkts beschreibt.
Alle hier erwähnten Messungen wurden bei einer Frequenz von 6.28 rad⋅s-1 und
einer Anregungsspannung von 0.1 V durchgeführt. Zur Untersuchung der Fre-
quenzabhängigkeit des komplexen Schubmoduls wurde für eine Probe - in
einem Frequenzbereich von 1.256 rad⋅s-1 bis 37.68 rad⋅s-1 - die isotherme
Gelierung untersucht.
Die sich aus dem Ergebnis – und Diskussionsteil ergebenen neuen Auswerte-
verfahren sollen anhand eines Beispiels graphisch dargestellt und diskutiert
werden. Die Ergebnisse der übrigen Messungen sind in Tabellenform im
Anhang A-1 aufgeführt und werden im Text diskutiert.
Im Anhang A-3 sind die Messkurven aller simultan durchgeführten Messungen
aufgeführt.148
Die simultan durchgeführten Messungen hatten einerseits den Zweck, die wäh-
rend der Gelierung auftretende Änderungen der beiden Kenngrößen –
komplexer Schubmodul und optische Drehung - zu korrelieren (s. Kap. 8.4).
Andererseits sollten die rheologischen Kenngrößen hauptsächlich dazu benutzt
werden, um mit ihnen nach der Perkolationstheorie den Gelpunkt und die kriti-
148 Auf eine Diskussion dieser Gelierkurven soll verzichtet werden.

ERGEBNISSE UND DISKUSSION 52
schen Exponenten zu bestimmen. Die Kenntnis dieser Größen ist für eine qua-
litative und quantitative Beschreibung des kompletten Verlaufes des Speicher-
moduls von Bedeutung.
8 Die Bestimmung des Gelpunkts und der kritischen Exponenten.
Wie in Kap. 4.2 erwähnt, ist eine möglichst genaue Bestimmung des Gelpunkts
von großem industriellen Interesse. Anhand des rheologischen Verhaltens der
untersuchten Systeme lässt sich der Gelpunkt nach verschiedenen Methoden
bestimmen. Zwei Methoden sollen hier näher erläutert werden.
8.1 Frequenzabhängigkeit des komplexen Schubmoduls.
WINTER und Mitarbeiter fanden bei der Untersuchung der Gelierung chemisch
vernetzender Systeme, dass der zeitliche Verlauf des Relaxationsmoduls G(t)
durch ein Potenzgesetz beschrieben werden kann, welches streng genommen
nur in der Nähe des Gelpunkts Gültigkeit besitzt.149,150
∆−t~)t(G (8.1)
Für die Frequenzabhängigkeit des Speicher- und Verlustmoduls findet man
ebenfalls ein Potenzgesetz, das am Gelpunkt denselben Exponenten ∆ liefert.
( ) ∆ωωω ~)(''G~'G (8.2)
Hieraus ergibt sich, dass der Quotient aus Verlust– und Speichermodul, der
sogenannte Verlustwinkel δ, am Gelpunkt frequenzunabhängig ist:151,152,153
0
'G''G
tan ω==δ . (8.3)
149 Chambon F, Winter HH (1985) Polym Bull 13:499150 Winter HH, Chambon F (1986) J Rheol 30:67151 Winter HH, Chambon F (1986) J Rheol 30:67152 Chambon F, Winter HH (1987) J Rheol 31(8):683153 Cuvelier G, Launay B (1990) Makromol Chem, Macromol Symp 40:23
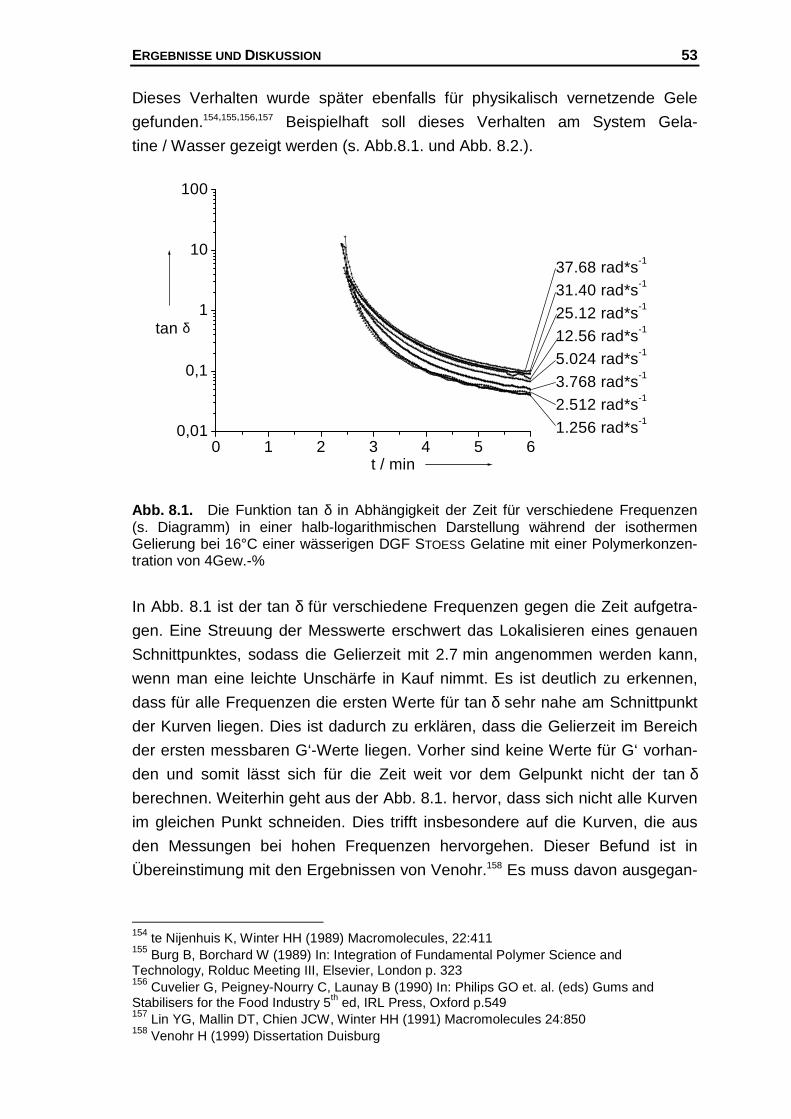
ERGEBNISSE UND DISKUSSION 53
Dieses Verhalten wurde später ebenfalls für physikalisch vernetzende Gele
gefunden.154,155,156,157 Beispielhaft soll dieses Verhalten am System Gela-
tine / Wasser gezeigt werden (s. Abb.8.1. und Abb. 8.2.).
Abb. 8.1. Die Funktion tan δ in Abhängigkeit der Zeit für verschiedene Frequenzen(s. Diagramm) in einer halb-logarithmischen Darstellung während der isothermenGelierung bei 16°C einer wässerigen DGF STOESS Gelatine mit einer Polymerkonzen-tration von 4Gew.-%
In Abb. 8.1 ist der tan δ für verschiedene Frequenzen gegen die Zeit aufgetra-
gen. Eine Streuung der Messwerte erschwert das Lokalisieren eines genauen
Schnittpunktes, sodass die Gelierzeit mit 2.7 min angenommen werden kann,
wenn man eine leichte Unschärfe in Kauf nimmt. Es ist deutlich zu erkennen,
dass für alle Frequenzen die ersten Werte für tan δ sehr nahe am Schnittpunkt
der Kurven liegen. Dies ist dadurch zu erklären, dass die Gelierzeit im Bereich
der ersten messbaren G‘-Werte liegen. Vorher sind keine Werte für G‘ vorhan-
den und somit lässt sich für die Zeit weit vor dem Gelpunkt nicht der tan δberechnen. Weiterhin geht aus der Abb. 8.1. hervor, dass sich nicht alle Kurven
im gleichen Punkt schneiden. Dies trifft insbesondere auf die Kurven, die aus
den Messungen bei hohen Frequenzen hervorgehen. Dieser Befund ist in
Übereinstimung mit den Ergebnissen von Venohr.158 Es muss davon ausgegan-
154 te Nijenhuis K, Winter HH (1989) Macromolecules, 22:411155 Burg B, Borchard W (1989) In: Integration of Fundamental Polymer Science andTechnology, Rolduc Meeting III, Elsevier, London p. 323156 Cuvelier G, Peigney-Nourry C, Launay B (1990) In: Philips GO et. al. (eds) Gums andStabilisers for the Food Industry 5th ed, IRL Press, Oxford p.549157 Lin YG, Mallin DT, Chien JCW, Winter HH (1991) Macromolecules 24:850158 Venohr H (1999) Dissertation Duisburg
0 1 2 3 4 5 60,01
0,1
1
10
100
37.68 rad*s-1
31.40 rad*s-1
25.12 rad*s-1
12.56 rad*s-1
5.024 rad*s-1
3.768 rad*s-1
2.512 rad*s-1
1.256 rad*s-1
tan δ
t / min

ERGEBNISSE UND DISKUSSION 54
gen werden, dass die Frequenzunabhängigkeit von tan δ am Gelpunkt nur für
geringe Frequenzen Gültigkeit besitzt.
Für die Zeit t = 2.7 min soll nun überprüft werden, ob die Beziehung in Gl. (8.2)
erfüllt ist. Hierzu ist in Abb. 8.2. der Logarithmus des Speicher -
bzw. Verlustmoduls gegen den Logarithmus der Frequenz aufgetragen.
Durch diese Darstellung wird deutlich, dass zum Zeitpunkt t = 2.7 min, eine
lineare Beziehung zwischen dem Speicher - bzw. Verlustmodul in Abhängigkeit
von der Frequenz vorliegt. Bis auf eine geringe Abweichung ergeben sich iden-
tische Werte für die Exponenten ∆G' und ∆G''. Durch die Mittelwertbildung wird ∆mit 0.695 berechnet. Diese Auswertungen bestätigen, dass die von WINTER für
chemisch vernetzende Systeme gemachte Theorie ebenso für physikalisch ver-
netzende Systeme Gültigkeit besitzt.
Abb.8.2. Der Verlustmodul G’’ und der Speichermodul G’ als Funktion der Frequenzω bei der Zeit t = 2.7 min in einer doppeltlogarithmischen Darstellung für eine wässeri-ge DGF STOESS Gelatine mit einer Polymerkonzentration von 4 Gew.-% bei 16°C
8.2 Auswertung nach der Perkolationstheorie.
Eine weitere Methode ist die Bestimmung des Gelpunkts gelierender Systeme
mit Hilfe der Perkolationstheorie. Grundlage für die Auswertung sind die in Kap.
4.2 aufgeführten Gln. (4.5) und (4.6). Diese beiden Gleichungen können nicht
0,1 1 10
0,01
0,1
1
∆G'
= 0.71∆
G''= 0.68
G' /
Pa
G''
/ Pa
ω / rad*s-1

ERGEBNISSE UND DISKUSSION 55
dazu herangezogen werden, den kompletten zeitlichen Verlauf des Speicher-
und Verlustmoduls zu beschreiben, denn die Perkolationstheorie besitzt nur
Gültigkeit in der Nähe der Perkolationsschwelle. Im hier vorliegenden Fall der
Gelierung, ist dies der Bereich um den Gelpunkt (s. Kap. 4). Für die Auswertung
sind folgende Fragen relevant: Wie nah ist "in der Nähe" der Perkolations-
schwelle?159 Welche experimentell ermittelten Werte dürfen bzw. müssen zur
Auswertung herangezogen werden?
MICHALCZYK und später VENOHR begegneten dem Problem auf folgende Weise.
Sie überführten die Perkolationsansätze Gln. (4.5) und (4.6) durch Logarithmie-
ren in eine linearisierte Form
)tt(logKlog''Glog ,gel −ν−= ηη (8.4)
)tt(logKlog'Glog G,gelG −µ+= (8.5)
und berücksichtigten bei ihren Auswertungen den Teil an Messwerten, der
durch Regressionsrechnungen nach Gln. (8.4) bzw. (8.5) den größten
Korrelationskoeffizienten lieferten. MICHALCZYK gab bei diesem Verfahren zu
bedenken, dass der Korrelationskoeffizient mit zunehmender Länge der für die
Regressionsrechnung verwendeten Zeitintervalle, in welchen die Messwerte
liegen, ein Maximum durchläuft.160 Es muss also hierbei die Anzahl der zur
Regressionsrechnung verwendeten Messwerte und damit auch die Länge des
Zeitintervalls solange variiert werden, bis der Korrelationskoeffizient maximal
wird. Im Falle einer Auswertung nach Gl. (8.5) beginnt das zu
berücksichtigende Zeitintervall immer mit dem ersten gemessenen Wert von G'.
Dieses birgt ein Problem, denn die Bestimmung des ersten Wertes von G' ist
abhängig von der Empfindlichkeit des verwendeten Rheometers. Wie oben
erwähnt, liegt die Empfindlichkeit des Rheometers bei 0.02 Pa. Wäre man in
der Lage, ein sensibleres Rheometer zu konstruieren, so würde das zur
Regression verwendete Zeitintervall vergrößert, was nicht ohne Einfluss auf die
Ergebnisse bliebe. Anders ausgedrückt: Man macht den Gültigkeitsbereich der
Perkolationstheorie abhängig von der Empfindlichkeit der verwendeten
Messeinrichtung.
An dieser Stelle könnte vorsichtig die Frage formuliert werden, ob der erste
bestimmte G'-Wert auf den reversibel gespeicherten Anteil der Arbeit, bedingt
durch ein bereits gebildetes Netzwerk, zurückzuführen ist, oder ob zu diesem
159 Venohr H (1999) Dissertation Duisburg160 Michalczyk A (1993) Dissertation Duisburg

ERGEBNISSE UND DISKUSSION 56
Zeitpunkt noch gar kein Netzwerk gebildet ist, und der geringe Beitrag des Spei-
chermoduls durch Verschlaufungen der Moleküle im Sol geleistet wird?161 Der
Gültigkeitsbereich der Perkolationstheorie würde hier ebenfalls verletzt, denn
der Einfluss der entanglements wird in der Theorie nicht berücksichtigt.
Welchen Einfluss die Breite des gewählten Zeitintervalls, bzw. die Anzahl der
Messwerte, auf die Bestimmung der Gelierzeit hat, lässt sich nur abschätzen.
Inwiefern sich die Gelierzeit auf die kritischen Exponenten auswirkt, wurde von
BORCHARD und Mitarbeitern am System Gelatine / Wasser untersucht.162 Sie
bestimmten für ein Zeitintervall, das wohlgemerkt über Regressionsrechnungen
nach den Gln. (8.4) und (8.5) den besten Korrelationskoeffizienten lieferte, eine
Gelierzeit von 7.0 min. Diese Zeit hoben sie um 10 s an, fixierten diesen Wert
und führten einen weiteren Itterationsschritt durch. Die kritischen Exponenten µund ν veränderten sich dabei um 10%. Hieraus lässt sich ableiten, dass zur
genauen Bestimmung der Gelierzeit der genaue Perkolationsbereich bekannt
sein muss. Eine genau bestimmte Gelierzeit ist wiederum Voraussetzung für
eine genaue Bestimmung der kritischen Exponenten.
Im folgenden Kapitel wird eine Methode beschrieben, die es ermöglicht, den
Gültigkeitsbereich der Perkolationstheorie scharf einzugrenzen und dabei nur
eine Gelierzeit tgel als Lösung zu liefern.
8.2.1 Die normierten Perkolationsansätze.
Bezieht man die Klammerausdrücke in den Gln. (4.5) und (4.6) auf eine
bestimmte Gelierzeit tgel, dann gehen die Gleichungen in sogenannte normierte
Perkolationsansätze über:
gelgelgel
ttfürttt
1K''G <⋅
−= ν−
ν−
η (8.6)
.ttfürt1tt
K'G gelgelgel
G >⋅
−= µ
µ
(8.7)
Die logarithmierten Ableitungen der Gln. (8.6) und (8.7) nach der Zeit liefern die
linearisierten Formen der normierten Perkolationsansätze:
161 Kästner S (1979) Polymer 20:1329162 Borchard W, Lechtenfeld M (1999) Colloid Polym Sci eingereicht

ERGEBNISSE UND DISKUSSION 57
( )
−+ν−
⋅
ν=
ν−η
gelgel
gel tt
1ln1tt
Kln
dt''dG
ln (8.8)
( ) .1tt
ln1tt
Kln
dt'dG
lngel
gelgel
G
−−µ+
⋅
µ=
µ (8.9)
Auswerteverfahren. Trägt man nach Gl. (8.8) den rechten Teil der Gleichung
gegen den linken Teil auf, dann weist die Kurve in ihrem Verlauf erst durch die
Vorgabe physikalisch sinnvoller Gelierzeiten einen linearen Bereich auf.163 tgel
wird solange variiert, bis der lineare Bereich maximal groß ist. Dieser Wert für
tgel wird dann als die Gelierzeit angenommen, die Messwerte die zu dem
linearen Bereich gehören bilden den Perkolationsbereich. In gleicher Weise
verfährt man bei der Auswertung nach Gl. (8.9). Es ist bemerkenswert, dass
man sowohl für die Auswertung des Verlust- als auch des Speichermoduls
einen maximalen linearen Bereich für die gleiche Gelierzeit erhält. Hinzukommt,
dass in beiden Fällen der lineare Bereich durch die gleiche Anzahl an
Messwerten gebildet wird. Dies gilt für alle in Tabelle A-1.1. aufgeführten
Systeme. Die Auswertung für eine 4 Gew.%ige Gelatine-Lösung ist in Abb. 8.3
bzw. Abb. 8.4 graphisch dargestellt.
Im Gegensatz zu den Auswertungen nach der Methode von MICHALCZYK bzw.
VENOHR, die den Perkolationsbereich ebenfalls aus einer linearisierten Form der
Perkolationsansätze ermitteln (s. S. 55), wird hier deutlich, dass die ersten
Werte für G' nicht dem Perkolationsbereich zugehören. Dies bestätigt die auf
Seite 55 aufgestellte Vermutung, dass die ersten Werte von G' durch Ver-
schlaufungen der Polymerketten (entanglements) hervorgerufen werden und
damit nicht zur Auswertung nach der Perkolationstheorie herangezogen werden
können.
163 Physikalisch sinnvolle Werte für die Gelierzeit sind solche im Zeitbereich um den erstenmessbaren Wert von G'. Es sollten keine Werte aus dem Bereich genommen werden, inwelchem die Viskosität noch keine deutlichen Anstiege zeigt, bzw. solche oberhalb desSchnittpunktes der G' und G'' Kurven.

ERGEBNISSE UND DISKUSSION 58
Abb. 8.3. Der natürliche Logarithmus der Ableitung des Verlustmoduls G'' nach derZeit gegen den natürlichen Logarithmus der normierten Zeitdifferenz entsprechend Gl.(8.8) für eine wässerige DGF STOESS Gelatine mit einer Polymerkonzentration von 4Gew.-% während der isothermen Gelierung bei 16°C. Die beiden von der Geradendeutlich abweichenden Werte wurden bei der Anpassung der Messwerte durch dieGerade nicht berücksichtigt
Abb. 8.4. Der natürliche Logarithmus der Ableitung des Speichermoduls G' nach derZeit gegen den natürlichen Logarithmus der normierten Zeitdifferenz entsprechend Gl.(8.9) für eine wässerige DGF Stoess Gelatine mit einer Polymerkonzentration von4 Gew.-% während der isothermen Gelierung bei 16°C. Der von der Geraden deutlichabweichende Wert wurde bei der Anpassung der Messwerte mit der Geraden nichtberücksichtigt
-1,4 -1,3 -1,2 -1,1 -1,0 -0,9
-3,0
-2,9
-2,8
-2,7
-2,6
-2,5
-2,4
-2,3
-2,2
1.63min
2.02min
tgel
= 2.65min
ln (
dG''/
dt)
ln (1 - t / tgel
)
-1,5 -1,4 -1,3 -1,2 -1,1 -1,0 -0,9-0,1
0,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8 3.67min
3.28min
tgel
= 2.65min
ln(d
G'/d
t)
ln (t / tgel
-1)

ERGEBNISSE UND DISKUSSION 59
Die linearen Bereiche in Abb. 8.3 und 8.4 beginnen in einer Entfernung von
0.63 min vor dem Gelpunkt und werden jeweils durch 12 Messwerte gebildet.
Dies entspricht einem Zeitbereich für den Verlauf von G'' von 1.63 bis 2.02 min
und für den Verlauf von G' von 3.28 bis 3.67 min. Diese schmalen Bereiche
stellen den Gültigkeitsbereich der Perkolationstheorie dar und müssen zur
Bestimmung der kritischen Exponenten herangezogen werden, will man nach
den Perkolationsansätzen gemäß der Gln. (4.5) und (4.6) auswerten.
Diese Auswertung ist in Abb. 8.5. bzw. 8.6. dargestellt und liefert als Lösung die
kritischen Exponenten ν = 0.58±0.01 bzw. µ = 2.07±0.05, gibt man für tgel
2.65 min vor. Die Diskussion der kritischen Exponenten anhand von Literatur-
werten bzw. anhand der Ergebnisse der anderen untersuchten Systemen soll
geschlossen in Kap. 8.3 erfolgen.
Für alle weiteren untersuchten Systeme ist im Anhang A-1 in der letzten Spalte
der Tab. A-1.1. in Betragsstrichen die Zeit aufgeführt, in welcher Entfernung
zum Gelpunkt der Perkolationsbereich anfängt und, angedeutet durch das
Pluszeichen, über welchen Zeitbereich sich der Perkolationsbereich erstreckt.
Abb. 8.5. Auswertung des zeitlichen Verlaufes des Verlustmoduls G" nach Gl. (4.5)für eine wässerige DGF STOESS Gelatine mit einer Polymerkonzentration von4 Gew.-% während der isothermen Gelierung bei 16°C
0,0 0,2 0,4 0,6 0,8 1,00,0
0,1
0,2
0,3
0,4
0,5
ν = 0.58 +/- 0.01tgel
= 2.65 min (vorgegeben)
G''
/ Pa
(tgel
- t) / min

ERGEBNISSE UND DISKUSSION 60
Abb. 8.6. Auswertung des zeitlichen Verlaufes des Speichermoduls G' nach Gl. (4.6)für eine wässerige DGF STOESS Gelatine mit einer Polymerkonzentration von 4 Gew.-% während der isothermen Gelierung bei 16°C
Durch die normierten Perkolationsansätze ist man nun in der Lage eine Gelier-
zeit zu ermitteln und den für die Perkolationstheorie relevanten oder gültigen
Bereich vor und nach dem Gelpunkt anzugeben. Dies soll im folgenden Kapitel
schematisch dargestellt werden. Vorerst sollen aber die in diesem Kapitel
bestimmten Gelierzeiten verglichen werden.
Vergleich der Gelierzeiten. Anhand der rheologischen Messgrößen wurde für
das gleiche System Gelatine / Wasser mit gleicher Polymerkonzentration bei
der gleichen Geliertemperatur der Gelpunkt aus der Frequenzabhängigkeit des
komplexen Schubmoduls mit ca. 2.7 min bzw. aus den normierten Perkola-
tionsansätzen mit 2.65 min bestimmt. Berücksichtigt man die Schwierigkeiten
bei der Schnittpunktsbestimmung der Messkurven in Abb. 8.1., so kann man
von einer sehr guten Übereinstimmung der Gelierzeiten sprechen.
In der vorliegenden Arbeit wurde lediglich für ein Gelierexperiment die Gelierzeit
aus der Frequenzabhhängigkeit ermittelt, um einen Vergleich mit der aus den
normierten Perkolationsansätzen bestimmten Gelierzeit aufzustellen. Für alle
weiteren untersuchten Systeme wurden die Gelierzeiten nach den normierten
Perkolationsansätzen bestimmt und die Ergebnisse im Anhang A-1 in Tab. A-
1.1. aufgeführt.
0,0 0,2 0,4 0,6 0,8 1,00,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
µ = 2.07 +/- 0.05tgel
= 2.65 min (vorgegeben)
G' /
Pa
(t - tgel
) / min
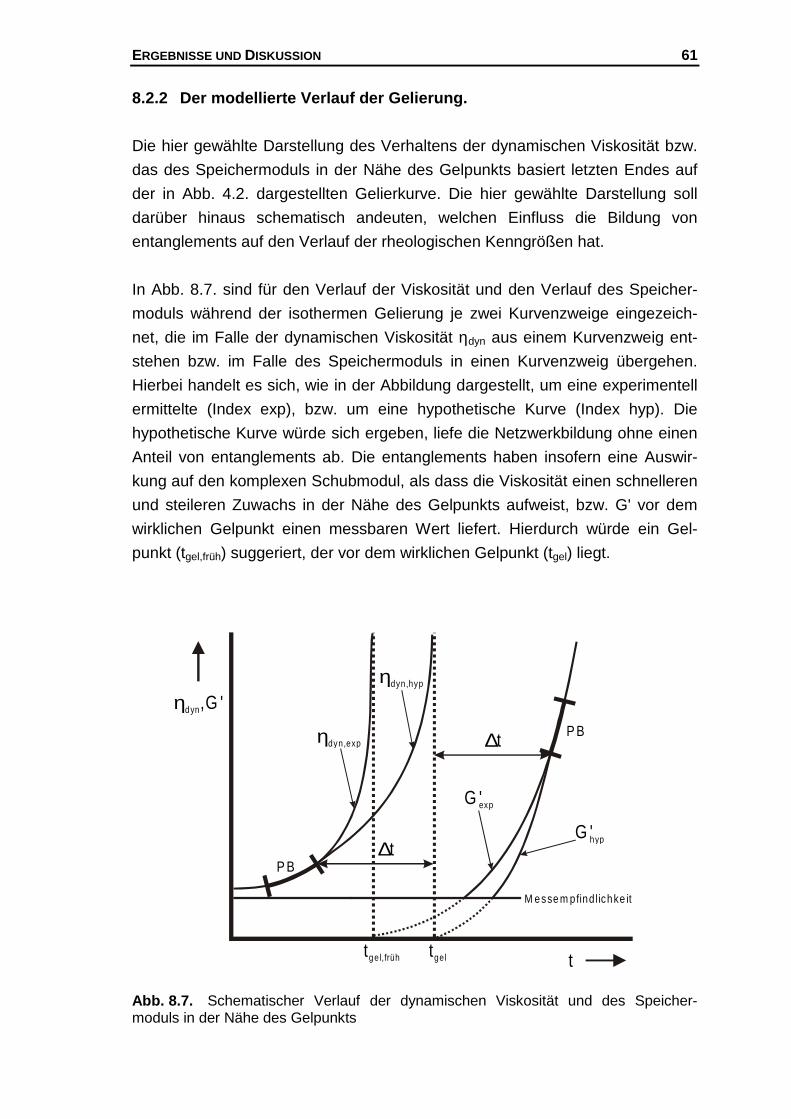
ERGEBNISSE UND DISKUSSION 61
8.2.2 Der modellierte Verlauf der Gelierung.
Die hier gewählte Darstellung des Verhaltens der dynamischen Viskosität bzw.
das des Speichermoduls in der Nähe des Gelpunkts basiert letzten Endes auf
der in Abb. 4.2. dargestellten Gelierkurve. Die hier gewählte Darstellung soll
darüber hinaus schematisch andeuten, welchen Einfluss die Bildung von
entanglements auf den Verlauf der rheologischen Kenngrößen hat.
In Abb. 8.7. sind für den Verlauf der Viskosität und den Verlauf des Speicher-
moduls während der isothermen Gelierung je zwei Kurvenzweige eingezeich-
net, die im Falle der dynamischen Viskosität ηdyn aus einem Kurvenzweig ent-
stehen bzw. im Falle des Speichermoduls in einen Kurvenzweig übergehen.
Hierbei handelt es sich, wie in der Abbildung dargestellt, um eine experimentell
ermittelte (Index exp), bzw. um eine hypothetische Kurve (Index hyp). Die
hypothetische Kurve würde sich ergeben, liefe die Netzwerkbildung ohne einen
Anteil von entanglements ab. Die entanglements haben insofern eine Auswir-
kung auf den komplexen Schubmodul, als dass die Viskosität einen schnelleren
und steileren Zuwachs in der Nähe des Gelpunkts aufweist, bzw. G' vor dem
wirklichen Gelpunkt einen messbaren Wert liefert. Hierdurch würde ein Gel-
punkt (tgel,früh) suggeriert, der vor dem wirklichen Gelpunkt (tgel) liegt.
Abb. 8.7. Schematischer Verlauf der dynamischen Viskosität und des Speicher-moduls in der Nähe des Gelpunkts
∆t
tgel,früh tgel t
ηdyn,G 'ηdyn,hyp
ηdyn,exp
G 'exp
G 'hyp∆t
P B
P B
M esse m pfind lichke it

ERGEBNISSE UND DISKUSSION 62
Zu Beginn des Gelierexperiments ist die Viskosität in der Solphase gering, d.h.
mögliche Verschlaufungen der Polymerketten können sich leicht wieder lösen,
denn ein Abgleiten der Ketten gegeneinander ist fast uneingeschränkt möglich.
Dazu kommt, dass die Konzentration der Helices zu diesem Zeitpunkt sehr
gering ist, und daher kaum Polymerketten in Netzwerkpunkten fixiert sind, was
deren freie Beweglichkeit einschränken würde. Zu diesem Zeitpunkt ist der Ein-
fluss der entanglements auf die Viskosität vernachlässigbar gering. Mit voran-
schreitender Reaktion nimmt die Helixkonzentration zu und immer mehr Poly-
merketten finden sich über die Aggregation der helikalen Bereiche zu Mole-
külclustern zusammen. Die Anzahl und Größe der Cluster wirken sich im ent-
sprechenden Maße auf die Viskosität aus. Zu diesem Zeitpunkt ist es durchaus
denkbar, dass eine oder mehrere Polymerketten, welche in einem Cluster fixiert
sind, einen benachbarten Cluster durchdringen, bzw. mit einer in diesem
Cluster eingebauten Polymerkette eine Verschlaufung (entanglement) bildet
und wieder in den urprünglichen Cluster zurückläuft bzw. in einen weiteren
benachbarten Cluster eindringt. Dieser Effekt würde sich entscheidend auf die
Viskosität auswirken, was durch eine Abweichung des experimentellen Kurven-
verlaufes von dem hypothetischen in einem Abstand ∆t vom Gelpunkt (tgel) ver-
deutlicht werden soll. Es sei bereits an dieser Stelle erwähnt, dass die Perkola-
tionstheorie den Einfluss der entanglements nicht berücksichtigt.
Im Gel liegen die Dinge genau anders. Hier wird der erste detektierbare Wert
des Speichermoduls, der das erste Auftreten eines unendlich großen Mole-
külclusters signalisiert, durch den Einfluss der entanglements beeinflusst.
Gerade in der Nähe des Gelpunkts ist es denkbar, dass die entanglements den
Verbund der unterschiedlichen Cluster, die letztlich den unendlich großen
Cluster bilden, besonders stärken. Hat sich durch die immer weiter steigende
Anzahl an Netzwerkpunkten das Gel immer engmaschiger vernetzt, dann dürfte
die Netzwerkstabilität so hoch sein, dass der Einfluss der entanglements ver-
nachlässigbar wird. Dieses Verhalten wird durch das Zusammenlaufen der
experimentellen bzw. hypothetischen Kurven in einer Entfernung ∆t zum Gel-
punkt angedeutet.
Dieser ∆t- Bereich entspricht der in Kap. 8.2.1 bestimmten Zeit, welche die Ent-
fernung des maximalen linearen Bereiches vom Gelpunkt angibt. Oberhalb bzw.
unterhalb dieser Zeit können die Einflüsse der entanglements vernachlässigt
werden, sodass von diesem Zeitpunkt an nach der Perkolationstheorie ausge-
wertet werden kann bzw. sollte. Dies macht Sinn, denn wie oben erwähnt
berücksichtigt die Perkolationstheorie einzig und alleine nur Cluster, die durch
das Knüpfen von neuen Bindungen (in diesem Fall Aggregation der Helices)

ERGEBNISSE UND DISKUSSION 63
zwischen Molekülen zustandekommen. Alle weiteren Einflüsse werden nicht
berücksichtigt. Dies gilt für das Clusterwachstum im Sol wie auch im Gel.
Der in den Kurven angegebene Bereich PB ist die Länge (Größe) des Perkola-
tionsbereiches, bestimmt durch die Linearisierung der normierten Perkolations-
ansätze Kap.8.2.1. Für alle weiteren untersuchten Systeme sind die Perkola-
tionsbereiche im Anhang A-1 in Tab. A-1.1. aufgeführt. Beim Vergleich dieser
Werte wird deutlich, dass mit steigender Gelierzeit der Perkolationsbereich grö-
ßer wird. Entsprechendes gilt auch für den Abstand des Perkolationsbereiches
zum Gelpunkt. Dies sei jedoch nur summarisch erwähnt, denn bei den 2- und 4-
Gew.-%igen Gelatine-Lösung trifft dies nicht 100%ig zu. Ob dies nur zufälligen
Charakter hat, kann nur geklärt werden, wenn Auswertungen aus zusätzlichen
Messungen in einem umfangreicheren Temperaturbereich erfolgen.
Weiterhin ist in Abb. 8.7. die durch das Rheometer vorgegebene Empfindlich-
keit von 0.02 Pa (s. Kap. 6.1.1) durch einen Strich angedeutet. Es zeigt sich
also, dass der Gelpunkt vor dem ersten Messwert der G' Kurve liegen muss,
was im Experiment auch gefunden wird.
An dieser Stelle sei erwähnt, dass die Perkolationstheorie streng nur für Mes-
sungen bei einer Frequenz von ω→0 Gültigkeit besitzt. Wertet man Messungen
bei endlichen Frequenzen nach der Perkolationstheorie aus, begeht man dem-
nach einen Fehler. Dieser Fehler ist unumgänglich, denn es kann kein dynami-
sches Experiment durchgeführt werden, ohne mit einer endlichen Frequenz
anzuregen. Den Einfluss der Frequenz auf die Ergebnisse der Auswertung nach
der Perkolationstheorie wurde von BORCHARD und Mitarbeiter untersucht. Sie
konnten zeigen, dass die Reduzierung der in dieser Arbeit verwendeten
Frequenz von 6.28 rad⋅s-1 auf 1.256 rad⋅s-1 keinen signifikanten Einfluss auf die
Gelierzeit hat.164 Die Durchführung weiterer Messungen bei noch niedrigeren
Frequenzen ist durch die in Kap. 6.1.1 erwähnten Schwierigkeiten beim Ver-
suchsaufbau mit der erforderlichen Qualität nicht möglich.
8.2.3 Die kombinierten Perkolationsansätze.
Werten man die Gelierkurven nach den in Kap. 8.2.1 erwähnten normierten
Perkolationsansätzen aus, erhält man die überaus wichtige Information über
den gültigen Perkolationsbereich sowie eine einzige Lösung für die Gelierzeit.
164 Borchard W, Lechtenfeld M (2001) Mat Res Innovat II Vol 4 No.5-6 p. 381

ERGEBNISSE UND DISKUSSION 64
Hieraus lassen sich dann weiterhin die kritischen Exponenten ν bzw. µ bestim-
men.
Eine weitere elegante Methode zur Bestimmung der kritischen Exponenten
basiert auf der Kombination des Speicher – und Verlustmoduls symmetrisch um
den Gelpunkt. Es handelt sich bei diesen sogenannten kombinierten Perkola-
tionsansätzen (Combined Funktions) um eine Kombination der Größen in äqui-
distanten Abständen zum Gelpunkt innerhalb der zuvor bestimmten Perkola-
tionsbereiche.165,166 Dies führt zu neuen Bestimmungsgleichungen, aus denen
die kritischen Exponenten bestimmt werden können.
Für die Formulierung der CF werden die äquidistanten Abstände zum Gelpunkt
durch den Betrag It-tgelI ausgedrückt. Es soll gelten:
( ) ( ) .tttttt gelgelgel −=−=− (8.10)
Hierdurch gehen die Gln. (4.5) und (4.6), unter der Annahme das tgel,η = tgel,G ist,
über in:ν−
η −= gelttK''G (8.11)
.ttK'G gelGµ−= (8.12)
Bildet man sowohl für Gl. (8.11) als auch für Gl. (8.12) den natürlichen Log-
arithmus und leitet die Ausdrücke nach der Zeit ab, so erhält man:
geltt
1dt
''Glnd
−⋅ν−= (8.13)
.tt
1dt
'Glnd
gel−⋅µ= (8.14)
Setzt man die Gln. (8.13) und (8.14) gleich, so erhält man den Ausdruck:167,168
.dt
''Glnddt
'Glnd ⋅νµ−= (8.15)
165 Borchard W, Lechtenfeld M (2001) Mat Res Innovat II Vol 4 No.5-6 p. 381166 Borchard W, Lechtenfeld M (1999) Colloid Polym Sci eingereicht167 Borchard W, Lechtenfeld M (2001) Mat Res Innovat II Vol 4 No.5-6 p. 381168 Borchard W, Lechtenfeld M (1999) Colloid Polym Sci eingereicht

ERGEBNISSE UND DISKUSSION 65
Man erwartet demnach eine lineare Relation mit einer Steigung (-µ/ν), wenn
man die linke Seite der Gl. (8.15) gegen die rechte aufträgt.
Aus dieser Relation alleine erhält man jedoch nur ein Verhältnis der kritischen
Exponenten, nicht deren einzelnen Wert. Hierzu ist die Formulierung einer wei-
teren Relation erforderlich.
Bildet man in Gl (8.16) die Differenz aus den Gln. (8.14) und (8.15) bzw. den
reziproken Wert Gl. (8.17)169,170
( )geltt
1dt
''Glnddt
'Glnd
−⋅ν−µ=− (8.16)
,)(
tt
dt''Glnd
dt'Glnd gel
1
ν−µ
−=
−
−(8.17)
dann erwartet man eine lineare Relation mit der Steigung 1/(µ-ν), wenn der
linke Teil der Gl. (8.17) gegen den rechten Teil aufgetragen wird. Für beide
Kurvenverläufe gilt, dass nahe bzw. direkt am Gelpunkt die Ausdrücke auf bei-
den Seiten der Gleichungen Null werden müssen, d.h. die Geraden müssen
durch den Koordinatenursprung verlaufen. Bezeichnet man die Steigung der
Geradengleichung (8.15) mit A = (µ/ν) und die Steigung der Geradengleichung
(8.17) mit B = 1/(µ-ν), so lassen sich zwei Ausdrücke angeben, mit denen die
kritischen Exponenten ausgerechnet werden können:
)1A(B1−
=ν und)1A(B
A−
=µ (8.18)
Die hier angeführten Relationen sollen auf die Gelierung einer wässerigen
Gelatine Lösung angewendet werden.
169 Borchard W, Lechtenfeld M (2001) Mat Res Innovat II Vol 4 No.5-6 p. 381170 Borchard W, Lechtenfeld M (1999) Colloid Polym Sci eingereicht
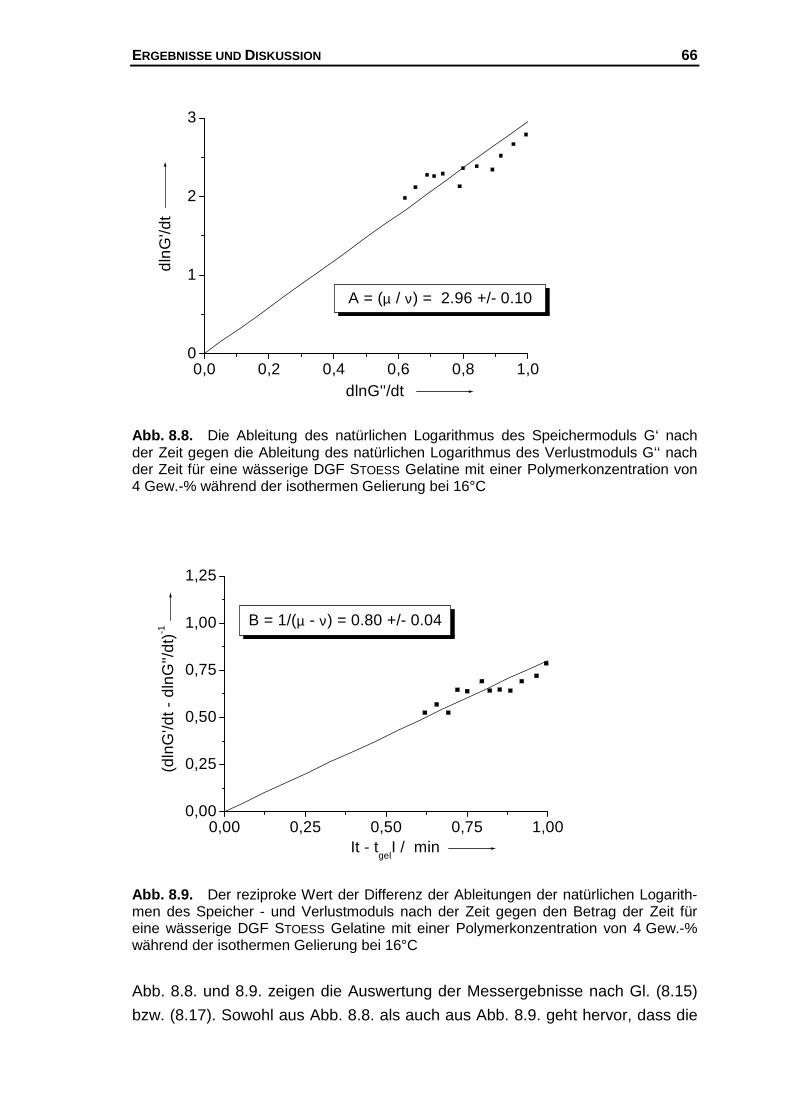
ERGEBNISSE UND DISKUSSION 66
Abb. 8.8. Die Ableitung des natürlichen Logarithmus des Speichermoduls G‘ nachder Zeit gegen die Ableitung des natürlichen Logarithmus des Verlustmoduls G‘‘ nachder Zeit für eine wässerige DGF STOESS Gelatine mit einer Polymerkonzentration von4 Gew.-% während der isothermen Gelierung bei 16°C
Abb. 8.9. Der reziproke Wert der Differenz der Ableitungen der natürlichen Logarith-men des Speicher - und Verlustmoduls nach der Zeit gegen den Betrag der Zeit füreine wässerige DGF STOESS Gelatine mit einer Polymerkonzentration von 4 Gew.-%während der isothermen Gelierung bei 16°C
Abb. 8.8. und 8.9. zeigen die Auswertung der Messergebnisse nach Gl. (8.15)
bzw. (8.17). Sowohl aus Abb. 8.8. als auch aus Abb. 8.9. geht hervor, dass die
0,0 0,2 0,4 0,6 0,8 1,00
1
2
3
A = (µ / ν) = 2.96 +/- 0.10
dlnG
'/dt
dlnG''/dt
0,00 0,25 0,50 0,75 1,000,00
0,25
0,50
0,75
1,00
1,25
B = 1/(µ - ν) = 0.80 +/- 0.04
(dln
G'/d
t - d
lnG
''/dt
)-1
It - tgel
I / min

ERGEBNISSE UND DISKUSSION 67
Erwartung eines linearen Kurvenverlaufs nicht optimal erfüllt wird, denn die
Werte streuen doch erheblich. Eine saubere Auswertung der Messkurven nach
den CF erfordert absolut rauschfreie Werte der rheologischen Kenngrößen G'
und G''. Insbesondere die G''- Werte unterliegen zu Beginn der Messung einem
sehr starken Rauschen d.h. die Messwerte nehmen nicht kontinuierlich zu.
Hieraus ergeben sich bei den Ableitungen nach der Zeit, viele negative Stei-
gungen von denen kein natürlicher Logarithmus gebildet werden kann. Die
Auswertung wäre daher nicht, oder zumindest nur äußerst bedingt möglich.
Dies wird weitestgehend verhindert, indem alle Rohdaten einer Glättungsproze-
dur unterzogen werden.171
In Tab. 8.1. sind die kritischen Exponenten, ermittelt aus den normierten Per-
kolationsansätzen und der aus den CF für das im Laufe der Arbeit ausgewählte
Beispiel für das System Gelatine / Wasser, gegenübergestellt.
Tab. 8.1. Vergleich der kritischen Exponenten ν und µ bestimmt für das SystemGelatine / Wasser mit einer Polymerkonzentration von 4 Gew.-% bei einer Temperaturvon 16°C nach den normierten Perkolationsansätzen bzw. der CF
ν µ
normierte
Perkolationsansätze0.58±0.01 2.07±0.05
CF 0.63±0.06 1.88±0.13
Berücksichtigt man die Fehler, kann man trotzdem von einer sehr guten Über-
einstimmung der kritischen Exponenten sprechen.
Die CF stellen demnach eine elegante Lösung zur Bestimmung der kritischen
Exponenten dar, wenn die Gelierzeit genau genug bestimmt wurde. Eine Kom-
bination der Größen um den Gelpunkt ist auf unterschiedlichen Wegen möglich.
Dies können Kombinationen höherer Ableitungen sein bzw. verschiedenartige
Verknüpfungen der Perkolationsansätze. Drei weitere Beispiele anderer CF
werden im Anhang A-2. berechnet.
Die hier aufgeführten, nach den verschiedenen Methoden bestimmten Expo-
nenten, sind neben weiteren für unterschiedliche Konzentrationen und Tempe-
raturen bestimmten Systemen im Anhang A-1 in den Tabellen A-1.1. und A-1.2.
im Anhang aufgeführt. Ein Vergleich der hier aufgelisteten kritischen Exponen-
171 Durch die Glättungsprozedur wird ein gleitender Durchschnitt durch die Bildung einesMittelwertes benachbarter Werte gebildet.

ERGEBNISSE UND DISKUSSION 68
ten zeigt, dass die Fehler bei dieser Methode wesentlich stärker ausfallen kön-
nen.
Ergebnisse der klassischen Theorie. Die Ergebnisse der klassischen Theorie
liefern die kritischen Exponenten ν = 0 und µ = 3, die Ergebnisse der Perkola-
tionstheorie liefern für die Exponenten Werte im Bereich 0.65 ≤ ν ≤ 1.35 und
1.3 ≤ µ ≤ 3.78 (s. Kap. 4.2 und 4.3). Hieraus lässt sich ableiten, dass nach der
klassischen Theorie der Wert für den Quotienten (µ/ν) divergieren müsste,
nach der Perkolationstheorie hingegen endliche Werte im Bereich von
1 ≤ (µ/ν) ≤ 5.8 liefern sollte.172 In Abb. 8.8. ist die Steigung der Geraden
(µ/ν) mit 2.96±0.09 berechnet worden. Der Wert liegt damit eindeutig im
Bereich der Werte, die von der Perkolationstheorie vorhergesagt wurden. In
keiner der untersuchten Fälle wird eine Gerade mit unendlicher Steigung
gefunden, so dass davon ausgegangen werden kann, dass die klassische
Theorie keine Gültigkeit besitzt, bzw. anders ausgedrückt, das kritische Phä-
nomen der Gelierung nicht beschreiben kann.173
Im folgenden Kapitel sollen die ermittelten kritischen Exponenten diskutiert wer-
den.
8.3 Diskussion der kritischen Exponenten.
Im Vergleich zu der Arbeit von VENOHR wurden in der vorliegenden Arbeit unter
anderen Gesichtspunkten die kritischen Exponenten nach der Perkolationstheo-
rie bestimmt. Es musste also davon ausgegangen werden, dass die Ergebnisse
möglicherweise stark voneinander abweichen. Tatsächlich sind die in dieser
Arbeit bestimmten Werte für ν und µ zu höheren Werten hin verschoben.
8.3.1 Der kritische Exponent νν.
Hier findet VENOHR Werte im Bereich von 0.04 bis 0.40. Die in dieser Arbeit
ermittelten Werte liegen mit einer Ausnahme bei dem für eine 4 Gew.-%ige
Gelatine-Lösung ν mit 1.06±0.02 bestimmt wurde, im Bereich von 0.55 bis 0.76.
Die Werte stimmen damit sehr gut mit dem von SAHIMI und ARBABI vorherge-
sagten Wert von ν ≈ 0.65, für das sogenannte ZIMM-Regime überein. Es ist
demnach davon auszugehen, dass am Gelpunkt starke hydrodynamische
172 Borchard W, Lechtenfeld M (2001) Mat Res Innovat II Vol 4 No.5-6 p. 381173 Borchard W, Lechtenfeld M (1999) Colloid Polym Sci eingereicht

ERGEBNISSE UND DISKUSSION 69
Wechselwirkungen zwischen den Gelatinemolekülen und nur geringe Diffusion
stattfinden.174,175 Die hier ermittelten kritischen Exponenten bestätigen die
Ergebnisse von BORCHARD, der ebenfalls für das System Gelatine / Wasser
einen kritischen Exponenten von ν = 0.61 bestimmt und in diesem Zusammen-
hang das ZIMM-Regime diskutiert hat.176
8.3.2 Der kritische Exponent µµ.
Hier findet VENOHR Werte im Bereich von 0.8 bis 2.25. Die in dieser Arbeit
ermittelten Werte liegen in einem Bereich zwischen 1.75 bis 2.27. Die Werte
von µ fallen demnach in den Bereich, des von DE GENNES für die Annahme
einer Analogie zwischen Elastizität und Leitfähigkeit eines Netzwerkes aus Iso-
latoren und Leitern vorhergesagten Wertes von µ = 1.7,177 bzw. dem von SAHIMI
vorhergesagten Wert von µ = 2.1,178 der sich für Modellrechnungen ergibt, wenn
das Dehnvermögen einer Bindung im Netzwerk berücksichtigt wird (s. Kap.
4.3).
Die hier ermittelten Werte für den kritischen Exponenten µ werden ebenfalls
durch Literaturwerte gestützt, bei denen für das System Gelatine / Wasser die
rheologischen Experimente nach der Perkolationstheorie ausgewertet wur-
den.179,180,181
Es muss demnach davon ausgegangen werden, dass sehr ähnliche molekulare
Abläufe während der Gelierung stattfinden und daher die Systeme einer Uni-
versalitätsklasse zuzuordnen sind.
Sehr kritisch betrachtet liegen die in dieser Arbeit ermittelten Werte für µ nicht
unbedeutend auseinander. Betrachtet man hierzu noch die Zusammenstellung
der Ergebnisse in Tab. A-1.1. wird deutlich, dass die Schwankungen für eine
Konzentration annähernd gleich groß sind. Für jede Konzentration gibt es
sowohl hohe als auch niedrige Werte für µ. Bei näherem Betrachten der Ergeb-
nisse deutet sich bei konstanter Konzentration ein Maximum des kritischen
174 Arbabi S, Sahimi M (1990) Phys Review Lett 65:725175 Sahimi M (1992) Mod Phys Lett B 6:507176 Borchard W (1998) Ber Bunsenges Phys Chem 102:1580177 de Gennes PG (1979) Scalling Concepts in Polymer Physics, Cornell University Press,Ithaca New York178 Sahimi M (1992) Mod Phys Lett B 6:507179 Djabourov M, Leblond J, Papon P (1988) J Phys (France) 49:333180 Djabourov M, Lechaire JP, Gaill F (1993) Biorheology 30:191181 Kumagai H, Fujii T, Inukai, T, Yano T (1993) Biosci Biotech Biochem 57(4):532

ERGEBNISSE UND DISKUSSION 70
Exponenten µ als Funktion der Geliertemperatur an, eine Tatsache, die von
VENOHR auch gefunden, aber nicht gedeutet wird.182 Eine Bestätigung dieses
Sachverhalts kann anhand der in Tab. A-1.1. aufgeführten geringen Anzahl
Messungen nur mit einem "unwohlen" Gefühl gemacht werden. In Kap. 9 wer-
den Langzeitmessungen am System Gelatine / Wasser diskutiert, die im Rah-
men der Arbeit nach dem alten Messprinzip erfolgten sowie nach der Perkola-
tionstheorie im Sinne von VENOHR ausgewertet wurden. Die Messergebnisse
sind in der Tab. A-1.3. im Anhang aufgeführt und zeigen ebenfalls, dass der
kritische Exponent µ als Funktion der Geliertemperatur ein Maximum durchläuft.
Hier soll nun eine "mögliche" Erklärung für diesen Befund gegeben werden, der
folgende Annahmen zu Grunde liegen.
1.) Für eine Polymerkonzentration existiert eine konstante Zahl potentieller
Bindungsstellen.
2.) Am Gelpunkt ist unabhängig von der Polymerkonzentration die Zahl der
Helices pro Volumen konstant.183,184
3.) Die Bildung von Doppel- und Dreifachhelices wird ausgeschlossen.185
Des Weiteren werden die aus Modellrechnungen vorhergesagten kritischen
Exponenten µ (s. Kap. 4.3) dazu benutzt, um folgendes molekulares Bild
bezüglich der Netzwerkstabilität am Gelpunkt abzuleiten. Es wird davon ausge-
gangen, dass bei großen kritischen Exponenten das Netzwerk am Gelpunkt
einen entsprechend großen Widerstand gegen eine äußere Krafteinwirkung lei-
sten kann. Insbesondere die von SAHIMI bzw. ARBABI und SAHIMI gemachten
Berücksichtigungen des Dehnvermögens elastischer Elemente, die einen Wert
von µ = 2.1 liefern können und bei Annahme von Biegeanteilen im Netzwerk
sogar bis hin zu µ = 3.75 ansteigen können, lassen diesen Schluss zu.
Es soll zwischen einer schnell ablaufenden Gelierung, die bei tiefen Temperatu-
ren zu erwarten ist, und einer mittelschnellen bzw. langsam ablaufenden Gelie-
rung (mittleren bzw. hohen Temperaturen) unterschieden werden.
Die Gelierung bei tiefen Temperaturen. Dies entspricht einer sehr raschen
Gelierung und einem kleinen Wert von µ, d.h. es liegt ein schwaches Netzwerk
am Gelpunkt vor. Es handelt sich hier um ein gummielastisches Netzwerk, bei
182 Venohr H (1999) Dissertation Duisburg183 Djabourov M, Leblond J, Papon P (1988) J Phys (France) 49:319184 Maibaum R (1998) Dissertation Duisburg185 siehe hierzu Kap. 1.3.2

ERGEBNISSE UND DISKUSSION 71
dem die Knotenpunkte nicht fluktuieren und im wesentlichen Konformations-
änderungen bei der Dehnung unterliegen.
Die schnelle Gelierung verhindert die ideale Ausbildung der Helices - sie errei-
chen u.U. nur einen Teil ihrer möglichen Länge, woraus ein geringer Überlap-
pungsgrad (Kontaktfläche der Helices im Aggregat) bei der Aggregation der
Helices resultiert.
Die durch die tiefen Temperaturen eingeschränkte Beweglichkeit der Polymer-
ketten unterbindet eine mögliche Fluktuation der Knotenpunkte. Es liegt keine
ausreichende Triebkraft vor, um zwei nicht ideal gestapelte (aggregierte, über-
lappte) helikale Bereiche zu lösen, die daraufhin erneut mit einem höheren
Überlappungsgrad durch einen Reifeprozess im Sinne einer Ostwaldreifung
aggregieren. Der Prozess der Ostwaldreifung kann sich dabei über einen länge-
ren Zeitraum erstrecken. BORCHARD und Mitarbeiter konnten zeigen, dass eine
Ostwaldreifung erst nach 4 Tagen beendet ist.186
helicale Bereiche
Abb. 8.10. Schematische Darstellung der Helixanordnung im System Gela-tine / Wasser am Gelpunkt in Folge einer schnellen Gelierung (tiefe Temperaturen)
Es ist anzunehmen, dass die Ausbildung intermolekularer H – Brückenbindun-
gen bei tiefen Temperaturen gegenüber den intramolekularen H –
Brückenbindungen, welche fluktuieren können, bevorteilt sind. Dieses Ver-
halten der Helixbildung bzw. deren Aggregation hätte ein schwächeres Netz-
werkgefüge am Gelpunkt zur Folge, was durch ein kleines µ experimentell
bestätigt wird. Diese Modellvorstellung kollidiert nicht mit der Tatsache, dass bei
tiefen Temperaturen die Gelierkurve schnell in einen entsprechend hohen Pla-
186 Borchard W, Bergmann K, Emberger A, Rehage G (1976) Progr. Colloid & Polymer Sci60:120

ERGEBNISSE UND DISKUSSION 72
teauwert für den Speichermodul übergeht, denn die Helixbildungsgeschwindig-
keit ist bei tiefen Temperaturen größer als bei höheren Geliertemperaturen.
Hinzu kommt, dass die Zeit nach dem Gelpunkt nicht mehr an die Bedingung
geknüpft ist, dass im Netzwerk die gleiche Helixkonzentration vorliegen muss,
wie es am Gelpunkt für Systeme unterschiedlicher Gelatinekonzentrationen
gegeben ist.
Die Gelierung bei mittleren Temperaturen. Hier findet man große Werte für µ,
d.h. es liegt ein festes Netzwerk am Gelpunkt vor, in dem u.U. die Biegeanteile
bei der Deformation von Bedeutung sind. Das Netzwerk besteht hier aus relativ
steifen Ketten, die bei der Deformation einer Biegung unterliegen. Die Helices
können sich hier, im Verhältnis zu denen bei sehr kurzen Gelierzeiten, fast
uneingeschränkt ausbilden. Es liegt hier offensichtlich ein ideales Wechselspiel
zwischen intermolekularen und intramolekularen H - Brückenbindungen vor.
Einerseits führt jetzt das erhöhte Bestreben, intramolekulare H -
Brückenbindungen auszubilden, im Vergleich zu dem Netzwerk bei tiefen
Temperaturen, zu einer erhöhten Fluktuation der Knotenpunkte, woraus eine
größere Kontaktfläche der aggregierten Helices (hoher Überlappungsgrad)
resultiert. Andererseits sind die Helixaggregate aufgrund der noch beträchtlich
ausgebildeten intermolekularen H - Brückenbindungen dicht und sehr stabil
gestapelt. Demnach besitzt das so entstandenen Netzwerk am Gelpunkt eine
nicht zu vernachlässigende Biegefestigkeit, was durch ein teilweise sehr hohes
µ von fast 2.3 bestätigt wird. Eine ideale und uneingeschränkte Ausbildung gro-
ßer Helixstapel im Netzwerk ist unwahrscheinlich, da davon ausgegangen wer-
den muss, dass die Helices nicht immer ideal, d.h. über die gesamte Länge der
Helices, aggregieren können.
helicale Bereiche
Abb. 8.11. Schematische Darstellung der Helixanordnung im System Gela-tine / Wasser am Gelpunkt in Folge einer "mittelschnellen" Gelierung (mittlere Tempe-raturen)

ERGEBNISSE UND DISKUSSION 73
Die Gelierung bei hohen Temperaturen. Dies entspricht Gelierzeiten von
mehr als 50 min und einem kleinen µ, d.h. es liegt ein schwaches Netzwerk am
Gelpunkt vor. Das Netzwerk wird aus Polymerketten aufgebaut, in denen die
Bindungsbereiche stark fluktuieren - es liegen allerdings pro Kette im Mittel
mehr als zwei Bindungen vor.
Ähnlich dem Modell bei mittleren Temperaturen können sich hier die Helices
fast uneingeschränkt ausbilden. Die Fluktuation der Bindungsbereiche sind
durch die hohen Temperaturen und die damit verbundenen schwachen inter-
molekularen H - Brückenbindungen wesentlich ausgeprägter als im Modell bei
mittleren Temperaturen. Die Helices können sich fast uneingeschränkt zu relativ
großen Helixstapeln zusammenfinden, jedoch ist der Verbund der Helixstapel,
bedingt durch die schwachen intermolekularen H – Brückenbindungen, nicht so
stabil wie der im Modell bei mittleren Temperaturen. Geht man von der idealen
Stapelung der Helices und damit von einer hohen Helixstapelgröße verbunden
mit einer verbesserten Thermostabilität aus, so würden während der Deforma-
tion keine Biegeanteile in dem System auftreten. Das Netzwerk besäße am
Gelpunkt nicht die Stabilität wie das Netzwerk im Modell bei mittleren Tempe-
raturen. Im weiteren Verlauf der Gelierung wirken die langsame Helixbildung
und die ausgeprägten Fluktuationen der Bindungsbereiche einem schnellen
Erreichen eines Plateauwertes für den Speichermodul entgegen.
helicale Bereiche
Abb. 8.12. Schematische Darstellung der Helixanordnung im System Gela-tine / Wasser am Gelpunkt in Folge einer langsamen Gelierung (hohe Temperaturen)
Der Maximalwert für µ wird für ein Gelatine / Wasser System bei mittlerer Kon-
zentration gefunden. Es scheint demnach eine ideale Polymerkonzentration zu
geben, bei der einerseits durch die Bereitstellung einer erhöhten Menge an
Polymer und damit potentieller Netzwerkstellen die Wahrscheinlichkeit groß ist,
dass sich die Helices uneingeschränkt ideal stapeln bzw. aggregieren können.

ERGEBNISSE UND DISKUSSION 74
Andererseits ist die Polymerkonzentration immer noch so gering, dass lokal
keine Behinderung der Helicierung bzw. deren Stapelung durch die in der Sol-
phase befindlichen Polymermoleküle oder bereits helicierter Bereiche stattfin-
det. Ein erhöhter Anteil an Polymeren dürfte ebenfalls die Fluktuationen der
Bindungsstellen negativ beeinflussen. Die oben erwähnte Behinderung der
Helixbildung könnte sich durch die Rückfaltungen abgeknickter Helices, z.B. die
Ausbildung von Hairpins äußern, die zwar eine Erhöhung der Viskosität der
Solphase zur Folge haben, jedoch nicht zu Bildungen eines stabilen Netzwerks
beitragen. Die Erwartung, dass sich mit zunehmender Gelatinekonzentration die
dynamische Viskosität der Solphase erhöht, wird durch die Experimente bestä-
tigt.
Anmerkung. Aufgrund der von KISTERS mit Hilfe der analytischen Ultrazentri-
fuge ermittelten Daten wird eine Entmischung der Gele mit niedrigen Polymer-
konzentrationen gefunden.187 Die stark von den übrigen kritischen Exponenten
abweichenden Werte für ein 2 Gew.-%iges Gelatine / Wasser System
(µCF = 4.02±0.25 und νCF = 1.58±0.19; s. Tab. A-1.2) lassen demnach darauf
schließen, dass diese Systeme keine homogenen Phasen bilden.
8.3.3 Das Skalenverhalten der kritischen Exponenten.
Untereinander können die kritischen Exponenten über sogenannte Skalen-
gesetze (scalling laws) miteinander verknüpft sein.188 Hierbei handelt es sich um
Ausdrücke, die, gehören die kritischen Exponenten einer Universalitätsklasse
an, ebenfalls Universalitätscharakter besitzen. Ein scaling law beschreibt die
Größe ∆, sie verknüpft µ und ν in folgender Form:189,190,191
ν+µµ=∆ (8.19)
Die Berechnungen dieser Größe aus den in dieser Arbeit ermittelten kritischen
Exponenten sind in Anhang A-1.1 und A-1.2 aufgeführt. Im Falle der aus den
normierten Perkolationsansätzen bestimmten kritischen Exponenten liegt der
Wert für ∆ in einem Bereich von 0.62±0.01 bis 0.79±0.01, für die kritischen
187 Borchard W, Cölfen H, Kisters D, Straatmann A (2001) In: Borchard W (ed) Sonderband,Progress in Ultracentrifugation, in preparation188 Liegt ein solcher Fall vor, spricht man auch von einem "scaling" der Größen189 Clerc JP, Girand G, Laugier JM, Luck M (1985) J. Phys. A 18:2565190 Michon C, Cuvelier G, Launay B (1993) Rheol Acta 32:94191 Sahimi M (1994) Applications of Percolation Theory, Taylor & Francis

ERGEBNISSE UND DISKUSSION 75
Exponenten bestimmt nach den CF in einem Bereich von 0.61±0.01 bis
0.82±0.01, wobei in beiden Fällen für den größten Teil der Messungen der Wert
im Bereich 0.75±0.02 liegt. In diesem Bereich sind die Ergebnisse in guter
Übereinstimmung mit denen aus der Literatur. Für das System Gela-
tine / Wasser wurde von BORCHARD und Mitarbeitern bereits der Wert
∆ = 0.74±0.03 bestimmt,192 für chemisch vernetzte Gele ermittelte DURAND und
Mitarbeiter den Wert ∆ = 0.72±0.02.193. Zweifelsfrei dürften die Systeme, für die
diese Werte bestimmt werden, einer Universalitätsklasse angehören. Ausnah-
men bilden aber nach wie vor Systeme, die unter den sogenannten "Randbe-
dingungen" gelieren, Systeme für die ∆ - Werte oberhalb 0.80 bzw. unterhalb
0.65 bestimmt werden.
Die Größe ∆ wurde bereits in Kap. 8.1 bei die Behandlung der Frequenzabhän-
gigkeiten des komplexen Schubmoduls am Gelpunkt erwähnt. Dass diese
Größe in einem direkten Zusammenhang mit dem scaling law steht, ist aus der
Literatur bekannt,194,195,196 einen mathematischen Beweis findet man jedoch
nicht. Nach BORCHARD kann der Beweis in folgender Weise geführt werden.197
Es gelten die Bedingungen aus Kap. 8.1, dass am Gelpunkt der Verlustwinkel
frequenzunabhängig ist Gl. (8.20), der Verlust- und Speichermodul als Funktion
der Frequenz dem selben Potenzgesetz mit gleichem Exponenten gehorchen
Gl. (8.21)
0
'G''G
tan ω==δ (8.20)
∆ω~''G~'G (8.21)
( ) ( ) ( )pp~''Gpp~''G
c1
c
1−ω⋅=−
ω−−ν− ν (8.22)
( ) ( ) ( ).pp~'Gpp~'G cc
1−=− µµ (8.23)
Die Klammerausdrücke in den Gln. (8.22) und (8.23) sollen vom Betrag her
gleich sein, es gilt:
192 Borchard W, Lechtenfeld M (2001) Mat Res Innovat II Vol 4 No.5-6 p. 381193 Durand D, Delsanti M, Adam M, Luck M (1987) Europhys Lett 3:297194 Clerc JP, Girand G, Laugier JM, Luck M (1985) J. Phys. A 18:2565195 Michon C, Cuvelier G, Launay B (1993) Rheol Acta 32:94196 Sahimi M (1994) Applications of Percolation Theory, Taylor & Francis197 Borchard W persönliche Mitteilung

ERGEBNISSE UND DISKUSSION 76
.Zpppp cc =−=− (8.24)
Durch Einsetzen der Gln. (8.22) und (8.23) unter Berücksichtigung von Gl.
(8.24) in Gl. (8.20) ergibt sich:
( ).ZZ
Ztan µ+ν−
µ
ν−ω=ω=δ (8.25)
Mit Gl. (8.23) folgt:
( )( )
.'Gtan1 µ+ν−
ω=δ µ (8.26)
Aus Gl. (8.21) geht hervor, dass G' am Gelpunkt proportional ω∆ ist. Berück-
sichtigt man dies neben der Bedingung dass tanδ = ω0 ist, dann geht Gl. (8.26)
über in:
( )( )
01
ω=
ωω
µ+ν−∆ µ (8.27)
Nach Auflösen der Klammerausdrücke folgt:
01
ω=ω
µ
µ+ν−⋅∆+(8.28)
Ein Potenzenvergleich liefert als Lösung für ∆:
.ν+µ
µ=∆ (8.29)
Hieraus lässt sich schlussfolgern, dass ∆ bestimmt aus den Ergebnissen der
Perkolationstheorie den selben Wert haben muss wie ∆ aus den frequenzab-
hängigen Messungen des komplexen Schubmoduls. In Kap. 8.1 wurde ein Mit-
telwert für ∆ mit 0.695 bestimmt, der damit sehr gut mit denen aus der Theorie
erwähnten übereinstimmt (s.o).

ERGEBNISSE UND DISKUSSION 77
8.4 Zusammenhang zwischen dem komplexen Schubmodul und der
optischen Drehung.
In den vorangegangenen Auswertungen wurde eine Substitution der Wahr-
scheinlichkeiten p in den Perkolationsansätzen Gln. (4.3) und (4.4) durch die
Zeiten t vorgenommen, um nach der Perkolationstheorie auszuwerten. Inwie-
fern eine Substitution der Wahrscheinlichkeiten durch die optische Drehung
erlaubt ist, soll anhand der simultanen Messungen der rheologischen Kenngrö-
ßen und der optischen Drehung während der isothermen Gelierung geklärt
werden.
Geht man davon aus, dass eine solche Substitution erlaubt ist, dann müsste
eine Proportionalität zwischen der Zeit und der optischen Drehung in der Nähe
des Gelpunkts existieren.
Zur Überprüfung dieser Annahme ist es von Vorteil, eine aus der Literatur
bekannte Größe, die sogenannte Konvertierungsvariable zu benutzen, die sich
auf den Helixanteil einer Probe bezieht.198,199,200,201,
.][][
][][
0
0t
0t,t,
0t,t,
α−αα−α
=α−α
α−α=Φ
∞=λ∞=λ
=λλ(8.30)
αt , im weiteren Verlauf der Arbeit als α bezeichnet, ist hierbei die optische Dre-
hung der untersuchten Gelatine Lösung zum Zeitpunkt t, α0 ist der Anteil der
Drehung, hervorgerufen durch die asymetrischen C-Atome in der Gelatine
(siehe dazu Kap. 3) und α∞ der Wert der Drehung für Kollagen, also der theore-
tische Wert, der resultieren würde, wenn eine Renaturierung des Kollagens
stattgefunden hätte. Hat keine Helixbildung stattgefunden, wie es bei einer
Temperatur von 40°C der Fall ist, ist α = α0 und damit Φ = 0. Für den oben
erwähnten Fall der Renaturierung wird α = α∞ und damit Φ = 1.
Ersetzt man nun die Konvertierungsvariable Gl. (8.30) durch die Zeit in den Gln.
(4.5) und (4.6), dann ergeben sich folgende Ausdrücke in Gln. (8.31) und (8.32),
bei denen Φgel der kritische Helixanteil am Gelpunkt ist:
198 v. Hippel PH, Harrington WF (1960) In: Protein Structure and Function, BrookhavenSymposia in Biol 13:213199 Harrington WF, v. Hippel PH (1962) Advan Protein Chem 16:1200 Djabourov M; Maquet J, Theveneau H, Leblond J, Papon P (1985) British Polymer J Vol 17,2:169201 Djabourov M, Leblond J, Papon P (1988) J Phys (France) 49:319

ERGEBNISSE UND DISKUSSION 78
( ) gelgel fürK''G Φ<ΦΦ−Φ=∗ν−∗
η (8.31)
( ) .fürK'G gelgelG Φ>ΦΦ−Φ=∗µ∗ (8.32)
Die Konstanten sowie die kritischen Exponenten sind, um sie von den bereits
bekannten kritischen Exponentent unterscheiden zu können, mit einem Stern
gekennzeichnet.202
Ausgehend von Gl. (8.30) lässt sich folgender Ausdruck für Φgel formulieren:
.0
0gelgel α−α
α−α=Φ
∞(8.33)
αgel ist der kritische Wert der optischen Drehung am Gelpunkt, also zum Zeit-
punkt t = tgel.
Durch Einsetzen der Gln. (8.30) und (8.33) in Gl. (8.31) bzw. (8.32) erhält man
für den Verlust- bzw. Speichermodul:203
gel0
gel fürK''G α<α
α−α
α−α=
∗ν−
∞
∗η (8.34)
.fürK'G gel0
gelG α>α
α−α
α−α=
∗µ
∞
∗ (8.35)
Die optische Drehung α ist bei den Untersuchungen des Systems Gela-
tine / Wasser stets eine negative Größe, die im Laufe der Gelierung weiter
abnimmt. Die Betragsstriche sorgen dafür, dass die Klammerausdrücke in den
Gln. (8.34) und (8.35) keine negativen Werte liefern und somit am Gelpunkt
weiterhin der Verlustmodul divergiert bzw. der Speichermodul Null wird.
Durch Gleichsetzten von Gl. (4.5) mit Gl. (8.34) und Gl. (4.6) mit (8.35) erhält
man zwei Ausdrücke:204
( ) gel*gelgel ttfürttK <−=α−α νν
η$ (8.36)
202 Borchard W, Lechtenfeld M (1999) Macromolecules eingereicht203 Borchard W, Lechtenfeld M (1999) Macromolecules eingereicht204 Borchard W, Lechtenfeld M (1999) Macromolecules eingereicht
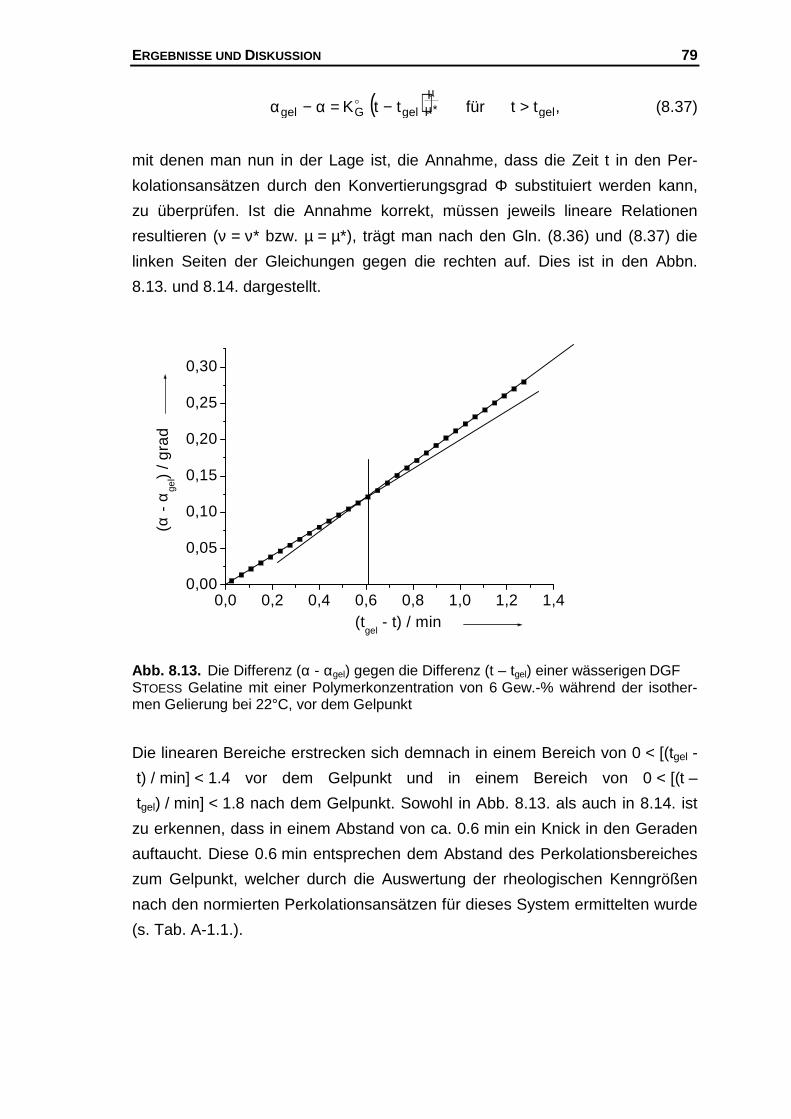
ERGEBNISSE UND DISKUSSION 79
( ) ,ttfürttK gel*gelGgel >−=α−α µµ$ (8.37)
mit denen man nun in der Lage ist, die Annahme, dass die Zeit t in den Per-
kolationsansätzen durch den Konvertierungsgrad Φ substituiert werden kann,
zu überprüfen. Ist die Annahme korrekt, müssen jeweils lineare Relationen
resultieren (ν = ν* bzw. µ = µ*), trägt man nach den Gln. (8.36) und (8.37) die
linken Seiten der Gleichungen gegen die rechten auf. Dies ist in den Abbn.
8.13. und 8.14. dargestellt.
Abb. 8.13. Die Differenz (α - αgel) gegen die Differenz (t – tgel) einer wässerigen DGFSTOESS Gelatine mit einer Polymerkonzentration von 6 Gew.-% während der isother-men Gelierung bei 22°C, vor dem Gelpunkt
Die linearen Bereiche erstrecken sich demnach in einem Bereich von 0 < [(tgel -
t) / min] < 1.4 vor dem Gelpunkt und in einem Bereich von 0 < [(t –
tgel) / min] < 1.8 nach dem Gelpunkt. Sowohl in Abb. 8.13. als auch in 8.14. ist
zu erkennen, dass in einem Abstand von ca. 0.6 min ein Knick in den Geraden
auftaucht. Diese 0.6 min entsprechen dem Abstand des Perkolationsbereiches
zum Gelpunkt, welcher durch die Auswertung der rheologischen Kenngrößen
nach den normierten Perkolationsansätzen für dieses System ermittelten wurde
(s. Tab. A-1.1.).
0,0 0,2 0,4 0,6 0,8 1,0 1,2 1,40,00
0,05
0,10
0,15
0,20
0,25
0,30
(α-
αge
l) / g
rad
(tgel
- t) / min

ERGEBNISSE UND DISKUSSION 80
Abb. 8.14. Die Differenz (αgel - α) gegen die Differenz (tgel – t) einer wässerigen DGFSTOESS Gelatine mit einer Polymerkonzentration von 6 Gew.-% während der isother-men Gelierung bei 22°C, nach dem Gelpunkt
Mit Hilfe der Abkürzungen in Gln. (8.38a) bzw. (8.39a) lassen sich die Gln.
(8.34) und (8.35) umstellen in die Gln. (8.38) bzw.(8.39):205
( ) ( ) gelgel fürK''G1
α<αα−α⋅= ⊗η
−∗ν (8.38)
( )0
1KK
1
α−α⋅=
∞
−∗η
⊗η
ν (8.38a)
( ) ( ) ∗⊗ α>αα−α⋅=∗µ fürK'G gelG
1
(8.39)
( ) .1
KKo
GG
1
∞
∗⊗
α−α⋅= ∗µ (8.39a)
Hieraus lässt sich die Bedingung (ν = ν* bzw. µ = µ*) testen, wenn man für µ*bzw. ν* die Werte einsetzt, die im Kap. 8.2.1 nach den normierten Perkolations-ansätzen bestimmt wurden. Durch Auftragen der rechten gegen die linken Sei-ten der Gln. (8.38) und (8.39) müssten lineare Beziehungen resultieren. Dies istin den Abbn. 8.15. und 8.16. wiedergegeben.
205 Borchard W, Lechtenfeld M (1999) Macromolecules eingereicht
0,0 0,3 0,6 0,9 1,2 1,5 1,80,0
0,1
0,2
0,3
0,4
0,5
(αge
l-
α)
/ gr
ad
(t - tgel
) / min
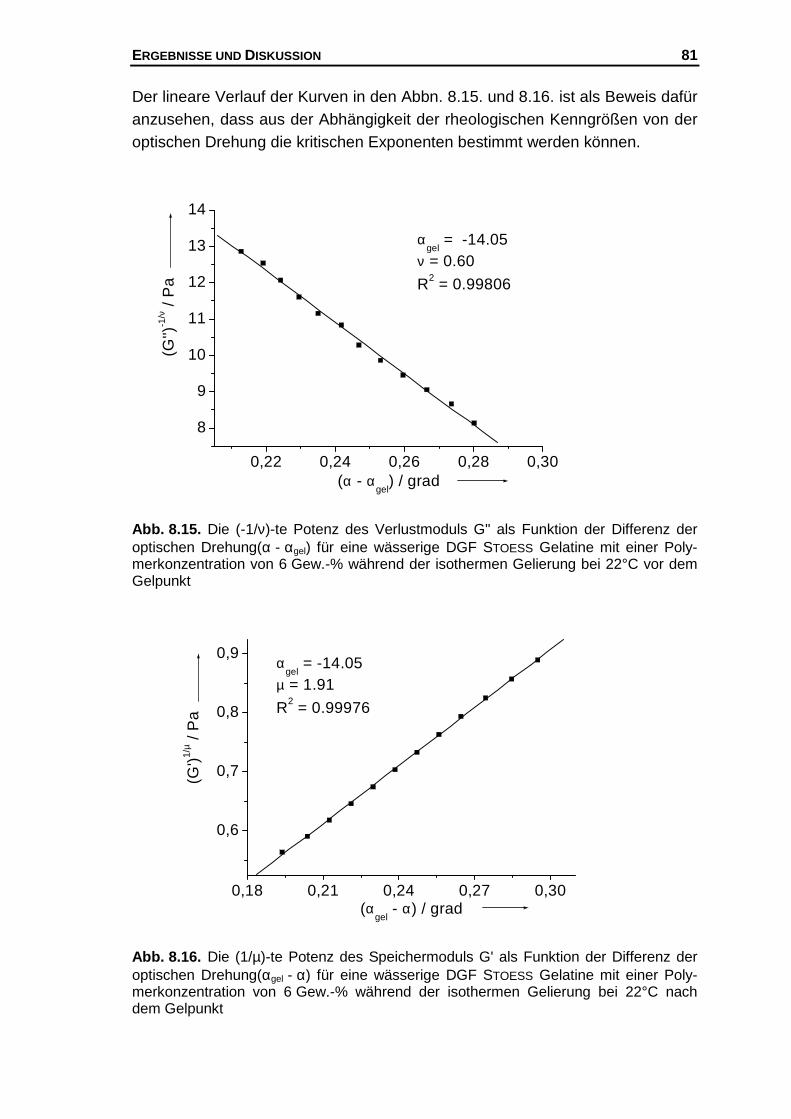
ERGEBNISSE UND DISKUSSION 81
Der lineare Verlauf der Kurven in den Abbn. 8.15. und 8.16. ist als Beweis dafüranzusehen, dass aus der Abhängigkeit der rheologischen Kenngrößen von deroptischen Drehung die kritischen Exponenten bestimmt werden können.
Abb. 8.15. Die (-1/ν)-te Potenz des Verlustmoduls G" als Funktion der Differenz deroptischen Drehung(α - αgel) für eine wässerige DGF STOESS Gelatine mit einer Poly-merkonzentration von 6 Gew.-% während der isothermen Gelierung bei 22°C vor demGelpunkt
Abb. 8.16. Die (1/µ)-te Potenz des Speichermoduls G' als Funktion der Differenz deroptischen Drehung(αgel - α) für eine wässerige DGF STOESS Gelatine mit einer Poly-merkonzentration von 6 Gew.-% während der isothermen Gelierung bei 22°C nachdem Gelpunkt
0,18 0,21 0,24 0,27 0,30
0,6
0,7
0,8
0,9α
gel= -14.05
µ = 1.91
R2 = 0.99976
(G')1/
µ/ P
a
(αgel
- α) / grad
0,22 0,24 0,26 0,28 0,30
8
9
10
11
12
13
14
αgel
= -14.05ν = 0.60
R2 = 0.99806
(G'')
-1/ν
/ Pa
(α - αgel
) / grad

ERGEBNISSE UND DISKUSSION 82
Exkurs. Gl. (8.39) entspricht der vor 40 Jahren von TODD gefundenen Relation
zwischen der optischen Drehung und dem Speichermodul.206 Für die Abhängig-
keit des Speichermoduls von der optischen Drehung fand TODD den Wert
µ* = 2.
Bildet man den natürlichen Logarithmus der Gl. (8.39), so müsste ein lineares
Verhältnis der Größen resultieren mit einer Steigung von 0.5. In Abb. 8.17. ist
dieser Sachverhalt aufgetragen.
Aus Abb. 8.17. ist eindeutig ersichtlich, dass die TODD-Relation voll erfüllt ist.
Lediglich für den Bereich sehr nahe am Gelpunkt ergibt sich ein stärkerer
Anstieg des Kurvenverlaufes. Das diese Bedingung ebenfalls für verschiedene
Konzentrationen bei unterschiedlichen Temperaturen erfüllt wird, konnte inzwi-
schen von BORCHARD und Mitarbeitern gezeigt werden.207
Abb. 8.17. Auftragung des natürlichen Logarithmus der Differenzen (αgel - α) gegenden natürlichen Logarithmus des Speichermoduls G‘ für eine wässerige ROUSSELOTGelatine mit einer Polymerkonzentration von 4 Gew.-% während der isothermen Gelie-rung bei 16°C, G+ = 1Pa, α+ = 1 Grad
206 Todd A (1961) Nature 191:567207 Borchard W, Lechtenfeld M unveröffentlichte Ergebnisse
-6 -4 -2 0 2 4 6-2
-1
0
1
2
3
4(1 / µ*) = 0.74
(1 / µ*) = 0.49
ln(α
gel-
α)
/ α+
ln G' / G+

ERGEBNISSE UND DISKUSSION 83
9 Die mathematische Beschreibung der G'(t) Funktion
- Das Aggregationsmodell -
Im gleichen Maße wie die Wissenschaftler sich mit der Frage beschäftigen:
"Wie beschreibt man das Verhalten eines Systems am Gelpunkt mathema-
tisch?", beschäftigt sie die Frage: "Welches Modell beschreibt den Verlauf des
Speichermoduls vollständig?" Der G'(t) Verlauf, der an den Verlauf einer Wur-
zelfunktion erinnert, lässt sich mathematisch problemlos durch eine Vielzahl
unterschiedlicher Funktionen beschreiben,208 jedoch "lediglich" mathematisch,
aber nicht physikalisch sinnvoll.209
VENOHR wendete die von V. SMOLUCHOWSKI210 aufgestellte Theorie, die, kurz
erklärt, auf der langsamen Koagulation von Teilchen basiert, in der jeder Kon-
takt zwischen zwei Teilchen irreversibel zu einem Aggregat führt, erfolglos auf
seine für das System Gelatine / Wasser Systemen experimentell ermittelten
G'(t) Funktionen an.211 Als Hauptgrund für das Misslingen gibt VENOHR die in der
Theorie nicht berücksichtigten Rückreaktionen an, die bei der Gelierung des
Systems Gelatine / Wasser auftreten.
In einem weiteren Vorhaben versuchte VENOHR die Pearson Verteilungsfunk-
tion212 auf den Verlauf der G'(t)-Kurven anzuwenden. Hieraus konnten zwar für
den Endwert des Speichermoduls Werte extrapoliert werden, jedoch gibt dieser
empirische Ansatz nicht die Entwicklung des Speichermoduls bei kleinen Zeiten
wieder.213
DJABOUROV und Mitarbieter formulierten einen Ansatz, der sich aus der Summe
zweier logarithmischer Terme zusammensetzt, um den Verlauf des Konvertie-
rungsgrades214 während der Gelierung des Systems Gelatine / Wasser als
Funktion der Zeit zu beschreiben.215 Hiermit lässt sich der Verlauf der Mess-
kurve gut beschreiben, jedoch liefert die Funktion aufgrund ihres logarithmi-
schen Terms einen stetigen Anstieg der Funktion und, für sehr lange Zeiten
Werte für den Konvertierungsgrad, die größer als eins und damit mathematisch
sowie physikalisch unmöglich sind. DJABOUROV vertritt die Meinung, dass der
208 Venohr H (1999) Dissertation Duisburg209 Borchard W persönliche Mitteilung210 v. Smoluchowski M (1917) Z phys Chem 92:129211 Venohr H (1999) Dissertation Duisburg212 Huismann R, Heuvel HM (1989) J Appl Polym Sci 37:595213 Venohr H (1999) Dissertation Duisburg214 siehe Kap. 8.4 hier Gl. (8.30)215 Djabourov M, Leblond J, Papon P (1988) J Phys (France) 49:319

ERGEBNISSE UND DISKUSSION 84
Helixanteil in "Tausenden von Stunden" keinen Gleichgewichtswert liefert.216 Für
die Anwendung auf die zeitliche Änderung des Konvertierungsgrades während
der Gelierung müsste die Funktion jedoch in einen Sättigungswert übergehen.
Der Versuch, den DJABOUROV - Ansatz auf den G'(t)-Verlauf anzuwenden
scheiterte, da der Kurvenverlauf von G'(t) in der Nähe des Gelpunkts nur
schlecht wiedergegeben wird.217
In den Arbeiten MAIBAUM218 und VENOHR219 wird auf einen Ansatz hingewiesen,
der auf BORCHARD zurückgeht und zum ersten Mal auf der Bunsentagung 1998
in Münster vorgetragen wurde.220
Der Verlauf der G'(t)-Kurven erinnert an die Langmuirsche Adsorptionsiso-
therme, bei denen die Kinetik der Adsorption sowie die der Desorption eine ent-
scheidende Rolle spielt. Die Aggregation der gebildeten Helices, was die Vor-
aussetzung für die Gelierung ist, kann als Adsorption verstanden werden,
ebenso wie die Desaggregation der Helixstapel als Desorption (siehe Kap.
1.3.2). BORCHARD macht in seinem Modell die Annahme, dass ein Gleichge-
wicht zwischen partiell helicierten Molekülketten in der Solphase und Molekül-
ketten, die bereits in das Netzwerk eingebaut wurden, herrscht.
[partiell helicierte Molekülketten]Sol Φ [Netzwerkketten]Gel
Entsprechend dem Adsorptions- bzw. Desorptionsverhalten an Oberflächen
formuliert er für die Aggregationsgeschwindigkeit der Helices, va bzw. für die
Dessaggregationsgeschwindigkeit, vd,
( ) ( )hc2h2aa xx1kv −Θ−= (9.1)
.kv dd Θ= (9.2)
Hierbei bedeuten:
Θ = Kontaktflächenanteil bereits aggregierter Helices in der Gelphase
1 - Θ = Kontaktflächenanteil der freien, nicht aggregierten Helices in der
Gelphase
216 Djabourov M, Lechaire JP, Gaill F (1993) Biorheology 30:191217 Borchard W, Lechtenfeld M unveröffentlichte Ergebnisse218 Maibaum R (1998) Dissertation Duisburg219 Venohr H (1999) Dissertation Duisburg220 Borchard W (1998) Ber Bunsenges Phys Chem 102:1580

ERGEBNISSE UND DISKUSSION 85
X2h = Zahlenanteil der Helices im Sol
X2hc = kritischer Zahlenanteil der Helices im Sol
Im Gleichgewicht gilt:
da vv = (9.3)
Für den Kontaktflächenanteil bereits aggregierter Helices in der Gelphase ergibt
sich:
( )( )hc2h2ad
hc2h2a
xxkk
xxk
−+−
=Θ (9.4)
Wohlweislich, dass die Perkolationstheorie das Verhalten am Gelpunkt sehr gut
beschreibt, berücksichtigt BORCHARD den ursprünglichen Perkolationsansatz
Gl.(4.4) und nimmt folgende Bedingungen an:
( ) Θ=− 1c kpp (9.5)
( ) ( ).ttkxx gel2hc2h2 −=− (9.6)
Hieraus lässt sich jetzt für den zeitabhängigen Verlauf des Speichermoduls fol-
gender Ausdruck Gl. (9.7) bzw. Gl. (9.8) formulieren, wobei die Konstanten K1
und K2 in den Gln. (9.9a) und (9.9b) wiedergeben sind.
( ) ( )( )
µ
µ
−+
−=
gela2d
gela12
ttkkk
ttkkkKt'G (9.7)
( ) ( )( )
µ
−+
−=
gel2
gel1
ttK1
ttKt'G (9.8)
( )µµ≡1
Kk
kkkK
d
d121 (9.9a)
.k
kkK
d
a22 ≡ (9.9b)

ERGEBNISSE UND DISKUSSION 86
Der alles entscheidende "Clou" an Gl. (9.8) ist, dass für den Fall, t→tgel, die
Größe K2 (t – tgel) im Nenner der Gl. (9.8) sehr klein wird und der Nenner damit
annähernd 1. Gl. (9.8) geht mit den Annahmen in Gl. (9.10) in den Perkola-
tionsansatz Gl. (4.6) bzw. (9.11) über:
G,gelgeld
a21G ttund
kkkk
KK =
≡
µ
µ (9.10)
( ) ( ) .ttKt'G gelGµ−= (9.11)
Für den Fall t→∞, wird K2(t - tgel) >> 1 und Gl. (9.8) liefert eine Konstante. Es
lässt sich also ein Endwert für den Speichermodul G'∞ Gl. (9.12) formulieren
bzw. durch ein Experiment bei endlichen Zeiten bestimmen:
.KK
'G2
1µ
∞
= (9.12)
Die Auswertungsprozedur gestaltete sich folgendermaßen. Die aus den Perko-
lationsansätzen bestimmten Gelierzeiten bzw. kritische Exponenten wurden in
der Auswertegleichung vorgeben und die Größen K1 und K2 mit Hilfe einer
nichtlinearen Kurvenanpassung berechnet.221 Die ersten Versuche, die G'(t)-
Kurven nach Gl. (9.8) auszuwerten lieferten Ausgleichskurven, die zu späteren
Zeit von den Messwerten deutlich abwichen und keinem konstanten Wert zuzu-
streben schienen. Dies konnte dadurch behoben werden, indem die Zeitdif-
ferenzen in Gl. (9.8) zu einer Potenz α erhoben wurden, die ebenfalls durch die
nichtlineare Kurvenanpassung berechnet wurde. Gl. (9.8) geht damit in Gl.
(9.13) über, in der µ' durch Gl. (9.14) wiedergegeben ist:
( ) ( )( )
'
gel2
gel1
ttK1
ttKt'G
µ
α
α
−+
−= (9.13)
.'αµ=µ (9.14)
221 Die Berechnung wurden mit Hilfe des Programms Origin 6.0 der Fa. MicrocalTM
durchgeführt. Die nichtlinearen Approximation erfolgt nach der Methode der kleinstenAbweichungsquadrate, basierent auf dem Levenberg – Marquardt – Algorithmus.
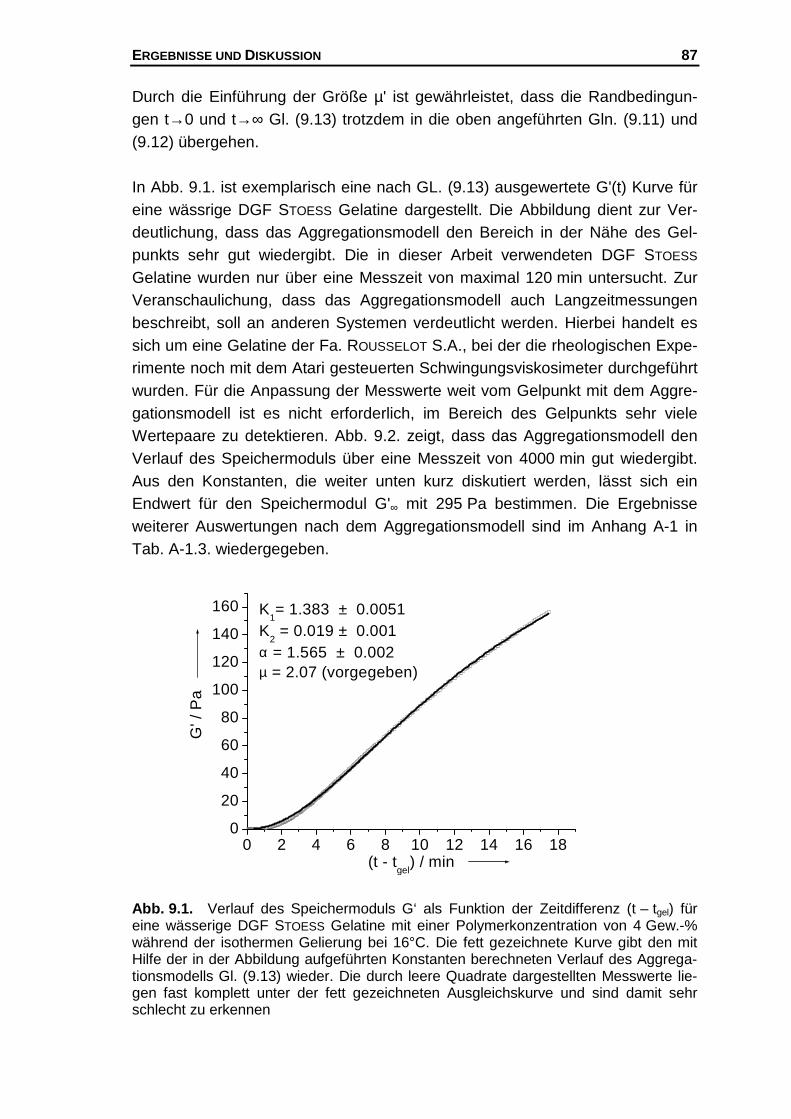
ERGEBNISSE UND DISKUSSION 87
Durch die Einführung der Größe µ' ist gewährleistet, dass die Randbedingun-gen t→0 und t→∞ Gl. (9.13) trotzdem in die oben angeführten Gln. (9.11) und(9.12) übergehen.
In Abb. 9.1. ist exemplarisch eine nach GL. (9.13) ausgewertete G'(t) Kurve füreine wässrige DGF STOESS Gelatine dargestellt. Die Abbildung dient zur Ver-deutlichung, dass das Aggregationsmodell den Bereich in der Nähe des Gel-punkts sehr gut wiedergibt. Die in dieser Arbeit verwendeten DGF STOESS
Gelatine wurden nur über eine Messzeit von maximal 120 min untersucht. ZurVeranschaulichung, dass das Aggregationsmodell auch Langzeitmessungenbeschreibt, soll an anderen Systemen verdeutlicht werden. Hierbei handelt essich um eine Gelatine der Fa. ROUSSELOT S.A., bei der die rheologischen Expe-rimente noch mit dem Atari gesteuerten Schwingungsviskosimeter durchgeführtwurden. Für die Anpassung der Messwerte weit vom Gelpunkt mit dem Aggre-gationsmodell ist es nicht erforderlich, im Bereich des Gelpunkts sehr vieleWertepaare zu detektieren. Abb. 9.2. zeigt, dass das Aggregationsmodell denVerlauf des Speichermoduls über eine Messzeit von 4000 min gut wiedergibt.Aus den Konstanten, die weiter unten kurz diskutiert werden, lässt sich einEndwert für den Speichermodul G'∞ mit 295 Pa bestimmen. Die Ergebnisseweiterer Auswertungen nach dem Aggregationsmodell sind im Anhang A-1 inTab. A-1.3. wiedergegeben.
Abb. 9.1. Verlauf des Speichermoduls G‘ als Funktion der Zeitdifferenz (t – tgel) füreine wässerige DGF STOESS Gelatine mit einer Polymerkonzentration von 4 Gew.-%während der isothermen Gelierung bei 16°C. Die fett gezeichnete Kurve gibt den mitHilfe der in der Abbildung aufgeführten Konstanten berechneten Verlauf des Aggrega-tionsmodells Gl. (9.13) wieder. Die durch leere Quadrate dargestellten Messwerte lie-gen fast komplett unter der fett gezeichneten Ausgleichskurve und sind damit sehrschlecht zu erkennen
0 2 4 6 8 10 12 14 16 180
20
40
60
80
100
120
140
160 K1= 1.383 ± 0.0051
K2
= 0.019 ± 0.001α = 1.565 ± 0.002µ = 2.07 (vorgegeben)
G' /
Pa
(t - tgel
) / min

ERGEBNISSE UND DISKUSSION 88
Abb. 9.2. Verlauf des Speichermoduls G‘ als Funktion der Zeitdifferenz (t – tgel) füreine wässerige ROUSSELOT Gelatine mit einer Polymerkonzentration von 4 Gew.%während der isothermen Gelierung bei 26°C. Die fett gezeichnete Kurve gibt den mitHilfe der in der Abbildung aufgeführten Konstanten berechneten Verlauf des Aggrega-tionsmodells Gl. (9.13) wieder. Die durch leere Quadrate dargestellten Messwerte lie-gen fast komplett unter der fett gezeichneten Ausgleichskurve und sind damit sehrschlecht zu erkennen
Diskussion der Konstanten. K1 beschreibt die Hinreaktion. K1 nimmt mit stei-
gender Temperatur ab. Das ist insofern verständlich, als mit steigender Tem-
peratur die Helixbildung langsamer abläuft. Hieraus folgt, dass K2, die
Geschwindigkeitskonstante für die Rückreaktion, ebenfalls abnehmen muss, da
bei einer mäßig schnell ablaufenden Helixbildung auch die Rückreaktion mäßig
schnell abläuft.
0 1000 2000 3000 40000
20
40
60
80
100
120
140
160
K1
0.231 ± 0.004α 0.590 ± 0.004K
20.033 ± 0.001
µ 1.70 (vorgegeben)
G' /
Pa
(t - tgel
) / min

ZUSAMMENFASSUNG 89
ZUSAMMENFASSUNG
In der vorliegenden Arbeit wurde die thermoreversible Gelierung des SystemsGelatine / Wasser anhand von rheologischen und optischen Methoden unter-sucht. Es kamen hierbei ein dynamisches Schwingungsviskosimeter sowie einPräzisionspolarimeter zum Einsatz. Beide Geräte wurden dabei in eine Ver-suchseinrichtung integriert, mit deren Hilfe eine simultane Bestimmung derrheologischen und optischen Kenngrößen möglich wurde. Zur Untersuchungschnell ablaufender Gelierungen schienen zwei experimentelle Veränderungendes dynamischen Schwingungsviskosimeters erforderlich zu sein. Zum einenschien die Zeit von mindestens 18 Sekunden zwischen zwei Messwerten nichtausreichend, um schnell ablaufende Reaktionen anhand der rheologischenKenngrößen im Anfangsbereich mit genügender Genauigkeit zu beobachten.Daher wurde die Steuereinrichtung des dynamischen Schwingungsviskosime-ters von Atari auf PC umgestellt, sodass nun Messungen in Sekundenschrittenerfolgen konnten. Zum anderen sollte, entgegen der früheren Temperatur-sprungmethode mit Hilfe zweier Thermostate, einem anderen Verfahren derVorzug gegeben werden, welches eine "Direkteinspritzung" des Probematerialsin die vortemperierte Messzelle zeitgleich mit dem Start der Messungen vor-sieht. Erst durch die Direkteinspritzung konnten von Anbeginn der Messung diezwingend erforderlichen isothermen Bedingungen zugrunde gelegt werden.
Die rheologischen Kenngrößen hatten die Aufgabe, das kritische Phänomen derGelierung mit Hilfe der Perkolationstheorie zu beschreiben. Zur Ermittlung desGelpunkts und der kritischen Exponenten wurde das von STAUFFER und DE
GENNES gefundene Potenzverhalten der rheologischen Kenngrößen in derNähe des Gelpunkts durch sogenannte normierte Perkolationsansätze zumAusdruck gebracht. Eine Auswertung der durchgeführten Experimente wurdeerst mit einer Substitution der Wahrscheinlichkeit, die für eine Auswertung nachder Perkolationstheorie mit Hilfe von Computersimulationen relevant ist, durchdie Zeit ermöglicht. Durch die Auswertung nach den normierten Perkolationsan-sätzen erhielt man als Lösung eine sehr genaue Gelierzeit, des weiteren dieüberaus wichtige Information, in welchem Bereich eine Auswertung nach derPerkolationstheorie erfolgen darf und sollte.
Aus der Frequenzabhängigkeit des komplexen Schubmoduls lässt sich nachWINTER für chemisch vernetzte Systeme der Gelpunkt bestimmen. Dies wurdein der vorliegenden Arbeit anhand eines Beispiels überprüft. Die so ermittelteGelierzeit ist in guter Übereinstimmung mit der Gelierzeit, die aus der normier-ten Perkolationstheorie bestimmt wurde.

90
Der aus der normierten Perkolationstheorie bestimmte kritische Exponent νstimmt sehr gut mit dem von SAHIMI und ARBABI vorhergesagten Wert für dassogenannte ZIMM-Verhalten überein. Es ist demnach davon auszugehen, dassam Gelpunkt starke hydrodynamische Wechselwirkungen zwischen den Gela-tinemolekülen und nur geringe Diffusion stattfinden. Der kritische Exponent µliegt in einem Bereich, der von DE GENNES für die Annahme einer Analogie zwi-schen Elastizität und Leitfähigkeit eines Netzwerkes aus Isolatoren und Leiternvorhergesagten Wertes von µ = 1.7, bzw. dem von SAHIMI vorhergesagten Wertvon µ = 2.1, der sich für Modellrechnungen unter Berücksichtigung des Dehn-vermögens einer Bindung im Netzwerk ergibt. Diese Annahmen dienten derveranschaulichten Darstellung eines molekularen Bildes der Gelierung, insbe-sondere die Aggregation der Helices am Gelpunkt.
Eine weitere Methode zur Bestimmung der kritischen Exponenten wurde durchdie Kombination des Speicher – und Verlustmoduls symmetrisch um den Gel-punkt geschaffen. Es handelt sich bei diesen sogenannten kombinierten Per-kolationsansätzen (Combined Functions) um eine Kombination der Größen inäquidistanten Abständen zum Gelpunkt innerhalb der zuvor durch die normier-ten Perkolationsansätze bestimmten Perkolationsbereiche. Anhand der Aus-wertungen nach den CF wird deutlich, dass die klassische Theorie nicht in Lageist, das Phänomen der Gelierung zu beschreiben.
Durch die simultane Bestimmung der rheologischen und optischen Kenngrößenwurde deutlich, dass eine Proportionalität zwischen der Zeit und der optischenDrehung während der thermoreversiblen Gelierung am Gelpunkt existiert. Es istdemnach möglich, nicht nur den Verlauf der rheologischen Kenngrößen alsFunktion der Zeit, sondern auch als Funktion der optischen Drehung im Sinneder Perkolationstheorie auszuwerten. Es wurde gezeigt, dass beide Möglich-keiten die gleichen kritischen Exponenten liefern.
Die Gelierzeit sowie der kritische Exponent dienten bei der mathematischenBeschreibung des kompletten Verlaufs des Speichermoduls als Funktion derZeit. Es wurde ein mathematisches und physikalisches Aggregationsmodell for-muliert, das auf den reaktionskinetischen Schritten der Langmuirschen Adsorp-tionsisotherme beruht. Das Modell ist so aufgebaut, dass es in der Nähe desGelpunkts komplett in den typischen Perkolationsansatz übergeht, bzw. fürZeiten in weiter Entfernung zum Gelpunkt einen Gleichgewichtswert für denSpeichermodul liefert. Beide Bereiche werden von dem Aggregationsmodellexakt beschrieben.

ANHANG 91
ANHANG
Anhang A-1 Ergebnisse der Auswertung nach der Perkolationstheorie.
Tab. A-1.1. Ergebnisse der Auswertung nach den normierten Perkolationsansät-zen.
wGelatine/ϑ(°C) tgel (min) µ ν ∆ PB(min)
0.02 / 14 8.42 1.76±0.01 0.64±0.01 0.64±0.01 I1.36I+0.45
0.02 / 15 11.19 2.22±0.01 0.72±0.01 0.72±0.01 I1.22I+0.50
0.02 / 16 14.70 1.79±0.01 1.06±0.02 0.62±0.01 I3.00I+0.67
0.04 / 16 2.65 2.07±0.05 0.58±0.01 0.78±0.01 I0.63I+0.39
0.04 / 18 3.67 2.26±0.02 0.72±0.02 0.76±0.01 I0.85I+0.61
0.04 / 20 6.38 1.75±0.02 0.55±0.02 0.76±0.01 I0.60I+0.36
0.06 / 20 2.00 2.27±0.01 0.61±0.02 0.79±0.01 I0.54I+0.18
0.06 / 22 4.64 1.91±0.02 0.60±0.01 0.76±0.01 I0.68I+0.40
0.06 / 24 15.08 1.77±0.01 0.61±0.01 0.73±0.01 I2.92I+0.65
0.08 / 18 0.54 1.96±0.08 0.61±0.01 0.76±0.01 I0.14I+0.25
0.08 / 20 1.10 2.11±0.06 0.76±0.01 0.76±0.01 I0.21I+0.39
0.08 / 22 2.22 1.86±0.05 0.68±0.07 0.73±0.03 I0.31I+0.47
wGelatine = Massenbruch des Polymeren im System Gelatine / Wasser
ϑ = Geliertemperatur in °C
tgel = Gelierzeit in Minuten
µ bzw. ν = kritische Exponenten
∆ = Scaling law, berechnet sich nach: ν+µ
µ=∆
PB = Perkolationsbereich, in dem nach der Perkolationstheorie ausge-
wertet wurde. Der Wert in Betragsstrichen gibt die zeitliche Entfer-
nung des Perkolationsbereiches zum Gelpunkt an, der Wert
dahinter gibt an, über welchen Zeitbereich sich der Perkolations-
bereich erstreckt. Beides sind Minutenangaben.

ANHANG 92
Tab. A-1.2. Ergebnisse der Auswertung nach den kombinierten Perkolationsan-sätzen (Combined Functions)
wGelatine ϑ(°C) µCF νCF ∆CF
0.02 14 4.02±0.25 1.58±0.19 0.72±0.04
0.02 15 2.17±0.15 0.61±0.08 0.78±0.03
0.02 16 1.81±0.28 1.17±0.23 0.61±0.08
0.04 16 1.88±0.13 0.63±0.06 0.75±0.03
0.04 18 1.79±0.12 0.62±0.07 0.74±0.03
0.04 20 2.98±0.10 0.74±0.05 0.81±0.02
0.06 20 2.95±0.15 0.61±0.05 0.82±0.02
0.06 22 1.95±0.01 0.61±0.05 0.76±0.01
0.06 24 1.72±0.17 0.65±0.11 0.73±0.05
0.08 18 2.20±0.26 0.76±0.14 0.74±0.06
0.08 20 2.20±0.16 0.82±0.08 0.73±0.03
0.08 22 3.10±0.18 1.06±0.08 0.75±0.03
wGelatine = Massenbruch des Polymeren im System Gelatine / Wasser
ϑ = Geliertemperatur in °C
µCF bzw. νCF = kritische Exponenten bestimmt aus den Combined Functions
∆CF = Scaling law, berechnet sich nach: CFCF
CFCF
ν+µ
µ=∆

ANHANG 93
Tab. A-1.3. Ergebnisse der Auswertung nach dem Aggregationsmodell für einewässerige M92 Gelatine.(3 Gew.-%; Ergebnisse und Messkurvensind von Venohr, 4 Gew.-%; Messkurven von Lechtenfeld, Bestim-mung der Gelierzeiten und der kritischen Exponenten nach derMethode von Venohr)
wGelatine ϑ tgel/ min K1 K2 α µ G'∞
0.03 18 3.52 4.59388 0.99633 0.29504 1.34 908
0.03 20 4.36 3.08363 0.7597 0.33521 1.40 768
0.03 22 8.45 1.85372 0.35674 0.3798 1.47 615
0.03 24 21.56 0.739 0.08837 0.4712 1.40 459
0.03 25 50.93 0.36165 0.03584 0.53157 1.30 354
0.03 26 114,61 0.10277 0.00493 0.70511 1.29 238
0.04 18 2.41 6.74376 0.6846 0.3332 1.01 1027
0.04 20 2.83 4.89106 0.69107 0.32148 1.12 914
0.04 22 4.12 2.6836 0.674 0.34652 1.65 720
0.04 24 9.44 1.23844 0.31118 0.40268 1.83 532
0.04 25 16.80 0.57174 0.1919 0.46849 2.49 331
0.04 26 39.50 0.23093 0.0332 0.59046 1.70 295
0.04 27 114.15 0.0014 5.1E-6 1.60671 1.12 50
wGelatine = Massenbruch des Polymeren im System Gelatine / Wasser
ϑ = Geliertemperatur in °C
tgel = Gelierzeit in Minuten
K1, K2 und α = Konstanten, ermittelt aus den Berechnungen des
Aggregationsmodells (Gl. 9.13)
µ = kritischer Exponenten
∞'G = Endwert für den Speichermodul

ANHANG 94
Anhang A-2 Ableitung weiterer CF.
Ausgehend von den Gln. (8.11) und (8.12) lässt sich eine Proportionalität zwi-
schen den Modulen G' und G'' und dem Betrag der Zeitdifferenz It - tgelI her-
stellen.
( )geltt
1~''G
1
−ν (A-2.1)
( ) geltt~'G1
−µ (A-2.2)
Die Kombination dieser beiden Ausdrücke liefert eine Funktion F1,222
( ) ( ) .const''G'G1F11
=≡ νµ (A-2.3)
die, hat man tgel, µ und ν richtig bestimmt, eine konstanten Wert am Gelpunkt
liefen soll.
Abb. A-2.1. Funktion F1 gegen die Zeitdifferenz It – tgelI für eine wässerige DGFSTOESS Gelatine mit einer Polymerkonzentration von 4 Gew.-% während der isother-men Gelierung bei 16°C
222 Borchard W persönliche Mitteilung
0,0 0,2 0,4 0,6 0,8 1,00,000
0,005
0,010
0,015
0,020
0,025
0,030
F1
It - tgel
I / min

ANHANG 95
Stellt man die Gln. (A-3.1) und (A-3.2) zu:
µ− geltt~'G (A-2.4)
ν− geltt
1~''G (A-2.5)
um, dann ergibt sich nach Multiplikation der beiden Relationen eine weitere
Funktion F2,223
( ) geltt~''G'G2F1
−⋅≡ ν−µ (A-2.6)
die am Gelpunkt It – tgelI → 0 verschwinden sollte.
Abb. A-2.2. Funktion F2 gegen die Zeitdifferenz It – tgelI für eine wässerige DGFStoess Gelatine mit einer Polymerkonzentration von 4 Gew.-% während der isother-men Gelierung bei 16°C
223 Borchard W persönliche Mitteilung
0,0 0,1 0,2 0,3 0,4 0,5 0,6 0,7 0,8 0,9 1,00,00
0,02
0,04
0,06
0,08
0,10
0,12
0,14
F2
It - tgel
I / min

ANHANG 96
Eine weitere Möglichkeit besteht in der Kombination der Zeitableitungen der
Gln. (8.11) und (8.12),
geltt~
dt
''dG 11
−ν−
ν+(A-2.7)
geltt~dt
'dG 11
−⋅µ
−µ(A-2.8)
zu einer Funktion F3,224
.const~dt
''dG
dt
'dG3F
11
11
ν+−µ
⋅
≡ (A-2.9)
für die ein konstanter Wert am Gelpunkt erwartet wird.
Abb. A-2.3. Funktion F3 gegen die Zeitdifferenz It – tgelI für eine wässerige DGFSTOESS Gelatine mit einer Polymerkonzentration von 4 Gew.-% während der isother-men Gelierung bei 16°C
224 Borchard W persönliche Mitteilung
0,0 0,2 0,4 0,6 0,8 1,00
4
8
12
16
20
24
28
F3
It - tgel
I / min
ANHANG 97
Anhang A-3 Messkurven der untersuchten Systeme.
Abb. A-3.1.a. Verlauf des Speicher- und Verlustmoduls als Funktion der Zeit wäh-rend der isothermen Gelierung bei 14°C einer wässerigen DGF STOESS Gelatine miteiner Polymerkonzentration von 2 Gew.-%
Abb. A-3.1.b. Verlauf der optischen Drehung als Funktion der Zeit während derisothermen Gelierung bei 14°C einer wässerigen DGF STOESS Gelatine mit einerPolymerkonzentration von 2 Gew.-%
0,1 1 10 100 10000,01
0,1
1
10
100G
' / P
aG
'' / P
a
t / min
-9
-8
-7
-6
-5
-4
0 1000 2000 3000 4000 5000 6000t / min
α/ g
rad

ANHANG 98
Abb. A-3.2.a. Verlauf des Speicher- und Verlustmoduls als Funktion der Zeit wäh-rend der isothermen Gelierung bei 15°C einer wässerigen DGF STOESS Gelatine miteiner Polymerkonzentration von 2 Gew.-%
Abb. A-3.2.b. Verlauf der optischen Drehung als Funktion der Zeit während derisothermen Gelierung bei 15°C einer wässerigen DGF STOESS Gelatine mit einerPolymerkonzentration von 2 Gew.-%
0,1 1 10 1000,01
0,1
1
10
100
G' /
Pa
G''
/ Pa
t / min
-7,5
-7,0
-6,5
-6,0
-5,5
-5,0
-4,5
-4,0
0 20 40 60 80 100 120t / min
α/
gra
d
ANHANG 99
Abb. A-3.3.a. Verlauf des Speicher- und Verlustmoduls als Funktion der Zeit wäh-rend der isothermen Gelierung bei 16°C einer wässerigen DGF STOESS Gelatine miteiner Polymerkonzentration von 2 Gew.-%
Abb. A-3.3.b. Verlauf der optischen Drehung als Funktion der Zeit während derisothermen Gelierung bei 16C einer wässerigen DGF STOESS Gelatine mit einer Poly-merkonzentration von 2 Gew.-%
-7,5
-7,0
-6,5
-6,0
-5,5
-5,0
-4,5
0 20 40 60 80 100 120t / min
α/ g
rad
0,1 1 10 1000,01
0,1
1
10
100
G' /
Pa
G''
/ Pa
t / min
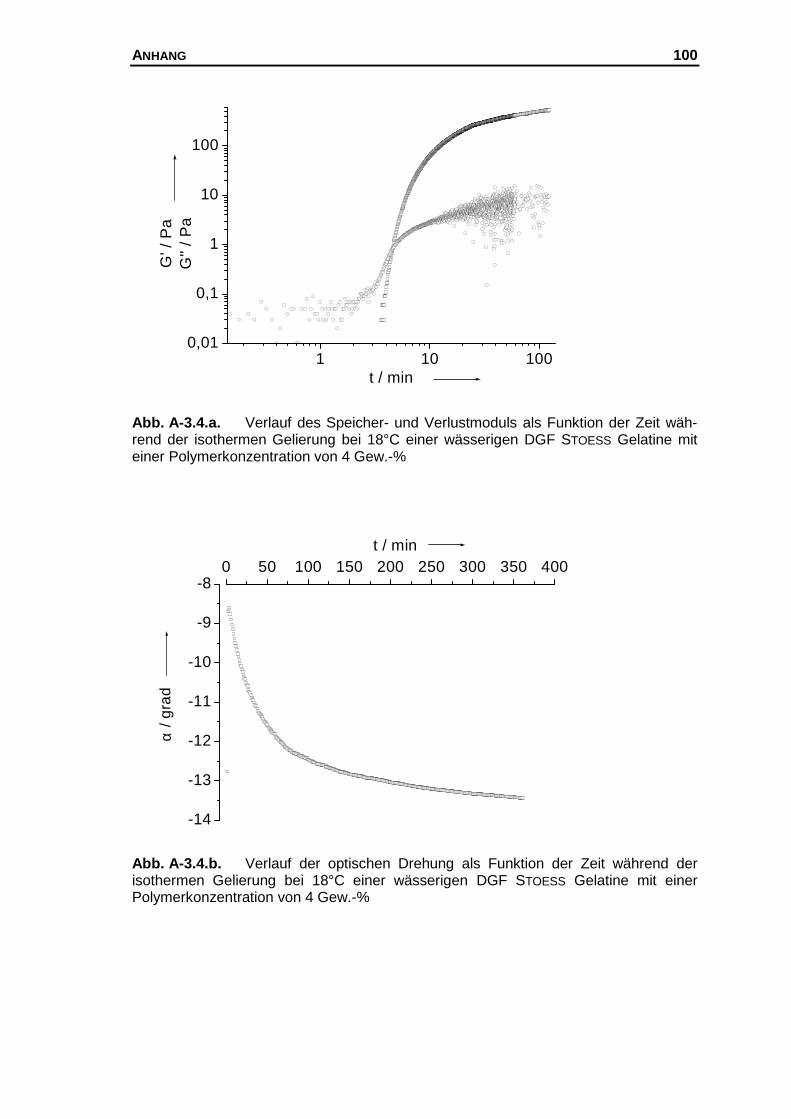
ANHANG 100
Abb. A-3.4.a. Verlauf des Speicher- und Verlustmoduls als Funktion der Zeit wäh-rend der isothermen Gelierung bei 18°C einer wässerigen DGF STOESS Gelatine miteiner Polymerkonzentration von 4 Gew.-%
Abb. A-3.4.b. Verlauf der optischen Drehung als Funktion der Zeit während derisothermen Gelierung bei 18°C einer wässerigen DGF STOESS Gelatine mit einerPolymerkonzentration von 4 Gew.-%
1 10 1000,01
0,1
1
10
100G
' / P
aG
'' / P
a
t / min
-14
-13
-12
-11
-10
-9
-80 50 100 150 200 250 300 350 400
α/ g
rad
t / min

ANHANG 101
Abb. A-3.5.a. Verlauf des Speicher- und Verlustmoduls als Funktion der Zeit wäh-rend der isothermen Gelierung bei 20°C einer wässerigen DGF STOESS Gelatine miteiner Polymerkonzentration von 4 Gew.-%
Abb. A-3.5.b. Verlauf der optischen Drehung als Funktion der Zeit während derisothermen Gelierung bei 20°C einer wässerigen DGF STOESS Gelatine mit einerPolymerkonzentration von 4 Gew.-%
1 10 1000,01
0,1
1
10
100
G' /
Pa
G''
/ Pa
t / min
-13
-12
-11
-10
-9
-80 20 40 60 80 100 120
t / min
α/
gra
d
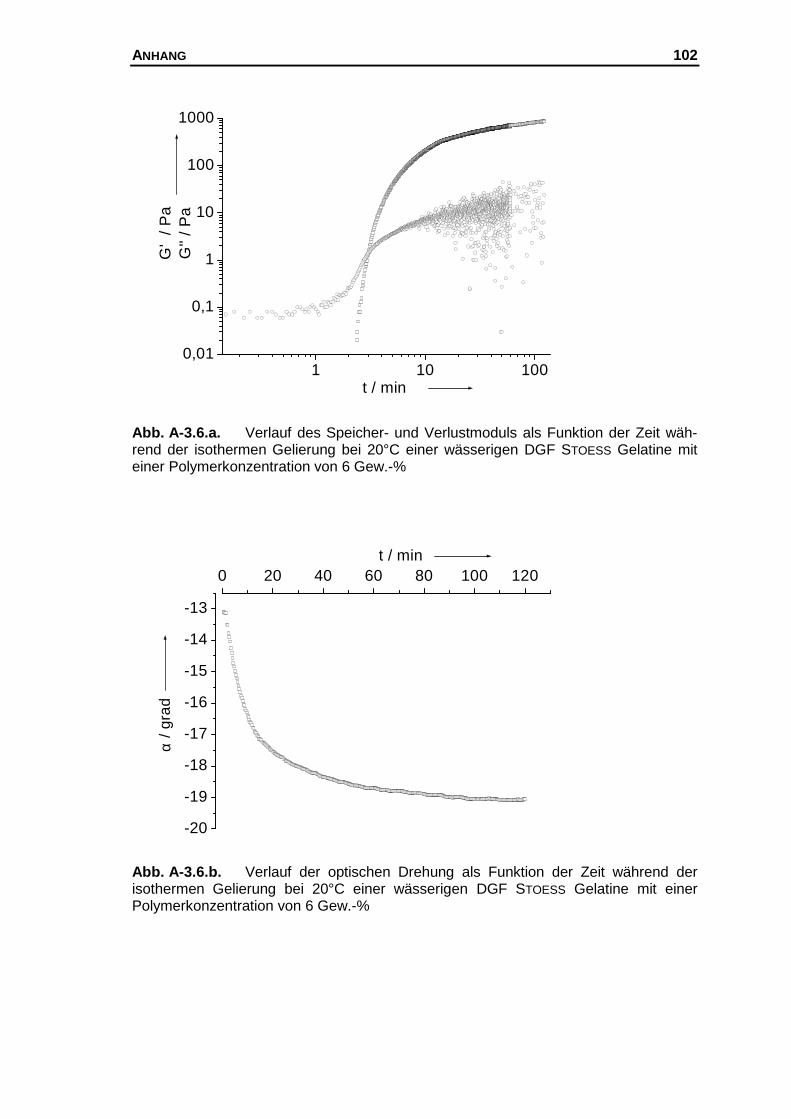
ANHANG 102
Abb. A-3.6.a. Verlauf des Speicher- und Verlustmoduls als Funktion der Zeit wäh-rend der isothermen Gelierung bei 20°C einer wässerigen DGF STOESS Gelatine miteiner Polymerkonzentration von 6 Gew.-%
Abb. A-3.6.b. Verlauf der optischen Drehung als Funktion der Zeit während derisothermen Gelierung bei 20°C einer wässerigen DGF STOESS Gelatine mit einerPolymerkonzentration von 6 Gew.-%
1 10 1000,01
0,1
1
10
100
1000
G'
/ P
aG
'' / P
a
t / min
-20
-19
-18
-17
-16
-15
-14
-13
0 20 40 60 80 100 120t / min
α/
gra
d
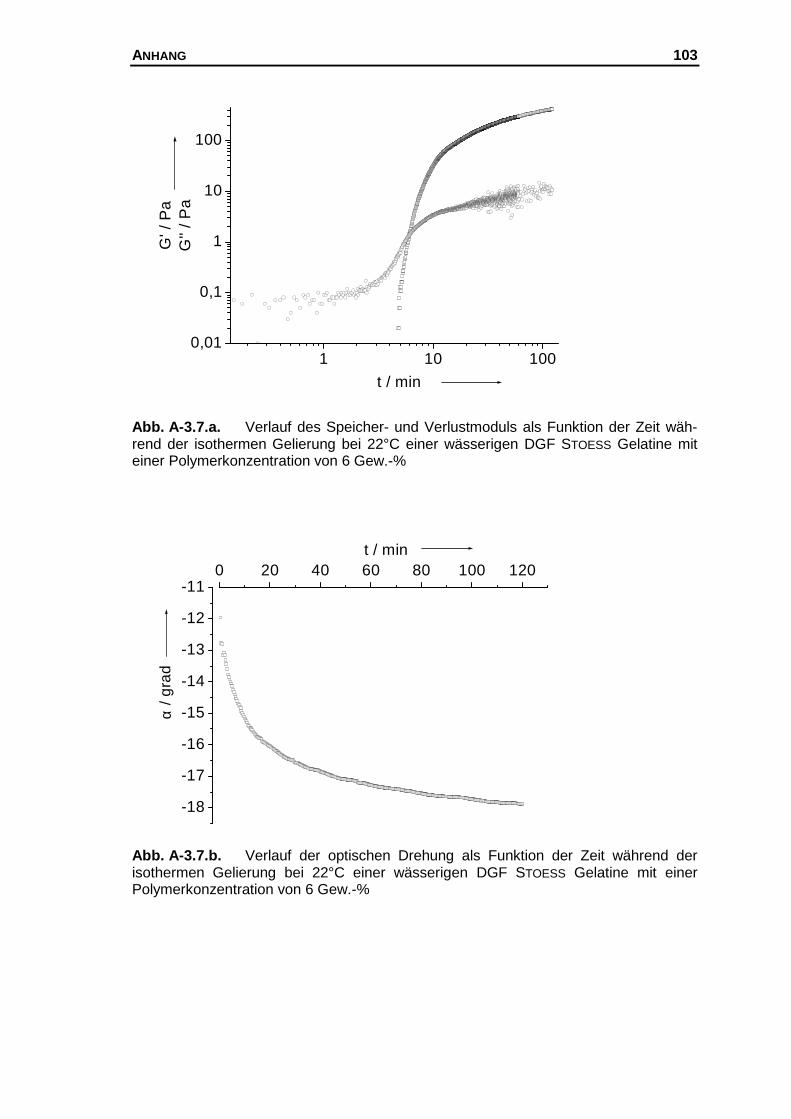
ANHANG 103
Abb. A-3.7.a. Verlauf des Speicher- und Verlustmoduls als Funktion der Zeit wäh-rend der isothermen Gelierung bei 22°C einer wässerigen DGF STOESS Gelatine miteiner Polymerkonzentration von 6 Gew.-%
Abb. A-3.7.b. Verlauf der optischen Drehung als Funktion der Zeit während derisothermen Gelierung bei 22°C einer wässerigen DGF STOESS Gelatine mit einerPolymerkonzentration von 6 Gew.-%
1 10 1000,01
0,1
1
10
100
G' /
Pa
G''
/ Pa
t / min
-18
-17
-16
-15
-14
-13
-12
-110 20 40 60 80 100 120
t / min
α/ g
rad

ANHANG 104
Abb. A-3.8.a. Verlauf des Speicher- und Verlustmoduls als Funktion der Zeit wäh-rend der isothermen Gelierung bei 24°C einer wässerigen DGF STOESS Gelatine miteiner Polymerkonzentration von 6 Gew.-%
Abb. A-3.8.b. Verlauf der optischen Drehung als Funktion der Zeit während derisothermen Gelierung bei 24°C einer wässerigen DGF STOESS Gelatine mit einerPolymerkonzentration von 6 Gew.-%
1 10 1000,01
0,1
1
10G
' / P
aG
'' /
Pa
t / min
-16,0
-15,5
-15,0
-14,5
-14,0
-13,5
-13,0
-12,5
-12,00 20 40 60 80 100 120
t / min
α/
gra
d

ANHANG 105
Abb. A-3.9.a. Verlauf des Speicher- und Verlustmoduls als Funktion der Zeit wäh-rend der isothermen Gelierung bei 18°C einer wässerigen DGF STOESS Gelatine miteiner Polymerkonzentration von 8 Gew.-%
Abb. A-3.9.b. Verlauf der optischen Drehung als Funktion der Zeit während derisothermen Gelierung bei 18°C einer wässerigen DGF STOESS Gelatine mit einerPolymerkonzentration von 8 Gew.-%
1 10 1000,01
0,1
1
10
100
1000
G' /
Pa
G''
/ Pa
t / min
-28
-26
-24
-22
-20
-18
-16
0 20 40 60 80 100 120t / min
α/
gra
d
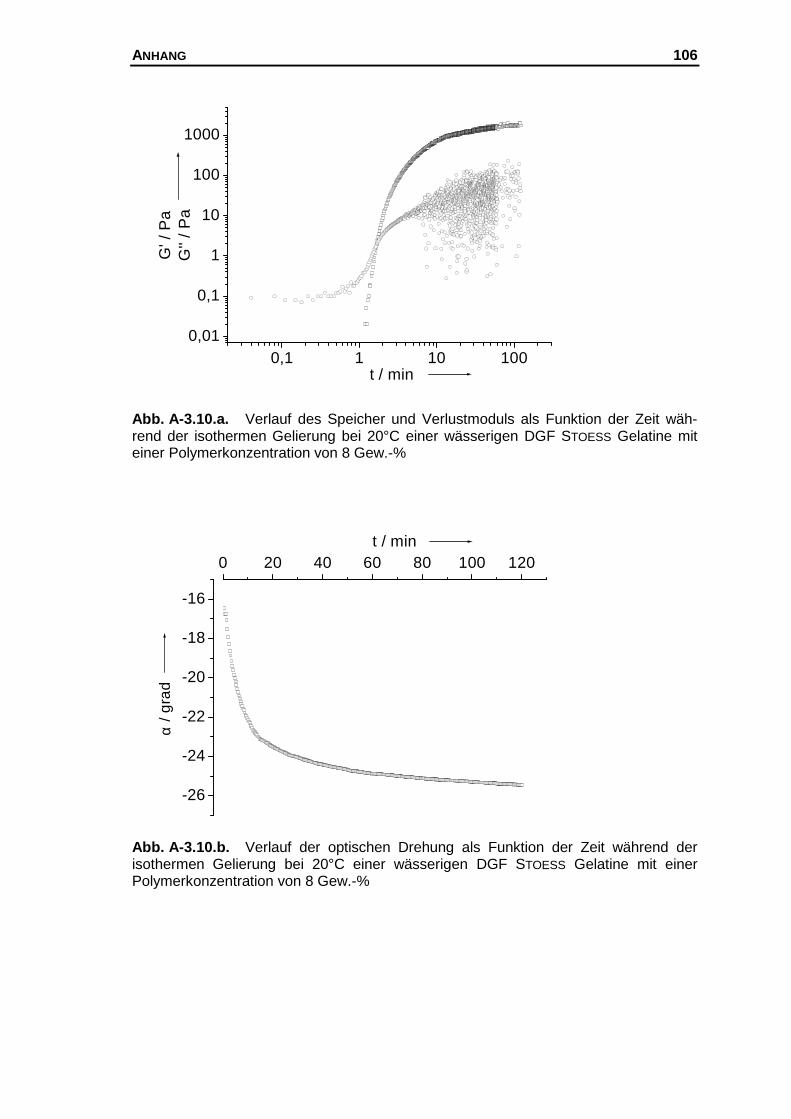
ANHANG 106
Abb. A-3.10.a. Verlauf des Speicher und Verlustmoduls als Funktion der Zeit wäh-rend der isothermen Gelierung bei 20°C einer wässerigen DGF STOESS Gelatine miteiner Polymerkonzentration von 8 Gew.-%
Abb. A-3.10.b. Verlauf der optischen Drehung als Funktion der Zeit während derisothermen Gelierung bei 20°C einer wässerigen DGF STOESS Gelatine mit einerPolymerkonzentration von 8 Gew.-%
0,1 1 10 1000,01
0,1
1
10
100
1000
G' /
Pa
G''
/ Pa
t / min
-26
-24
-22
-20
-18
-16
0 20 40 60 80 100 120t / min
α/ g
rad

ANHANG 107
Abb. A-3.11.a. Verlauf des Speicher- und Verlustmoduls als Funktion der Zeit wäh-rend der isothermen Gelierung bei 22°C einer wässerigen DGF STOESS Gelatine miteiner Polymerkonzentration von 8 Gew.-%
Abb. A-3.11.b. Verlauf der optischen Drehung als Funktion der Zeit während derisothermen Gelierung bei 22°C einer wässerigen DGF STOESS Gelatine mit einerPolymerkonzentration von 8 Gew.-%
0,01 0,1 1 10 1000,01
0,1
1
10
100
1000
G' /
Pa
G''
/ Pa
t / min
-24
-23
-22
-21
-20
-19
-18
-17
-16
0 20 40 60 80 100 120
t / min
α/
gra
d

LITERATURVERZEICHNIS 108
LITERATURVERZEICHNIS
Adam M, Delsanti M, Okasha R, Hild G (1979) J Phys Lett (Paris) 40:L 539
Almdal K, Dyre J, Hvidt S, Kramer O (1993) Polymer Gels Networks 1:5
Arbabi S, Sahimi M (1990) Phys Review Lett 65:725
Atkins PW (1990) Physikalische Chemie, VCH, Weinheim
Babel W (1996) Chemie in unserer Zeit 30:86
Benguigui L, Busnel JP, Durand D (1991) Polymer 32(14):2680
Bohidar HB, Jena SS (1993) J Chem Phys 8(11):8970
Boltzmann L (1874) Sitzber KGl Akad Wiss Wien 2. Abt. 70:225Borchard W (1983) In: Finch CA (ed) Thermoreversible Gelation, Cambridge
Residental School of Chemistry, Plenum PressBorchard W (1994) In: Water Based Polymers, Gels and Mesophases, The
Centre of Professional Advancement 5, ChicagoBorchard W (1994) In: Water Based Polymers, Rheology I: Dilute Polymer-
Water Systems, The Centre of Professional Advancement 5, ChicagoBorchard W (1998) Ber Bunsenges Phys Chem 102:1580
Borchard W, Burg B (1990) Progr Colloid Polym Sci 83:200Borchard W, Bergmann K, Emberger A, Rehage G (1976) Progr. Colloid &Polymer Sci 60:120Borchard W, Cölfen H, Kisters D, Straatmann A (2001) In: Borchard W (ed)Sonderband, Progress in Ultracentrifugation, in preparationBorchard W, Lechtenfeld M (1999) Colloid Polym Sci eingereicht
Borchard W, Lechtenfeld M (1999) Macromolecules eingereichtBorchard W, Lechtenfeld M (2001) Mat Res Innovat II Vol 4 No.5-6 p. 381Broadbent SR, Hammersley JM (1954) Proc Camp Phil Soc 53:629
Bunde A, Roman HE (1996) Physik in unserer Zeit 27(6):246
Burg B (1988) Dissertation DuisburgBurg B, Borchard W (1989) In: Integration of Fundamental Polymer Science and
Technology, Rolduc Meeting III, Elsevier, London p.323
Chambon F, Winter HH (1985) Polym Bull 13:499
Chambon F, Winter HH (1987) J Rheol 31(8):683
Clerc JP, Girand G, Laugier JM, Luck M (1985) J. Phys. A 18:2565
Cuvelier G, Launay B (1990) Makromol Chem, Macromol Symp 40:23Cuvelier G, Peigney-Nourry C, Launay B (1990) In: Philips GO et. al. (eds)
Gums and Stabilisers for the Food Industry 5th ed, IRL Press, Oxfordp.549
de Gennes PG (1976) J Phys (Paris) 37:L1
de Gennes PG (1979) J Physique (Paris) Lett 40:197

LITERATURVERZEICHNIS 109
de Gennes PG (1979) Scalling Concepts in Polymer Physics, Cornell UniversityPress, Ithaca New York
de Gennes PG (1980) Comp Rendus Acad Sci (Paris) 286B:131
Djabourov M (1988) Contemp Phys 29(3):273
Djabourov M, Leblond J, Papon P (1988) J Phys (France) 49:319
Djabourov M, Leblond J, Papon P (1988) J Phys (France) 49:333
Djabourov M, Lechaire JP, Gaill F (1993) Biorheology 30:191Djabourov M; Maquet J, Theveneau H, Leblond J, Papon P (1985) British
Polymer J Vol 17, 2:169Dobson GR, Gordon M (1965) J Chem Phys 43:705
Durand D, Delsanti M, Adam M, Luck M (1987) Europhys Lett 3:297
Engel J (1962) Arch Biochem Biophysics 97:150
Fa. LOT Oriel Instruments (2000) The book of photon toolsFerry JD (1970) Viscoelastic Properties of Polymers, John Wiley & Sons Inc.,
New YorkFlory PJ (1941) J Amer Chem Soc 63:3083,3091,3096
Flory PJ (1974) Disc Faraday Soc 57:1
Gauthier-Manuel B, Guyon E (1980) J Phys Lett (Paris) 41:L503Goodwin JW, Hughes RW (2000) Rheology for chemists, Royal Society of
Chemistry
Gordon M, Ross-Murphy SB (1979) J. Phys A 12:L155
Harrington WF, v. Hippel PH (1962) Advan Protein Chem 16:1Hermans PH (1949) In: Kruyt HR (ed) Colloid Science II, Elsevier, Amsterdam,
p.483
Hinsken H, Borchard W (1995) Colloid Polym Sci 273:913
Huismann R, Heuvel HM (1989) J Appl Polym Sci 37:595
Kantor Y, Webman I (1984) Phys Rev Lett 52:1891
Kästner S (1979) Polymer 20:1329
Kumagai H, Fujii T, Inukai, T, Yano T (1993) Biosci Biotech Biochem 57(4):532
Lechtenfeld M, Borchard W (1999) Phys Chem Chem Phys 1:3129
Lechtenfeld M, Michalczyk A, Borchard W (2001) Rheol Acta angenommen
Lin YG, Mallin DT, Chien JCW, Winter HH (1991) Macromolecules 24:850

LITERATURVERZEICHNIS 110
Lloyd JD (1926) In: Alexander J (ed) Colloid chemistry I. Chemical CatalogCompany, New York, p.767
Maibaum R (1999) Dissertation Duisburg
Martin JE, Adolf D, Wilcoxon JP (1989) Phys Rev A 39:1325
Michalczyk A (1993) Dissertation Duisburg
Michon C, Cuvelier G, Launay B (1993) Rheol Acta 32:94
Pahl MH (1983) Praktische Rheologie der Kunststoffschmelzen und Lösungen,VDI-Verlag
Parker TG, Dalgleish DG (1977) J Dairy Res 44:85Penich-Covas C, Dev SB, Gordon M, Judd M, Kajiwara K (1974) Discussion of
the Faraday Division on Gels and Gelling Processes 57:165Pezron I, Djabourov M (1990) J Polm Sci B, Polym Phys 28:1823Piez KA (1985) In: Encyclopedia of Polymer Science and Engeneering,
Collagen, 2nd Ed. Vol. 3, Wiley and Sons, London, p.699
Rehage G (1977) Berichte der Bunsengesellschaft Bd. 81, Nr.10
Ross-Murphy SB (1992) Polymer 33(12):2622Roux S, Guyon E (1986) In: Stanley HE, Ostrowski N (eds) On Growth and
Form, Martinus Nijhoff Boston
Sahimi M (1992) Mod Phys Lett B 6:507
Sahimi M (1994) Applications of Percolation Theory, Taylor & FrancisSchwarzl F, Staverman AJ (1956) In: Stuart HA (ed) Physik der Hochpolymere
IV, Springer Verlag, Berlin Göttingen HeidelbergSchwarzl FR (1990) Polymermechanik, Springer, Berlin Heidelberg New York
Stauffer D (1979) Physics Reports 54:1
Stauffer D, Aharony A (1995) Perkolationstheorie, VCH WeinheimStauffer D, Coniglio A, Adam M (1982) Advances in Polymer Science 44:103,
Springer Verlag, BerlinStockmayer WH (1944) J Chem Phys 11:45 ibid 12:125
te Nijenhuis K (1981) Colloid Polym Sci 259(5):522te Nijenhuis K (1997) Thermoreversible Networks, Springer, Berlin Heidelberg
New York, p.3te Nijenhuis K, Winter HH (1989) Macromolecules, 22:411
Todd A (1961) Nature 191:567Tschoegel NW (1989) The phenomenological theory of linear viscoelastic
behaviour, Springer, Berlin Heidelberg New York
v. Hippel PH, Harrington WF (1959) Biochem Biophys Acta 37:427v. Hippel PH, Harrington WF (1960) In: Protein Structure and Function,
Brookhaven Symposia in Biol 13:213

LITERATURVERZEICHNIS 111
v. Smoluchowski M (1917) Z phys Chem 92:129
Venohr H (1999) Dissertation Duisburg
Vollhardt KPC (1990) Organische Chemie, VCH Weinheim
Winter HH, Chambon F (1986) J Rheol 30:67

LEBENSLAUF
Name: Markus LechtenfeldGeboren am: 08.07.1969 in DuisburgFamilienstand: verheiratet, 2 Kinder
Schulbildung
1975 – 1979 Grundschule in Duisburg
1979 – 1985 Wilhelm-Lehmbruck-Realschule in DuisburgAbschluss: Fachoberschulreife
1987 – 1989 Städt. Kollegschule und Fachschule für Technik inDuisburgAbschluss: Fachhochschulreife (in Teilzeitform)
Berufliche Ausbildung
1985 – 1988 ChemielaborantThyssen Stahl AG, Duisburg
1989 Zivildienst in Duisburg
Wissenschaftliche Ausbildung
1989 – 1992 Fachhochschule NiederrheinStudienschwerpunkt: ChemieingenieurwesenDiplom: 4.12.1992
1993 – 1997 Gerhard-Mercator-Universität DuisburgStudienschwerpunkt: Chemie (DII) – AngewandtePhysikalische ChemieVordiplom: 4.3.1994Diplom: 14.3.1997
seit 1997 Gerhard-Mercator-Universität DuisburgWissenschaftliche Hilfskraft im Fachgebiet AngewandtePhysikalische Chemie unter Leitung von Prof. W.Borchard, Anfertigung der Dissertation (Beginn: 4/97)
Beruflicher Hintergrund
1988 – 1989 Thyssen Stahl AG, DuisburgChemielaborant
ab 4/2001 Anstellung als Chemiker in der F+E bei derDGF Stoess AG